

Abstract
In Warburg metabolism, suppression of mitochondrial metabolism contributes to a low cytosolic ATP/ADP ratio favoring enhanced aerobic glycolysis. Flux of metabolites across the mitochondrial outer membrane occurs through voltage-dependent anion channels (VDAC). In cancer cells, free dimeric tubulin induces VDAC closure and dynamically regulates mitochondrial membrane potential (ΔΨ). Erastin, a small molecule that binds to VDAC, antagonizes the inhibitory effect of tubulin on VDAC and hyperpolarizes mitochondria in intact cells. Here, our aim was to identify novel compounds from the ChemBridge DIVERSet library that block the inhibitory effect of tubulin on ΔΨ using cell-based screening. HCC4006 cells were treated with nocodazole (NCZ) to increase free tubulin and decrease ΔΨ in the presence or absence of library compounds. Tetramethylrhodamine methylester (TMRM) fluorescence was assessed by high-content imaging to determine changes in ΔΨ. Compounds were considered positive if ΔΨ increased in the presence of NCZ. Using confocal microscopy, we identified and validated six lead molecules that antagonized the depolarizing effect of NCZ. Lead compounds and erastin did not promote microtubule stabilization, so changes in ΔΨ were independent of tubulin dynamics. The most potent lead compound also decreased lactate formation. These novel small molecules represent a potential new class of anti-Warburg drugs.
Keywords: erastin, mitochondrial membrane potential, tubulin, voltage-dependent anion channel, Warburg metabolism
Introduction
In the 1920s, Otto Warburg described a unique metabolic phenotype of tumors characterized by enhanced aerobic glycolysis and apparently defective respiration.1,2 However, mitochondria isolated from tumors and mitochondria in intact cancer cells maintain ΔΨ, ATP, and NADH generation, among other functions.3–6 Aerobic glycolysis yields up to 90% of cellular ATP in cancer cells, with the remaining ATP production occurring in mitochondria.5,7 By contrast, in aerobic nonproliferative cells, glycolytic ATP generation contributes only ~5% of the total ATP produced. The predominance of a glycolytic phenotype is not a permanent feature. Environmental stimuli, including limitation of glucose availability and treatment with the pyruvate analog dichloroacetate to increase mitochondrial metabolism, promote a switch from glycolytic to predominantly oxidative metabolism.8–10 The metabolic flexibility of tumors provides a therapeutic opportunity to develop novel anti-Warburg drugs that increase oxidative mitochondrial metabolism and decrease pro-proliferative glycolysis. Pharmacological interventions to inhibit glycolysis or promote mitochondrial metabolism cause tumor cell death both in vitro and in vivo.11–16
Mitochondrial metabolism depends on the flux of metabolites across mitochondrial membranes. Respiratory substrates, ADP, and Pi cross the mitochondrial outer membrane through voltage-dependent anion channels (VDAC). VDAC, the most abundant protein in the mitochondrial outer membrane, has been proposed as a master regulator of mitochondrial metabolism. VDAC closure contributes to suppression of mitochondrial function in the Warburg phenotype.4,13,17,18 VDAC is a ~30 kDa protein comprising three isoforms (VDAC1, VDAC2, and VDAC3) with a high degree of homology.19–21 VDAC closure blocks the movement of most organic anions into mitochondria and prevents the exchange of ADP and Pi for ATP between the cytosol and mitochondria. Free dimeric α,β-tubulin closes VDAC inserted in lipid bilayers and suppresses respiration in isolated mitochondria and permeabilized cells.22,23
Proton pumping coupled to electron transfer through Complexes I, III, and IV of the respiratory chain in the mitochondrial inner membrane generates a protonmotive force (Δp) comprised chiefly of a mitochondrial membrane potential (ΔΨ). Δp in turn drives ATP synthesis through the F1-FO ATP synthase (Complex V), coupled to the movement of protons back into the mitochondrial matrix. Previously, we showed that free tubulin dynamically regulates ΔΨ in cancer cells.24 The relative closure of VDAC by elevated cytosolic free tubulin limits the availability of respiratory substrates to mitochondria, contributing to the suppression of mitochondrial metabolism in proliferating cells.4,24 We further showed that erastin, a VDAC-binding molecule,25 blocks the inhibitory effect of free tubulin on VDAC inserted into lipid bilayers and reverses the mitochondrial depolarization induced by elevated free dimeric tubulin in intact cancer cells.18 Thus, VDAC– tubulin interaction is a novel pharmacological target to regulate mitochondrial metabolism in cancer cells.
Here, we used high-content imaging to screen 50,080 compounds from the ChemBridge DIVERSet library to identify anti-Warburg small molecules that block tubulin-induced mitochondrial depolarization. We identified six lead compounds that increased ΔΨ in the presence of elevated free tubulin in cancer cells. We further determined dose–response for these compounds and validated the hyperpolarizing effect by confocal microscopy. We also showed that increased ΔΨ by erastin and lead compounds was not due to microtubule stabilization or depleted cytosolic free tubulin. In addition, the most potent lead compound decreased lactate formation, indicating an anti-Warburg, antiaerobic glycolysis effect.
Materials and Methods
Materials
RPMI 1640, F-12K, and Eagle’s minimum essential medium were purchased from American Tissue Culture Collection (Manassas, VA); ChemBridge DIVERSet screening library from ChemBridge Corporation (San Diego, CA); tetramethylrhodamine methylester (TMRM), CellTracker Green, and Hoechst 33342 from Invitrogen (Carlsbad, CA); erastin, nocodazole (NCZ), and paclitaxel (PTX) from Sigma-Aldrich (St. Louis, MO); Microtubules/Tubulin In Vivo Assay Kit from Cytoskeleton (Denver, CO); and L-Lactate Assay Kit I from Eton Bioscience (San Diego, CA). All other reagents were analytical grade.
Cell Culture
HepG2 human hepatoma cells (American Type Culture Collection) and Huh7 human hepatocarcinoma cells (courtesy of Jack R. Wands, Brown University) were grown in Eagle’s minimum essential medium. A549 human lung cancer (American Type Culture Collection) and HCC4006 lung adenocarcinoma (courtesy of Robert M. Gemmill, Medical University of South Carolina) cells were grown in F-12K and RPMI 1640 medium, respectively. All growth medium was supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. Cell lines were cultured in 5% CO2/air at 37 °C. For all experiments, cells were plated 48 h before experimentation. All experiments involving live cells were performed in 5% CO2/air at 37 °C in modified Hank’s balanced salt solution (HBSS) containing (in mM) NaCl 137, Na2HPO4 0.35, KCl 5.4, KH2PO4 1, MgSO4 0.81, CaCl2 0.95, glucose 5.5, NaHCO3 25, and HEPES 20, pH 7.4, as described.24
High-Content Imaging Small-Molecule Screen
Cells were loaded for 30 min at 37 °C with 200 nM TMRM, 5 μM CellTracker Green, and 3 μM Hoechst 33342. After loading and washing, subsequent incubations were performed with 50 nM TMRM to maintain equilibrium distribution of the fluorophore.26 High-content live cell imaging was performed in the GE IN Cell Analyzer 2000 wide-field cell imaging system (Pittsburgh, PA) using a Nikon 40× 0.6 N.A. air objective. TMRM, CellTracker Green, and Hoechst 33342 were excited at 555, 490, and 350 nm, respectively, and fluorescence was imaged using 605, 490, and 455 nm barrier filters from four different fields (65–125 cells per field) of one well for each compound group or individual compound screened. Images were analyzed using GE IN Cell 1000 Analyzer software. Hoechst-stained nuclei were used to identify and count cells. Individual cells were segmented by assigning a CellTracker Green border in association with each individual Hoechst-stained nucleus. After background subtraction, the mean TMRM fluorescence intensity was determined for all red (TMRM) pixels within the CellTracker Green border of each cell. The mean cellular TMRM fluorescence for all cells in a field was then calculated for comparison after various treatments. The Z factor (maximal value of 1), a measure of the likelihood that a high-throughput screening assay will be able to discriminate positive “hits,” was calculated to be 0.982 using TMRM fluorescence means and standard deviations of positive (erastin + NCZ) and negative (NCZ alone) experimental controls.27 This Z score indicates that the screening assay was very robust for lead identification.
Laser Scanning Confocal Microscopy
Cells were loaded with TMRM as described above. Red TMRM fluorescence was imaged with a Zeiss LSM 880 NLO inverted laser scanning confocal microscope (Thornwood, NY) with a 63× 1.4 N.A. planapochromat oil immersion lens. TMRM was excited at 543 nm, and fluorescence was detected with a Quasar multichannel spectral detector at 590–610 nm and a 1 Airy unit diameter pinhole. TMRM fluorescence after subtraction of background was quantified using Zeiss Zen software and Photoshop CS4 (Adobe Systems, San Jose, CA). Representative confocal and IN Cell 2000 images of the red fluorescence of TMRM were pseudocolored using a rainbow intensity scale. In individual experiments, images were collected before and after treatment from the same groups of cells.
Determination of Free and Polymerized Tubulin by Western Blotting
Free and polymerized tubulin were determined using a Microtubules/Tubulin In Vivo Assay Kit according to the manufacturer’s instructions. Briefly, cells were scraped into microtubule stabilization buffer containing (in mmol/L) MgCl2 5, EGTA 1, GTP 0.1, ATP 1, and PIPES buffer 100, and (in %) glycerol 30, Nonidet-P40 0.1, Triton X-100 0.1, Tween-20 0.1, beta-mercaptoethanol 0.1, antifoam 0.001, and BME 0.2, pH 7.4, with a protease inhibitor cocktail, and homogenized by aspirating five times through a 25 G needle. Homogenates were centrifuged at 2000g for 5 min at 37 °C to eliminate cell debris. Supernatants were then centrifuged at 100,000g for 30 min at 37 °C. Supernatants after the second centrifugation step contained free tubulin. The pellets contained polymerized tubulin and were resuspended in ice-cold 2 mM CaCl2.
Free and polymerized tubulin were loaded on 4%–12% Bis-Tris gels. Proteins were transferred using an iBlot Dry Blotting System (Invitrogen). Blots were blocked in 5% nonfat milk and probed with 1:500 anti-β-tubulin monoclonal antibody (Cytoskeleton) overnight at 4 °C. Immunoblots were detected by 1:2000 secondary antibodies conjugated to peroxidase (goat anti-mouse IgG-HRP: Sc-2005, Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature. Detection was conducted using a chemiluminescence reagent (Supersignal West Dura Extended Duration Substrate). Protein was quantified by the Lowry method (Bio-Rad DC Protein Assay, Hercules, CA). In these experiments, fractions were prepared from virtually identical numbers of cells and loaded onto gels in equal volume. Because the results were expressed as ratios of free to polymerized tubulin, any small variation of the number of cells extracted was offset.
Lactate Assay
Cells in HBSS were treated with vehicle or X1, and extracellular HBSS aliquots were collected at different time points. Lactate was measured with an L-Lactate Assay Kit I that yields a tetrazolium reaction product measured by absorbance at 490 nm following the manufacturer’s instructions using a BioTek ELX808IU absorbance plate reader (Winooski, Vermont).
Statistics
Differences between groups were analyzed by Student’s t-test using p < 0.05 as the criterion of significance. Data points are means ± standard error (SE) of at least three independent experiments with at least four fields surveyed per experiment. Images are representative of three or more independent experiments.
Results
High-Content Cell-Based Screening Identifies Small Molecules That Prevent Mitochondrial Depolarization by Elevated Cytosolic Free Tubulin
We used an IN Cell Analyzer 2000 wide-field cell imaging system to develop a high-content cell-based screen (Fig. 1). Previously, we characterized the effects of free tubulin and erastin on mitochondrial membrane potential in HepG2 human hepatoma cells and found that erastin is a VDAC– tubulin antagonist.18,24 In this study, one of our goals was to show that this effect occurred in other cancer cell lines. Accordingly, we began by using another cell line, HCC4006 lung cancer cells, to identify erastin-like small molecules by high-content screening. HCC4006 cells cultured for 48 h in 96-well plates were coloaded with Hoechst 33342, CellTracker Green, and TMRM to label nuclei, cell area, and mitochondria, respectively (Fig. 1A). Using IN Cell software, we identified individual cells by nuclear labeling, segmented cytoplasmic areas from CellTracker Green fluorescence, and then quantified TMRM fluorescence to determine the relative magnitude of ΔΨ within each cell (Fig. 1B). The mean cellular TMRM fluorescence (average pixel intensity per segmented cell) was determined in each field to assess changes in ΔΨ in response to treatments. In the initial screening, baseline images were collected before treatment for 1 h with the microtubule destabilizer NCZ (10 μM) to maximize cytosolic free tubulin, or NCZ plus mixtures of 10 small molecules (10 μM each) from the 50,080 DIVERSet ChemBridge compound library. NCZ alone decreased TMRM fluorescence by about 40%. Thus, mixtures of compounds that increased mean cellular TMRM fluorescence in the presence of NCZ by at least 45% relative to cells treated with NCZ alone were considered initial hits. The 10 compounds of each hit mixture were subsequently tested individually using the same methodology in a second screen (Fig. 1A). Individual compounds that increased TMRM fluorescence in the presence of NCZ by at least 45% relative to cells treated with NCZ were considered positive hits. After the second screening, we identified more than 30 positive hits. Dose–response relationships were then determined using the IN Cell Analyzer 2000 to identify the most potent individual compounds. These compounds were further validated by confocal microscopy.
Figure 1.
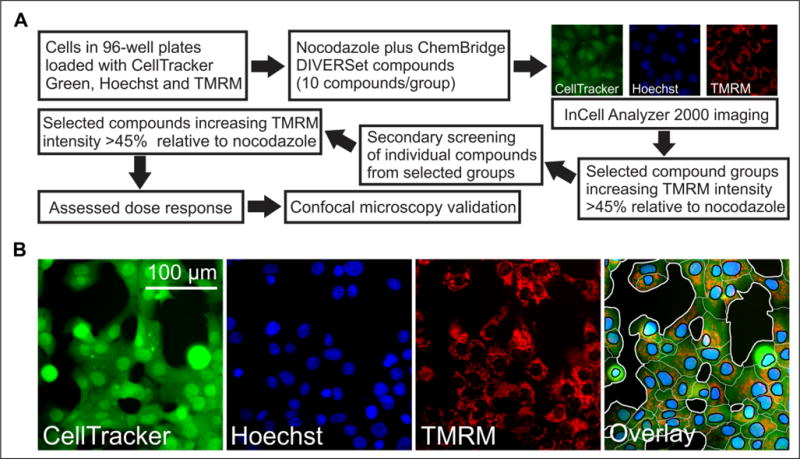
Strategy to identify small molecules that prevent mitochondrial depolarization after NCZ using high-content imaging. An IN Cell Analyzer 2000 was used to identify lead compounds that revert NCZ-induced mitochondrial depolarization. (A) The first screen was performed using mixtures of 10 library compounds. Mixtures increasing TMRM fluorescence in the presence of NCZ by ≥45% relative to cells treated with NCZ alone were considered positive. Compounds from positive mixtures were then assessed individually in a secondary screening. The top individual compounds were assessed for dose–response, and the most active compounds were selected and validated by confocal microscopy. (B) Representative images for each channel and the overlay are shown. A representative segmentation (far right panel) demonstrates how cells were segmented based on the fluorescent channels. Black areas are extracellular space, and white lines define cellular boundaries. Black lines segment blue nuclei from green cytoplasmic spaces. TMRM fluorescence for each cell was calculated by multiplying the mean cytoplasmic TMRM fluorescence (background subtracted) by the cytoplasmic area.
Erastin and Lead Compounds Block the Mitochondrial Depolarizing Effect of Nocodazole in HCC4006 Human Lung Cancer Cells
In HCC4006 cancer cells, NCZ decreased TMRM fluorescence by ~40% (Fig. 2A, upper middle panel), similar to other cancer cell lines studied previously.18,24 Erastin (10 μM) blocked NCZ-induced mitochondrial depolarization fully (Fig. 2A, upper right panel), as reported previously for other cancer cell lines.18,24 Similar to erastin, the six most active lead compounds (10 μM) identified from the screening increased TMRM fluorescence by 70%–225% compared with NCZ and 35%–90% compared with untreated cells (Fig. 2A). In this way, six erastin-like small molecules were identified that blocked tubulin-dependent mitochondrial depolarization in situ. At 10 μM, the most potent lead compound, X1, increased TMRM fluorescence three times as much as erastin (Fig. 2A,B).
Figure 2.
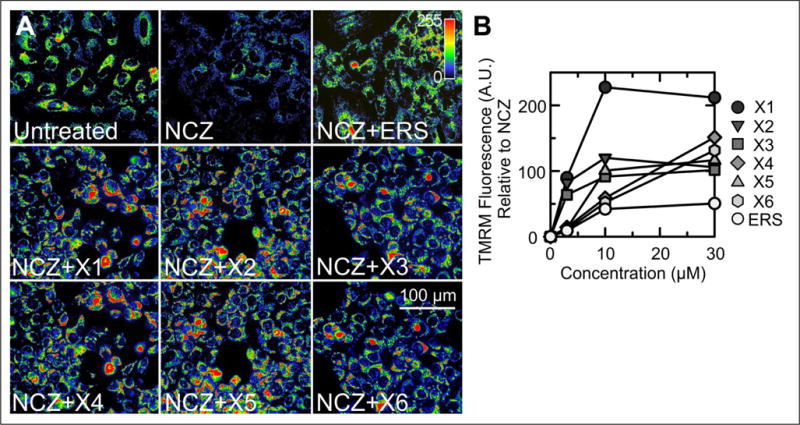
Erastin and ChemBridge small molecules block NCZ-induced mitochondrial depolarization in HCC4006 cells. (A) TMRM fluorescence imaged by an IN Cell Analyzer 2000 was assessed after 1 h of treatment with DMSO (untreated), NCZ (10 μM) alone, and NCZ plus 10 μM of erastin or lead compounds (X1–X6). An indicator bar (upper right) shows the color-coded intensity scale for TMRM fluorescence. Note the decreased TMRM fluorescence after NCZ in comparison with the untreated cells, which was reversed by erastin and X1–X6. (B) The percentage increase of TMRM fluorescence relative to NCZ alone is plotted for different concentrations of lead compounds X1–X6 and erastin. *p < 0.05 compared with NCZ alone (n = 3).
Dose Dependence of Lead Compounds and Erastin
The dose–response of the six most potent lead compounds and erastin was assessed using the IN Cell Analyzer 2000. HCC4006 cells loaded with Hoechst 33342, CellTracker Green, and TMRM were treated with lead compounds and erastin (0, 3, 10, and 30 μM) for 1 h in the presence of NCZ (10 μM). Lead compounds and erastin did not restore mitochondrial polarization in the presence of NCZ at concentrations less than 3 μM (data not shown). At 3 μM and higher, lead compounds and erastin increased mitochondrial polarization in the presence of NCZ dose dependently (Fig. 2B). At 3 μM, X1, X2, and X3 increased TMRM fluorescence by up to ~95% compared with NCZ alone. Mitochondrial hyperpolarization maximized at 10 μM for X1, X2, X3, X5, and erastin. For X4 and X6, the maximal increase of TMRM fluorescence occurred at 30 μM. At 100 μM, the four most potent compounds (X1–X4) caused mitochondrial release of TMRM (data not shown).
Structural Features of Lead Compounds
Compounds X1, X2, and X3 share a common core structure, namely, the 2-alkylsulfonyl-5-chloro-4-phenylcarboxamide scaffold (Fig. 3). Chemical diversity in these three analogs comes from varying aromatic substitutions at the 3- and 4-positions of the phenylcarboxamide moiety. Compounds X2 and X3 possess a two-dimensional Tanimoto similarity coefficient of 0.84, while X1 and X3 are more dissimilar (0.49). When compared with erastin, X1–X3 display Tanimoto coefficients of 0.13, 0.18, and 0.17, respectively. Importantly, X1–X3 all possess a terminal chlorophenyl moiety, as found in erastin. While the X4 chalcone and X5 have structures dissimilar from those of X1–X3, they share a common arylpropanone scaffold where the second ketone substituent contains a six-membered nitrogen-containing ring linked through a 2-carbon bridge. However, there is little similarity between X3 and X5 (Tanimoto = 0.13) and between X4 and X5 (Tanimoto = 0.11). X4 and X5 also have little similarity to erastin (Tanimoto = 0.15 and 0.16). Compound X6 has minimal similarity to X4 and X5 (aryl group and a nitrogen-containing six-membered ring separated by an alkyl spacer) with Tanimoto coefficients of 0.20 and 0.22, respectively. However, X6 does have moderate similarity to erastin (Tanimoto = 0.24). Compound X6 may not be a suitable lead for drug development due to the procarcinogenic pyrene nucleus. Erastin has a more complex structure than X1–X6 but does share a chlorophenyl substituent with X1 and X3 (Fig. 3).
Figure 3.
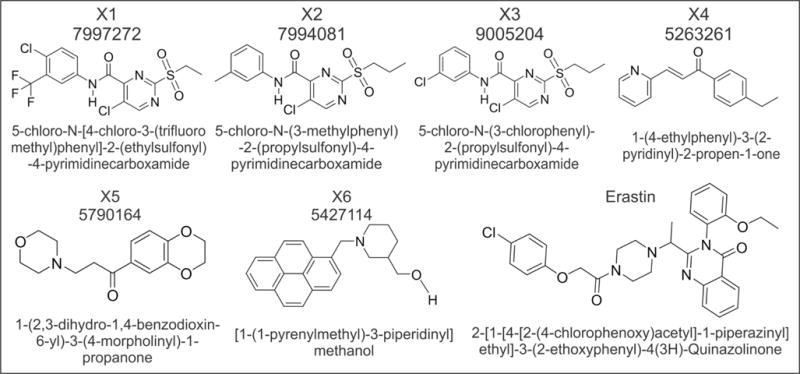
Structure of lead compounds. ChemBridge ID number, structure, and chemical nomenclature are shown for the six lead compounds identified from high-throughput screening and erastin.
Confocal Microscopy Validates the Hyperpolarizing Effects of Lead Compounds in Multiple Cancer Cell Lines
To confirm that lead compounds identified in the high-throughput screen increase ΔΨ in the presence of NCZ, and to extend our results to other cell lines, we assessed TMRM fluorescence by confocal microscopy in HepG2 human hepatoma, A549 human lung cancer, and Huh7 hepatocarcinoma cells both before and after addition of NCZ alone and NCZ plus lead compounds (Fig. 4). As expected, in HepG2 cells NCZ (10 μM) decreased TMRM intensity by about half (Fig. 4). X1, X2, X3, X4, and X6 (10 μM), but not X5, increased TMRM intensity by one- to threefold in the presence of NCZ (Fig. 4A,C). At 30 μM, all six lead compounds increased ΔΨ in the presence of NCZ by two- to fourfold (Fig. 4B,C). Time-lapse imaging showed that hyperpolarization of NCZ-treated HepG2 cells after X1 and X4 reached a plateau after about 30 min and increased little more after an hour (Suppl. Fig. S1). Thus, confocal microscopy of TMRM fluorescence in HepG2 cells confirmed the results obtained by automated wide-field fluorescence imaging of HCC4006 cells with the IN Cell Analyzer 2000. In the presence of NCZ, X1 also increased ΔΨ by ~3.4-fold in both A549 human lung cancer and Huh7 human hepatocarcinoma cells (data not shown). These last results illustrate that the effects of lead compounds were not restricted to the HCC4006 cells originally used in the screening. Overall, the blockage of the depolarizing effect of high free tubulin by the lead compounds was similar to and apparently even greater than that of erastin.
Figure 4.
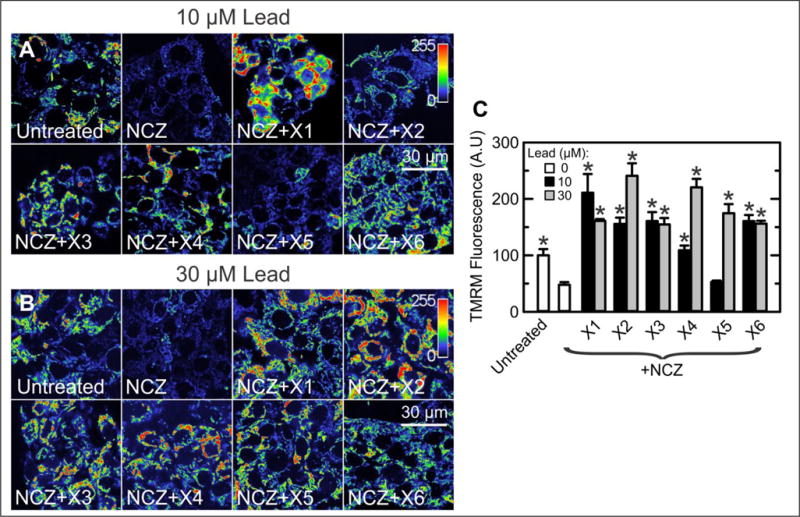
Lead validation by confocal microscopy in HepG2 human hepatoma cells. (A) HepG2 cells were untreated or treated with NCZ (10 μM) or NCZ (10 μM) plus lead compounds (10 μM), and confocal imaging was performed after 60 min. The indicator bar is a pseudo–color scale of the intensity of TMRM fluorescence. Note that X1–X4 and X6 reverted mitochondrial depolarization caused by NCZ (see also C). (B) Conditions are the same as in A except that the lead compound concentration was 30 μM. Note that all compounds reverted NCZ-induced mitochondrial depolarization and actually hyperpolarized mitochondria relative to untreated cells. (C) TMRM fluorescence under conditions of A and B is plotted. *p < 0.05 compared with NCZ alone (n = 3).
The Hyperpolarizing Effect of Lead Compounds and Erastin Is Independent of Changes of Tubulin Polymerization
The microtubule stabilizer PTX promotes tubulin polymerization and decreases free tubulin, whereas the microtubule destabilizer NCZ decreases tubulin polymerization and increases free tubulin. Because of these changes of free tubulin, PTX and NCZ hyperpolarized and depolarized mitochondria, respectively, due to relative opening and closing of VDAC.24 To assess if lead compounds and erastin hyperpolarize mitochondria by stabilizing microtubules and decreasing free tubulin, we treated HepG2 cells with vehicle, NCZ (10 μM), PTX (10 μM), erastin (10 μM), and compounds X1–X6 (10 μM) for 30 min. Each drug was added as a sole treatment. Free and polymerized tubulin were assessed by Western blotting (Fig. 5). As expected, NCZ increased free tubulin ~245% and decreased polymerized tubulin ~75%, thereby increasing the free/polymerized ratio by ~800% (Fig. 5B). In contrast, PTX decreased free tubulin ~90% and increased polymerized tubulin ~750%, thereby decreasing the free/polymerized ratio by ~99%. There was a trend for erastin and X1–X6 to produce small increases in free/polymerized tubulin ratios whose magnitude was far less than that after NCZ, but only increases after X1 and X5 were statistically significant (p < 0.05) (Fig. 5B). However, no lead compound stabilized microtubules as observed with PTX. To assess whether erastin and lead compounds antagonized NCZ-dependent tubulin depolymerization, we treated HepG2 cells with NCZ alone and with NCZ plus erastin, X1, or X4. In the presence of NCZ, erastin, X1, and X4 did not restore free/polymerized tubulin ratios to the levels of vehicle only–treated cells, although modest decreases of the ratios were observed compared with NCZ alone (Suppl. Fig. S2). Importantly, erastin, X1, and X4 failed to show the very strong microtubule-depolymerizing effect of PTX. These results demonstrate that mitochondrial hyperpolarization by X1–X6 and erastin was not caused by depletion of cytosolic free tubulin, as is the case for PTX-induced mitochondrial hyperpolarization. Thus, the mechanism of action of X1–X6 and erastin to increase ΔΨ was independent of changes of free tubulin.
Figure 5.
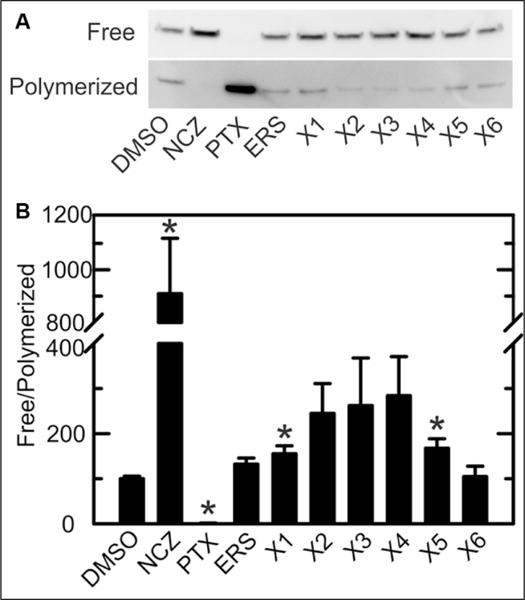
Free/polymerized tubulin ratios after treatment with erastin and lead compounds. (A) Western blotting of tubulin was performed after separating free and polymerized tubulin, as described in Materials and Methods. HepG2 cells were treated with vehicle (DMSO), NCZ, or 10 μM lead compounds X1–X6. Representative blots are shown from three independent experiments. (B) Free/polymerized tubulin ratios were calculated using relative band densities. *p < 0.05 compared with DMSO (n = 3).
X1 Decreases Lactate Production by HCC4006, HepG2, and Huh7 Human Cancer Cells
To determine whether enhanced mitochondrial metabolism by lead compounds occurs simultaneously with a decrease in glycolysis, we assessed the effect of X1 on lactate generation in HCC4006, HepG2, and Huh7 cancer cells. Without treatment, the rate of lactate formation was nearly constant for each cell line (Fig. 6). X1 (10 μM) had little to no effect on lactate production in HCC4006 and HepG2 cells but nearly completely inhibited lactate formation after 2 h in Huh7 cells (Fig. 6, lower panel). X1 (30 and 100 μM) decreased the rate of lactate formation progressively after 1 h, reaching maximum inhibition at 3 h (lactate formation rate close to zero) in HCC4006 (Fig. 6, upper panel), HepG2 (middle panel), and Huh7 (lower panel) cells. For all cell lines, inhibition of lactate production was dose dependent. Collectively, the findings were consistent with the conclusion that X1 is an anti-Warburg agent that both decreases aerobic glycolysis and increases mitochondrial ΔΨ in cancer cells.
Figure 6.
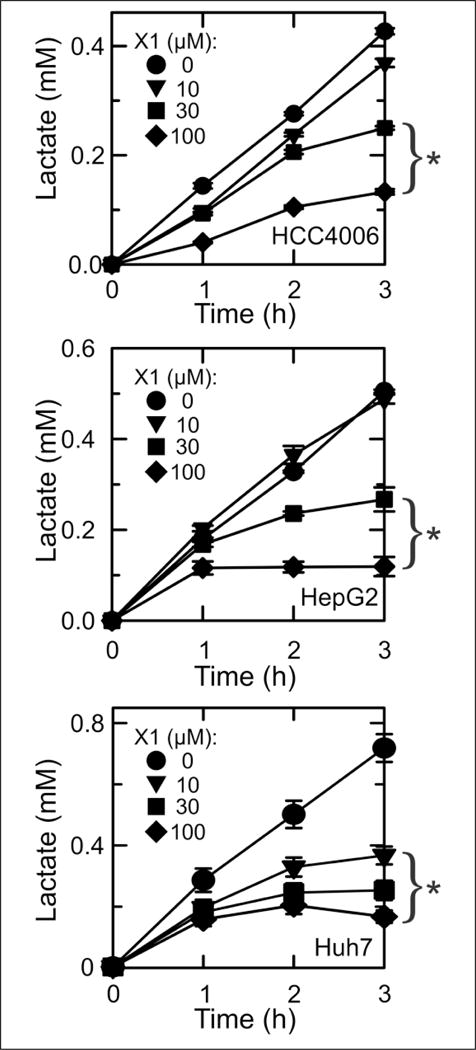
Inhibition of lactate formation by HCC4006, HepG2, and Huh7 human cancer cells after X1 treatment. HCC4006, HepG2, and Huh7 cells were treated with 0–100 μM X1, and lactate in the medium was measured. *p < 0.05 compared with 0 μM (n = 3).
Discussion
In cancer cells, suppressed mitochondrial metabolism contributes to a low cytosolic ATP/ADP ratio that favors glycolysis in the Warburg metabolic phenotype.28,29 We have proposed that VDAC closing by free tubulin contributes to the Warburg phenotype, and that antagonists of the inhibitory effect of free tubulin on VDAC promote mitochondrial metabolism and revert the Warburg effect to the metabolism of nontransformed postmitotic cells. We showed previously that cytosolic free tubulin dynamically modulates mitochondrial ΔΨ formation in cancer cells.24 More recently, we showed that erastin, a small molecule that binds to VDAC and kills RAS-harboring cancer cells,25 antagonizes tubulin-induced VDAC closure in vitro and restores ΔΨ in intact cells with elevated cytosolic free tubulin induced by microtubule destabilization.18 These findings suggest that the VDAC–tubulin interaction could be a potential pharmacological target to promote mitochondrial metabolism, suppress aerobic glycolysis, and revert the pro-proliferative Warburg phenotype in cancer cells.
Here, we designed a cell-based high-content drug screening assay to identify small molecules that, similar to erastin, revert the mitochondrial depolarization induced by high cytosolic free tubulin (Fig. 1). Microtubule destabilization by NCZ was used to increase cytosolic free tubulin levels and facilitate the identification of lead compounds capable of hyperpolarizing mitochondria even at high levels of free tubulin. We identified 30 small molecules that increased mitochondrial ΔΨ in the presence of elevated cytosolic free tubulin. The six hits that promoted the greatest increases of ΔΨ were selected as lead compounds (Fig. 2A). A dose– response was determined for each of these six leads (Fig. 2B), and their hyperpolarizing effects in the presence of NCZ were validated by confocal microscopy (Fig. 4). We named these compounds X1–X6 in rank order of potency to increase ΔΨ in cancer cells exposed to NCZ.
Although X1–X6 hyperpolarized mitochondria in the presence of high free tubulin, only the three most potent lead compounds (X1–X3) were structurally similar (Fig. 3). X1, X2, and X3 each contain a phenylpyrimidine carboxamide, a structure associated with cytotoxicity in several cancer cell lines.30 Notably, the inclusion of a phenylpyrimidine carboxamide moiety to foretinib, a c-Met inhibitor and experimental chemotherapeutic agent, increases cytotoxicity.31 Of the nonphenylpyrimidine carboxamide lead compounds, the most potent was X4, which has a chalcone core structure. Various analogs of chalcones have been reported to have anticancer properties,32 and some chalcones interact directly with tubulin.33–35 Little is known about how chalcone analogs affect cancer cell metabolism and bioenergetics.
Mitochondrial ΔΨ was heterogeneous between cells both before and after addition of erastin and lead compounds (Figs. 2 and 4 and Suppl. Fig. S1). The basis for such heterogeneity remains unknown, although we have previously suggested that differences in cell cycle progression may play a role.4 In general, lead compounds hyperpolarized mitochondria in virtually all cells, although the magnitude of the response varied (Figs. 2 and 4 and Suppl. Fig. S1). Whether there is a differential hyperpolarizing effect of erastin and lead compounds on cells with different basal ΔΨ remains to be determined. At the highest concentration tested (100 μM), X1 and X4 produced an initial hyperpolarization, followed by subsequent depolarization at 30–120 min (not shown). Thus, the mitochondrial hyperpolarization caused by our lead compounds may promote mitochondrial dysfunction, and we are currently investigating whether oxidative stress may contribute to this dysfunction, since previously erastin was shown to promote iron-dependent oxidative stress that culminates in the killing of cancer cells.36
We used NCZ as a tool to maximize cytosolic free tubulin and VDAC closure, allowing us to identify compounds that block this inhibitory effect and restore ΔΨ. This strategy allowed for the largest increase of ΔΨ in response to erastin-like small molecules and enhanced the sensitivity of our screening assay. As shown previously for erastin, the addition of lead compounds to untreated cells also caused mitochondrial hyperpolarization by reversing the partial blockade of VDAC by endogenous free tubulin (data not shown).18 However, once cells were treated with erastin or lead compounds, NCZ no longer caused significant mitochondrial depolarization. Thus, in erastin and lead compound-treated cells, ΔΨ was the same in the presence and absence of NCZ. Previous findings also showed that proliferating cancer cells have a substantially higher amount of αβ-heterodimeric free tubulin than differentiated postmitotic cells.24 For this reason, cancer cells hyperpolarize after erastin, but postmitotic cells do not.
To determine whether the hyperpolarizing effect of erastin and lead compounds was due to a decrease in free tubulin similar to PTX treatment, we measured free and polymerized tubulin after erastin and lead compounds in the presence and absence of NCZ. We found that erastin and X1–X6 alone did not decrease free tubulin, and that some hyperpolarizing compounds actually increased free tubulin, indicating that mitochondrial hyperpolarization induced by lead compounds was not caused by an effect similar to that of PTX. Moreover, erastin and X1–X6 did not prevent the microtubule-depolymerizing effect of NCZ. In previous work, PTX antagonized NCZ-induced mitochondrial depolarization, which was attributed to VDAC opening after decreased free tubulin from microtubule stabilization.24 Our findings that erastin and the lead compounds do not stabilize microtubules and decrease free tubulin similar to PTX indicate that mitochondrial hyperpolarization occurs by a mechanism independent of tubulin dynamics.
Previously, we showed that the respiratory inhibitor, myxothiazol, only slightly decreases ΔΨ, because consumption of glycolytically formed ATP maintains ΔΨ by the mitochondrial F1FO-ATP synthase working in reverse mode.24 Under such conditions, subsequent addition of oligomycin to inhibit the ATP synthase collapses ΔΨ. NCZ causes partial mitochondrial depolarization by a different mechanism, because increased free tubulin after NCZ blocks VDAC and the uptake of ΔΨ-generating metabolites, an effect antagonized by the microtubule-stabilizing agent, PTX. NCZ does not directly inhibit the respiratory chain. Erastin and presumably our erastin-like lead compounds antagonize the inhibitory effect of free tubulin on VDAC to hyperpolarize mitochondria.18 Nonetheless, off-target mechanisms different from the antagonism of tubulin-dependent VDAC inhibition cannot be completely excluded from increasing ΔΨ. For example, inhibition of the F1FO-ATPase or increased utilization of respiratory substrates could also increase ΔΨ. Future studies will be needed to exclude such possibilities and to show directly that the lead compounds antagonize tubulin-dependent inhibition of VDAC reconstituted in planar bilayers similarly to erastin.
To assess the effects on aerobic glycolysis, we measured lactate production by Huh7, HepG2, and HCC4006 cancer cells after 0–100 μM X1. At concentrations of >10 μM, X1 decreased the rate of lactate production within 1–2 h. By 3 h, rates approached zero (Fig. 6). Thus, X1 reverted the two major features characteristic of the pro-proliferative Warburg metabolic phenotype by inhibiting aerobic glycolysis and enhancing mitochondrial metabolism, as evidenced by increased ΔΨ. Future studies are planned to assess how these lead compounds affect cancer cell proliferation and viability, oxygen consumption, ATP/ADP ratios, and the redox status of NAD(P) as further indicators of mitochondrial metabolism.
In conclusion, we identified small molecules (X1–X6) that block NCZ-induced mitochondrial depolarization without decreasing the amount of free tubulin. In addition, the most potent lead compound (X1) decreases aerobic glycolysis (lactate production), acting as an anti-Warburg agent. In the future, we will seek to assess how and whether erastin and these lead compounds promote selective killing of cancer cells.
Supplementary Material
Acknowledgments
We thank Dr. Patrick M. Woster for help in analyzing the chemical similarity between lead compounds, Drs. Jack R. Wands and Robert M. Gemmill for generously providing cell lines, and Dr. Steve Titus of GE Healthcare for technical assistance with high-content imaging and analysis.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is in partial fulfillment of the requirements for the degree of doctor of philosophy to D.N.D. and was supported, in part, by Grants 4R25GM072643-12 (D.N.D.); T32DK083262 (D.N.D. and J.J.L.); R01 DK073336, R01 DK037034, and 14.Z50.31.0028 (J.J.L.); and R01 CA18445601, P50 CA058187, ACS 13-043-01, and COBRE Pilot Project GM103542 (E.N.M.). Imaging facilities were supported, in part, by P30 CA138313.
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8(6):519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburg O. On the Origin of Cancer Cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Mathupala SP, Ko YH, Pedersen PL. The Pivotal Roles of Mitochondria in Cancer: Warburg and Beyond and Encouraging Prospects for Effective Therapies. Biochim Biophys Acta. 2010;1797(6–7):1225–1230. doi: 10.1016/j.bbabio.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maldonado EN, Lemasters JJ. Warburg Revisited: Regulation of Mitochondrial Metabolism by Voltage-Dependent Anion Channels in Cancer Cells. J Pharmacol Exp Ther. 2012;342(3):637–641. doi: 10.1124/jpet.112.192153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakashima RA, Paggi MG, Pedersen PL. Contributions of Glycolysis and Oxidative Phosphorylation to Adenosine 5′-Triphosphate Production in AS-30D Hepatoma Cells. Cancer Res. 1984;44(12 Pt 1):5702–5706. [PubMed] [Google Scholar]
- 6.Pedersen PL. Tumor Mitochondria and the Bioenergetics of Cancer Cells. Prog Exp Tumor Res. 1978;22:190–274. doi: 10.1159/000401202. [DOI] [PubMed] [Google Scholar]
- 7.Gambhir SS. Molecular Imaging of Cancer with Positron Emission Tomography. Nat Rev Cancer. 2002;2(9):683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 8.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 Bound to Mitochondria: Cancer’s Stygian Link to the “Warburg Effect” and a Pivotal Target for Effective Therapy. Semin Cancer Biol. 2009;19(1):17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sottnik JL, Lori JC, Rose BJ, et al. Glycolysis Inhibition by 2-Deoxy-D-Glucose Reverts the Metastatic Phenotype In Vitro and In Vivo. Clin Exp Metastasis. 2011;28(8):865–875. doi: 10.1007/s10585-011-9417-5. [DOI] [PubMed] [Google Scholar]
- 10.Smolkova K, Bellance N, Scandurra F, et al. Mitochondrial Bioenergetic Adaptations of Breast Cancer Cells to Aglycemia and Hypoxia. J Bioenerg Biomembr. 2010;42(1):55–67. doi: 10.1007/s10863-009-9267-x. [DOI] [PubMed] [Google Scholar]
- 11.Haugrud AB, Zhuang Y, Coppock JD, et al. Dichloroacetate Enhances Apoptotic Cell Death via Oxidative Damage and Attenuates Lactate Production in Metformin-Treated Breast Cancer Cells. Breast Cancer Res Treat. 2014;147(3):539–550. doi: 10.1007/s10549-014-3128-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madhok BM, Yeluri S, Perry SL, et al. Dichloroacetate Induces Apoptosis and Cell-Cycle Arrest in Colorectal Cancer Cells. Br J Cancer. 2010;102(12):1746–1752. doi: 10.1038/sj.bjc.6605701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maldonado EN. VDAC-Tubulin, an Anti-Warburg Pro-Oxidant Switch. Front Oncol. 2017;7:4. doi: 10.3389/fonc.2017.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedersen PL. Warburg, Me and Hexokinase 2: Multiple Discoveries of Key Molecular Events Underlying One of Cancers’ Most Common Phenotypes, the “Warburg Effect”, i.e., Elevated Glycolysis in the Presence of Oxygen. J Bioenerg Biomembr. 2007;39(3):211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 15.Saed GM, Fletcher NM, Jiang ZL, et al. Dichloroacetate Induces Apoptosis of Epithelial Ovarian Cancer Cells through a Mechanism Involving Modulation of Oxidative Stress. Reprod Sci. 2011;18(12):1253–1261. doi: 10.1177/1933719111411731. [DOI] [PubMed] [Google Scholar]
- 16.Wong JY, Huggins GS, Debidda M, et al. Dichloroacetate Induces Apoptosis in Endometrial Cancer Cells. Gynecol Oncol. 2008;109(3):394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemasters JJ, Holmuhamedov E. Voltage-Dependent Anion Channel (VDAC) as Mitochondrial Governator— Thinking Outside the Box. Biochim Biophys Acta. 2006;1762(2):181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Maldonado EN, Sheldon KL, DeHart DN, et al. Voltage-Dependent Anion Channels Modulate Mitochondrial Metabolism in Cancer Cells: Regulation by Free Tubulin and Erastin. J Biol Chem. 2013;288(17):11920–11929. doi: 10.1074/jbc.M112.433847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blachly-Dyson E, Forte M. VDAC Channels. IUBMB Life. 2001;52(3–5):113–118. doi: 10.1080/15216540152845902. [DOI] [PubMed] [Google Scholar]
- 20.Colombini M. VDAC: The Channel at the Interface between Mitochondria and the Cytosol. Mol Cell Biochem. 2004;256–257(1–2):107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 21.Sampson MJ, Lovell RS, Craigen WJ. The Murine Voltage-Dependent Anion Channel Gene Family. Conserved Structure and Function. J Biol Chem. 1997;272(30):18966–18973. doi: 10.1074/jbc.272.30.18966. [DOI] [PubMed] [Google Scholar]
- 22.Rostovtseva TK, Sheldon KL, Hassanzadeh E, et al. Tubulin Binding Blocks Mitochondrial Voltage-Dependent Anion Channel and Regulates Respiration. Proc Natl Acad Sci USA. 2008;105(48):18746–18751. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Timohhina N, Guzun R, Tepp K, et al. Direct Measurement of Energy Fluxes from Mitochondria into Cytoplasm in Permeabilized Cardiac Cells In Situ: Some Evidence for Mitochondrial Interactosome. J Bioenerg Biomembr. 2009;41(3):259–275. doi: 10.1007/s10863-009-9224-8. [DOI] [PubMed] [Google Scholar]
- 24.Maldonado EN, Patnaik J, Mullins MR, et al. Free Tubulin Modulates Mitochondrial Membrane Potential in Cancer Cells. Cancer Res. 2010;70(24):10192–10201. doi: 10.1158/0008-5472.CAN-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yagoda N, von Rechenberg M, Zaganjor E, et al. RAS-RAF-MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature. 2007;447(7146):864–868. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemasters JJ, Ramshesh VK. Imaging of Mitochondrial Polarization and Depolarization with Cationic Fluorophores. Methods Cell Biol. 2007;80:283–295. doi: 10.1016/S0091-679X(06)80014-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 28.Maldonado EN, DeHart DN, Patnaik J, et al. ATP/ADP Turnover and Import of Glycolytic ATP into Mitochondria in Cancer Cells Is Independent of the Adenine Nucleotide Translocator. J Biol Chem. 2016;291(37):19642–19650. doi: 10.1074/jbc.M116.734814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maldonado EN, Lemasters JJ. ATP/ADP Ratio, the Missed Connection between Mitochondria and the Warburg Effect. Mitochondrion. 2014;19(Pt A):78–84. doi: 10.1016/j.mito.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu S, Sun C, Chen C, et al. Synthesis and Biological Evaluation of Novel 8-Morpholinoimidazo[1,2-a]pyrazine Derivatives Bearing Phenylpyridine/Phenylpyrimidine-Carboxamides. Molecules. 2017;22(2):310. doi: 10.3390/molecules22020310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu W, Wang W, Xu S, et al. Synthesis, and Docking Studies of Phenylpyrimidine-Carboxamide Derivatives Bearing 1H-Pyrrolo[2,3-b]pyridine Moiety as c-Met Inhibitors. Bioorg Med Chem. 2016;24(8):1749–1756. doi: 10.1016/j.bmc.2016.02.046. [DOI] [PubMed] [Google Scholar]
- 32.Mahapatra DK, Bharti SK, Asati V. Anti-Cancer Chalcones: Structural and Molecular Target Perspectives. Eur J Med Chem. 2015;98:69–114. doi: 10.1016/j.ejmech.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Chen J, Wang J, et al. Design, Synthesis, and Biological Evaluation of Stable Colchicine Binding Site Tubulin Inhibitors as Potential Anticancer Agents. J Med Chem. 2014;57(17):7355–7366. doi: 10.1021/jm500764v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y, Chen J, Xiao M, et al. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm Res. 2012;29(11):2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Wang Z, Li CM, et al. Discovery of Novel 2-Aryl-4-Benzoyl-Imidazoles Targeting the Colchicines Binding Site in Tubulin as Potential Anticancer Agents. J Med Chem. 2010;53(20):7414–7427. doi: 10.1021/jm100884b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.