

Abstract
Background & Aims
Campylobacter jejuni, a prevalent food-borne bacterial pathogen, exploits the host innate response to induce colitis. Little is known about the roles of microbiota in C jejuni-induced intestinal inflammation. We investigated interactions between microbiota and intestinal cells during C jejuni infection of mice.
Methods
Germ-free C57BL/6 Il10–/– mice were colonized with conventional microbiota and infected with a single dose of C jejuni (109 colony-forming units/mouse), via gavage. Conventional microbiota was cultured under aerobic, microaerobic, or anaerobic conditions and orally transplanted into GF Il10–/– mice. Colon tissues were collected from mice and analyzed by histology, real-time PCR, and immunoblotting. Fecal microbiota and bile acids were analyzed with 16S sequencing and high-performance liquid chromatography with mass spectrometry, respectively.
Results
Introduction of conventional microbiota reduced C jejuni-induced colitis in previously germ-free Il10–/– mice, independent of fecal load of C jejuni, accompanied by reduced activation of mTOR. Microbiota transplantation and 16S rDNA sequencing experiments showed that Clostridium XI, Bifidobacterium, and Lactobacillus were enriched in fecal samples from mice colonized with microbiota cultured in anaerobic conditions (which reduce colitis) compared to mice fed microbiota cultured under aerobic conditions (susceptible to colitis). Oral administration to mice of microbiota-derived secondary bile acid sodium deoxycholate, but not ursodeoxycholic acid or lithocholic acid, reduced C jejuni-induced colitis. Depletion of secondary bile acid-producing bacteria with antibiotics that kill anaerobic bacteria (clindamycin) promoted C jejuni-induced colitis in SPF Il10–/– mice compared to the non-specific antibiotic nalidixic acid; colitis induction by antibiotics was associated with reduced level of luminal deoxycholate.
Conclusions
We identified a mechanism by which the microbiota controls susceptibility to C jejuni infection in mice, via bacteria-derived secondary bile acids.
Keywords: Metabolism, DCA, infection, microbiome
Introduction
Campylobacter jejuni is one of the prevalent causes of bacterial-derived diarrheal illness in developed countries, and global incidence has been on the rise this past decade.1 Furthermore, C. jejuni causes serious post-infection complications, including arthritis, Guillian-Barré Syndrome, Irritable Bowel Syndrome and Inflammatory Bowel Diseases (IBD).1 Clinical symptoms of campylobacteriosis include abdominal cramps, watery to bloody diarrhea, fever and gastrointestinal inflammation.2 At the cellular level, the intestinal tract of C. jejuni-infected patients displays infiltration of immune cells such as neutrophils, crypt abscesses and presence of fecal leukocytes.3 Gnotobiotic technology applied to germ free (GF) Il10-/- mice (129 SvEv) showed that the human clinical C. jejuni strain 81-176 induces acute intestinal inflammation resembling key features of human campylobacteriosis (neutrophils infiltration, crypt abscesses, and bacterial invasion).4 Subsequent studies showed that innate immunity is critical for campylobacteriosis as the inflammatory response is similar between Il10-/- and Il10-/-; Rag2-/-mice.5 Moreover, phosphatidylinositol 3-kinases gamma (PI3Kγ) signaling mediated neutrophil migration into colonic tissues and is essential for C. jejuni-induced colitis.5 The mammalian target of rapamycin (mTOR), a downstream target of PI3K, has been implicated in many functions, including cell growth, proliferation, survival, and innate and adaptive immune responses.6-8 The mTOR inhibitor rapamycin prevents and treats C. jejuni-induced campylobacteriosis in Il10-/- mice.9 These findings highlight the important role of PI3K/mTOR in C. jejuni-induced colitis. However, the role of commensal gut microbiota in controlling host susceptibility to C. jejuni infection is unknown. Interestingly, C. jejuni colonic luminal colonization level is not associated with the bacterial ability to induce colitis,5, 9-11 suggesting a complex interaction between the pathogen, microbiota and host response.
The intestinal microbiota exerts numerous effects on the host, especially on immune response following infection. For example, the microbiota regulates granulocytosis and neonate response to Escherichia coli K1 and Klebsiella pneumoniae sepsis.12 In addition, the microbiota is also found to enhance myelopoiesis and protect against Listeria monocytogenes infection.13 At the gut level, acetate derived from Bifidobacteria metabolism protects host against enterohaemorrhagic E. coli O157:H7 infection by inhibiting its Shiga toxin translocation.14, 15 Commensal segmented filamentous bacterium induces IL17- and IL22-producing Th17 cells16 and mice with deficiency of IL22 succumb to Citrobacter rodentium infection.17 Recently, microbiota transplantation has shown tremendous success against recurrent Clostridium difficile infection,18 suggesting that microbial manipulation has the potential to treat infectious microorganisms. In addition, the biotransformation of secondary bile acids by C. scindens was found to inhibit C. difficile colonization and infection,19 suggesting a critical role of microbial metabolites on C. difficile pathogenesis. Intriguingly, bile acids, particularly secondary bile acid deoxycholic acid (DCA), are associated with various chronic diseases such as metabolic diseases, intestinal inflammation, and colorectal cancers.20-22 Whether the microbiota-derived bile acid metabolism prevents colonization of and host response to C. jejuni is unknown.
In this study, we hypothesized that specific groups of microbiota control C. jejuni-induced enteritis through the production of specific microbial metabolites that modulate host-derived inflammatory signaling. Our data indicate that specific anaerobic microbes and their metabolites protect Il10-/- mice against C. jejuni-induced intestinal inflammation, through modulation of host response. These metabolites and signaling pathways represent critical events in C jejuni–induced intestinal inflammation and could define potential new therapeutic targets.
Materials and Methods
Mouse experiments
Animal experiments were in accordance with the Animal Research: Reporting of In Vivo Experiments (https://www.nc3rs.org.uk/arrive-guidelines). All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Florida (201608025). Cohorts of 8-12 weeks old age matched male or female 4 to 9 mice/group were used, and the sample size was based on previous published reports showing significant colitis with that sample size.4, 9, 23 The sibling littermate mice were fed ad libitum chew diet and water in individually ventilated cage (IVC) with Alpha Dri bedding. All animal procedures were performed at light cycle. GF Il10-/- mice were transferred to SPF conditions for 3 or 14 days and 14-day stool was collected as conventionalized microbiota (CONV-Biota). Freshly collected stools were immediately suspended in 30% glycerol PBS stock, quantified (OD600 value of 1 was estimated as 108 CFU/ml), and stored at -80 °C. Before oral gavage, the stool preparation was thawed, diluted and immediately gavaged to mice at 108 CFU/mouse. The CONV-Biota was also cultured under aerobic (Aero-Biota), microaerobic (Microaero-Biota) or anaerobic (Anaero-Biota) conditions using Brain Hart Infusion (BHI) agar plates. For C. jejuni infection experiments, GF C57BL/6 Il10 -/- mice were transferred from GF isolators to SPF housing and immediately gavaged with 109 C. jejuni CFU/mouse (strain 81–17624) for 12 days and sacrificed as described before.9 For whole microbiota protection experiments, GF Il10 -/- mice were orally gavaged with a single dose of CONV-Biota (108 CFU/mouse) for 14 days before 12-day C. jejuni infection. For specific microbiota protection experiments, GF Il10 -/- mice were gavaged with a single dose of 108 CFU/mouse Aero-Biota, Microaero-Biota, Anaero-Biota or the three microbiota pooled. We began the 12-day C. jejuni infection 14 days post-gavage. To deplete mouse microbiota, C57BL/6 Il10 -/- mice in SPF housing were given an antibiotics cocktail in drinking water9 or clindamycin (Sigma-Aldrich) was gavaged at 67 mg/kg body weight (BW) or nalidixic acid (Sigma-Aldrich) was gavaged at 200 mg/kg BW for 7 days. One day after the antibiotic treatment, the mice were gavaged with a single dose of 109 C. jejuni CFU/mouse for 21 days. To investigate the impact of bile acids on C. jejuni-induced colitis, GF Il10-/- mice were infected as before and were gavaged daily with 30 mg/kg BW of deoxycholic acid, lithocholic acid, or ursodeoxycholic acid (Sigma-Aldrich) for 12 days. Although we observed intestinal inflammation at different time points of C. jejuni infection (day 4, 5, 6, and 12 post infection) in ex-GF Il10-/- mice, we opted for the 12-day post-infection time point in this study because of the consistency in host response (severe colitis), and also because this colonization time was used to determine efficacy of therapeutic intervention in previous studies (neutrophil depletion, mTOR inhibition, and PI3Kγ blockade).4, 5, 9 Mice were followed clinically for evidence of diarrhea, failure to thrive and mortality. At the end of experiments, tissue samples from mouse colon and stool were collected for protein, RNA, histology, and culture assay. For live C. jejuni counting, MLN and liver were aseptically resected. Colon tissue was opened, resected, and washed three times in sterile PBS. Colonic luminal content (stool) was also collected. The freshly collected tissues and stool were weighed, homogenized in PBS, serially diluted, and plated on Campylobacter-selective blood plates (Remel) for 48 h at 37°C using the GasPak system (BD Biosciences). C. jejuni colonies were counted, and data were presented as CFU per gram tissue or stool. Histopathological images were acquired using a DP71 camera and DP Controller 3.1.1.276 (Olympus) as described before.4 Intestinal inflammation was scored using a score from 0–4.
DNA extraction, 16S rDNA gene amplification and multiparallel sequencing
Mouse stool samples were collected and DNA was extracted using bead beater disruption and phenol : chloroform separation followed by DNeasy Blood & Tissue Kit (Qiagen) as described before.23, 25 The V1-V3 region hypervariable region of the 16S rDNA was amplified using primer pair 8F (5′- AGAGTTTGATCCTGGCTCAG -3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′). Both the forward and the reverse primers contained universal Illumina paired-end adapter sequences, as well as unique individual 4-6 nucleotide barcodes between PCR primer sequence and the Illumina adapter sequence to allow multiplex sequencing (Supplementary Table 5). PCR products were visualized on an agarose gel, before samples were purified using the Agencourt AMPure XP kit (A63881, Beckman Coulter) and quantified by qPCR with the KAPA Library Quantification Kit (KK4824, KAPA Biosystems). Equimolar amount of samples were then pooled and sequenced with an Illumina MiSeq.
Quantification of bile acid species
Bile acids in mouse stool were extracted using methanol. Briefly, vacuum-dried stool was suspended in methanol and sonicated. After incubated with occasional shakes, the supernatant was collected after the suspension was centrifuged. The pellet was re-extracted in methanol for an additional two times. The three pooled supernatants were subject to HPLC/MS analysis. Calibration was made with the addition of individual or pooled bile acid standards (Sigma-Aldrich) into GF mouse stools and the bile acids were extracted. Bile acids were quantified as previously described.26 Briefly, bile acid analysis was performed on liquid chromatography– tandem mass spectrometry (LC-MS, Agilent 6130 quadrupole) with an Agilent Zorbax SB-C18 1.8 μm (2.1 × 50 mm) column. Mobile phase A contained methanol/water (1:1,v/v) and mobile phase B consisted of methanol + 10mM ammonium acetate + 0.1% ammonia (pH=8.8). The running method was 100% A (0-1 min), linear increase to 50% B (1-9 min), then to 100% B (9-13 min) followed by 100% A (13-18 min). Flow rate was 0.2ml/min. The column was re-equilibrated from 18.1-24 min with 100% A. MS was performed using AP-ESI ionization technique and the spectra were analyzed in negative mode. Because tauromuricholic acid and TCA share same mass and retention time, we reported them as TCA.
Data analysis was performed with Agilent ChemStation-B.04.03 software. The data were exported to Excel spreadsheets and the relative bile acid (%) within a sample was calculated by normalizing with the combined bile acid extracted ion intensity.
Statistical Analysis
Values are shown as mean ± standard error of the mean as indicated. Differences between groups were analyzed using the nonparametric Mann–Whitney U test performed using Prism 5.0 software. Bile acid data were analyzed using ANOVA in R. Experiments were considered statistically significant if P values were <.05.
Detailed methods of 16S rDNA sequencing analysis, western blotting of infected splenocytes and colon tissues, fluorescence in situ hybridization (FISH), immunohistochemistry (IHC) and real time RT-PCR are in the Supplementary Material available online.
Results
Reduction of microbiota promotes C. jejuni-induced colitis
To define the interaction between microbiota and host susceptibility to campylobacteriosis, we manipulated the intestinal microbial content using antibiotics, fecal transfer and GF mice. First, we infected GF and specific pathogen free (SPF) Il10-/- mice with the human clinical C. jejuni isolate 81-176 (109 colony forming unit (CFU)/mouse). Interestingly, C. jejuni failed to induce colitis in SPF Il10-/- mice (Figure 1A, left panels and 1B), whereas ex-GF Il10-/- mice developed severe colitis at 12 days post infection (Figure 1A, right panels). In addition, microbiota-depletion using a broad spectrum antibiotic cocktail (Supplementary Figure 1A and B), rendered conventionalized Il10-/- mice susceptible to C. jejuni-induced colitis (Figure 1A, middle panels) albeit to a lower extent than GF mice (Figure 1A, right panels). In line with the colitis severity, C. jejuni induced proinflammatory gene expression of Il1β, Cxcl2 and Il17a mRNA in colonic tissue of ex-GF Il10-/- mice and SPF Il10-/- mice with microbiota depleted, compared to their respective uninfected mice (Figure 1C). C. jejuni induces colitis as early as 4-6 days post infection and these early time points have then been used for treatment intervention.4, 5, 9, 11 Therefore, we infected GF Il10-/- mice with C. jejuni and evaluated colitis 5 or 12 days post infection. Consistent with previous reports, C. jejuni induced colitis at 5 days post infection, although with less severity than 12-day infection (Supplementary Figure 2A and B). Interestingly, C. jejuni colonic luminal colonization level and invasion into liver and mesenteric lymph node (MLN) were comparable between 5- and 12-day infection (Supplementary Figure 2C), whereas Il6 and Cxcl1 mRNA expression was greater in colonic tissue of 5-day infection mice (Supplementary Figure 2D).
Figure 1. Pan-depletion of microbiota exacerbates C. jejuni-induced intestinal inflammation in Il10-/- mice.
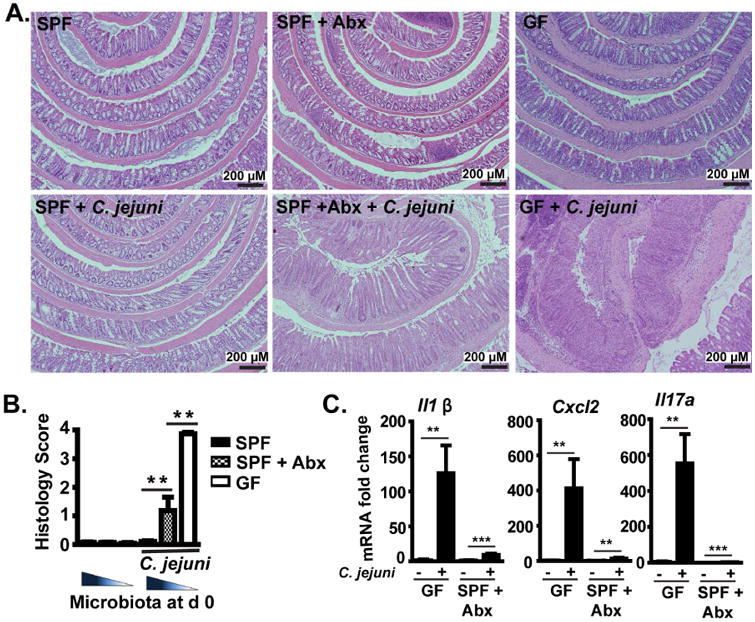
Cohorts of 5-9 GF or SPF Il10-/- mice were gavaged with a single dose of 109 CFU C. jejuni/mouse and were euthanized 12 or 21 days post-infection. SPF Il10-/-mice were treated with an antibiotics (Abx) cocktail in their drinking water for 7 days before infection. (A) H&E staining showing representative intestinal histology of C. jejuni-induced colitis in SPF (left), SPF+ Abx (middle) and GF (GF, right) Il10-/- mice. (B) Quantification of histological intestinal damage score. (C) Colonic Il1β, Cxcl2, and Il17a mRNA qPCR fold change relative to GF and normalized to Gapdh. **, P<0.01. Scale bar is 200 μm. Results are representative of 3 independent experiments.
Microbiota transplantation attenuates C. jejuni-induced colitis in GF mice
From above observations of increased campylobacteriosis susceptibility in microbiota-depleted SPF Il10-/- mice, we reasoned that component of the microbiota confers resistance to C. jejuni-induced colitis in Il10-/- mice. To gain a better understanding of the relationship between microbial acquisition and susceptibility to campylobacteriosis, we transferred (conventionalized) germ free Il10-/- mice to SPF housing for either 3 or 14 days, and then infected these cohorts with C. jejuni for 12 days. Interestingly, C. jejuni-induced colitis was attenuated in day 14, but not day 3 conventionalized Il10-/- mice (Supplementary Figure 3A and B), indicating the presence of protective microbiota. Therefore, we selected 14-day conventionalized microbiota (CONV-Biota) for the subsequent campylobacteriosis protection experiments. GF Il10-/- mice were transplanted by orogastric gavage with a single dose of CONV-Biota, and 14 days post inoculation the mice were infected with C. jejuni. Consistently, Il10-/- mice colonized with CONV-Biota resisted C. jejuni-induced intestinal inflammation compared to mono-associated Il10-/- mice (Figure 2A and B). We then asked whether colonization resistance was implicated in the protective effect of microbiota against campylobacteriosis. Surprisingly, luminal C. jejuni colonization levels were comparable between mono-associated GF and CONV-Biota Il10-/- mice (Figure 2C), although their inflammatory status is dramatically different (Figure 2B). These results indicate that the microbiota may exert a protective role against C. jejuni infection through a novel mechanism, independent of luminal colonization exclusion.
Figure 2. Microbiota prevents C. jejuni-induced intestinal inflammation in GF Il10-/- mice in a manner independent of luminal colonization exclusion.
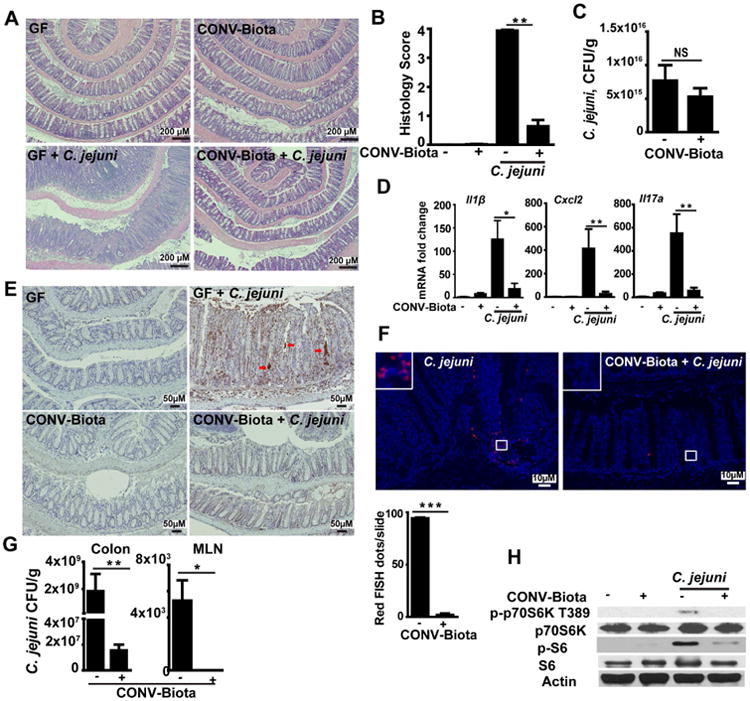
Cohorts of 5-6 GF Il10-/- mice were colonized with a conventionalized microbiota (CONV-Biota) for 14 days or left in GF conditions. The mice were then infected with a single dose of 109 CFU C. jejuni/mouse and were euthanized 12 days post-infection. (A) Representative intestinal histology images. (B) Quantification of histological intestinal damage score. (C) C. jejuni colonic luminal colonization level using culture. (D) Colonic Il1β, Cxcl2, and Il17a mRNA qPCR fold change relative to GF and normalized to Gapdh. (E) Immunohistochemistry of myeloperoxidase positive neutrophils (brown dots). Red arrows indicate neutrophil accumulation in the crypt lumen and formation of crypt abscesses. (F) Presence of C. jejuni (red dots, counted as dots/slide (lower panel)) in colonic sections of infected mice, detected using fluorescence in situ hybridization (FISH) assay. (G) Live C. jejuni count in the colon tissue (left panel) and MLN (right panel). Briefly, MLN and colon tissue were aseptically resected, weighed, homogenized in PBS, serially diluted, and plated on Campylobacter-selective blood plates. The bacteria were counted after 48 h at 37°C using the GasPak system. (H) Western blot of total and phosphorylated p70S6 (T389) and S-6 (S235/236) and Actin from resected colon tissues. Scale bar is 200 (Fig. A), 50 (Fig. E) or 10 (Fig. F) μm. All graphs depict mean ± SEM. NS, not significant, *, P<0.05; **, P<0.01. Results are representative of 3 independent experiments.
A strong inflammatory host response is observed following C. jejuni-infection,4, 5 thus we interrogated the impact of the microbiota on various proinflammatory mediators. C. jejuni strongly induced proinflammatory Il1β, Cxcl2 and Il17a mRNA accumulation in colonic tissue of ex GF Il10–/– mice, an effect attenuated by 85, 93, and 90%, respectively, in CONV-Biota mice (Figure 2D). Because neutrophils play a key role in C. jejuni-mediated colitis,5 we assessed the status of neutrophil infiltration using immunohistochemistry (IHC). CONV-Biota significantly reduced MPO positive neutrophil migration into the colon of C. jejuni infected Il10–/– mice (Figure 2E). In addition, fluorescence in situ hybridization (FISH) showed that while C. jejuni is present deeply in the inflamed crypts and in the lamina propria section of the intestine of ex GF Il10-/- mice, the bacterium was barely detectable (90% less) in colonic tissues of CONV-Biota Il10-/- mice (Figure 2F). Consistent with the FISH results, mice transplanted with CONV-Biota displayed reduced C. jejuni invasion in colonic tissue (by 99%) and MLN (non-detectable) compared to ex GF Il10-/- mice (Figure 2G). The inhibitory effect of CONV-Biota on campylobacteriosis and host responses resemble the effect seen with mTOR inhibition in Il10-/-mice.9 We then reasoned that the microbiota may attenuate C. jejuni-induced mTOR signaling. Interestingly, C. jejuni induced mTOR downstream target p-p70S6k T389 and p-S6 (S235/236) in colonic tissues of ex-GF Il10-/- mice was attenuated in mice colonized with CONV-Biota (Figure 2H). To further document the cellular distribution of C. jejuni-induced mTOR signaling and the effect of CONV-Biota, we determined p-S6 positive cells using IHC. Notably, p-S6 accumulation was detected in both epithelial and lamina propria immune cells of C. jejuni infected ex-GF mice, whereas tissues of mice colonized with CONV-Biota displayed strong reduction in p-S6 staining (Supplementary Figure 4). Collectively, these results suggest that C. jejuni exploits host mTOR signaling and inflammatory responses to invade colonic tissues, a process blocked by the CONV-Biota without decreasing C. jejuni luminal load.
Specific groups of anaerobic microbiota protect against C. jejuni-induced colitis
Encouraged by the protective feature of the CONV-Biota, we reasoned that a core bacterial community within the microbiota granted resistance to campylobacteriosis. To identify this essential population, we compared microbiota composition between campylobacteriosis-resistant CONV-Biota (14-day conventionalization) and colitis-permissive microbiota (3-day conventionalization) (Supplementary Figure 3) and correlated it with histological colitis level. Interestingly, PCoA analysis revealed distinct separation between microbiota from Il10-/- mice conventionalized for 3 (Light blue: severe colitis) and 14 days (Dark blue: mild colitis) (Figure 3A), whereas no differences in diversity and richness were observed between conditions (Figure 3 B-C). Further analysis showed that significant genera (FDR-P < 0.05) that were different between the 3 and 14 day groups were mostly anaerobic strains (Figure 3D and Supplementary Table 1), while no differences in Campylobacter relative abundance were detected. To functionally dissect the microbial population within the CONV-Biota providing protection against C. jejuni infection, we cultured the CONV-Biota under aerobic, microaerobic or anaerobic conditions using Brain Heart Infusion (BHI) agar plates. GF Il10-/- mice were then colonized with the respective microbiota and 14 days later we infected the mice with a single dose of C. jejuni and then measured inflammation 12 days later. Consistent with previous observations,4 C. jejuni induced severe intestinal inflammation in GF Il10-/- mice (Figure 4A and B). Il10-/- mice pre-colonized with either aerobic (Aero-Biota) or microaerobic (Microaero-Biota) microbes also developed severe and comparable intestinal inflammation following C. jejuni-infection. In contrast, mice colonized with anaerobic microbes (Anaero-Biota) were protected against C. jejuni-induced intestinal inflammation, while C. jejuni luminal colonization level was comparable between mice colonized with Aero- and Anaero-Biota (Figure 4C). In addition, the protective effect was also observed in mice colonized with the three microbial groups pooled together. Consistent with colitis difference, C. jejuni induced stronger proinflammatory gene expression of Il1β, Cxcl2 and Il17a mRNA in colonic tissue of mice colonized with Aero-Biota compared to mice colonized with Anaero-Biota (Figure 4D).
Figure 3. Anaerobic bacteria are enriched in campylobacteriosis resistant mice.
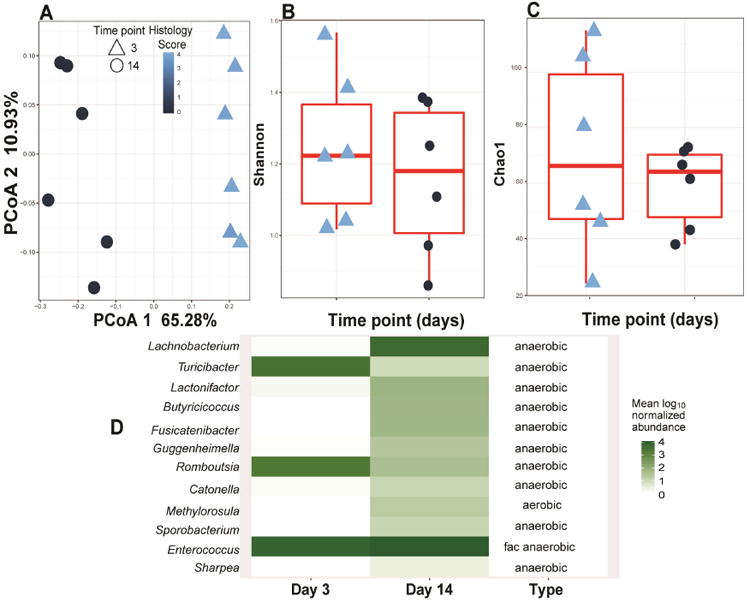
Stool samples from mice conventionalized for 3 or 14 days and then infected with C. jejuni were subjected to 16S rDNA sequencing 12 days after C. jejuni infection (see Supplementary Figure 3). (A) PCoA comparing the microbiome composition of C. jejuni infected mice conventionalized for 3 or 14 days. Histological inflammation scores are shown as color code. PCoA 1 FDR-P = 0.01 (linear mixed effect model) and 1.26e-05 (t-test) and cage FDR-P= 0.97 (linear mixed effect) (B) Shannon diversity and (C) Choa1 richness show no differences or correlation with histological inflammation scores (FDR-P > 0.05 linear mixed effect and t-test). Histological inflammation scores are shown as color code (D) Heatmap representation of genera significantly different (FDR-P < 0.05, t-test) between 3 days conventionalized and 14 days conventionalized Il10-/- mice prior to C. jejuni infection. Majority of the genera enriched in campylobacteriosis resistant mice are anaerobic. Fac anaerobic: facultative anaerobic.
Figure 4. Anaerobic microbiota isolated from CONV-Biota attenuates C. jejuni-induced colitis.
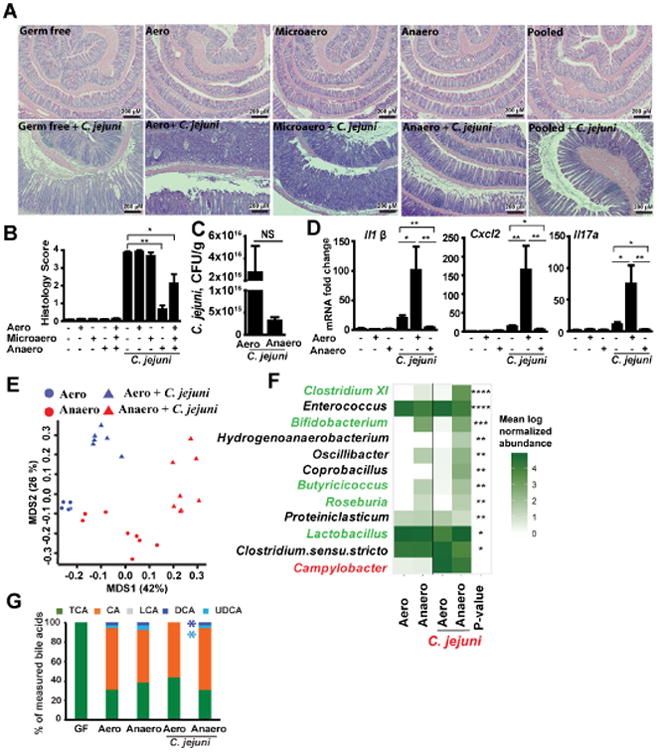
Cohorts of 4-8 GF Il10-/- mice were colonized with microbiota cultured under aerobic (Aero), microaerobic (Microaero), or anaerobic (Anaero) conditions, or all three groups pooled for 14 days. The mice were then gavaged with a single dose of 109 CFU C. jejuni/mouse and were euthanized 12 days post-infection. Stool samples from Anaero- and Aero-Biota mice were subjected to 16S rDNA sequencing and HPLC/MS analysis of bile acids. (A) H&E staining showing representative intestinal histology of C. jejuni-induced colitis in Il10-/- mice. (B) Quantification of histological intestinal damage score. (C) C. jejuni colonic luminal colonization level using culture in mice colonized with Aero- or Anaero-Biota. (D) Colonic Il1β, Cxcl2, and Il17a mRNA qPCR fold change relative to GF and normalized to Gapdh. (E) PCoA comparing Anaero- and Aero-Biota microbiome composition pre- and post-C. jejuni infection, based on 16S rDNA sequencing. (F) Heatmap representation of genera significantly different between Anaero-and Aero-Biota-colonized Il10-/- mice following C. jejuni infection, plus Campylobacter (red), which was not significant. Their abundance prior to infection is also shown. Green font indicates genera associated with anti-inflammatory response. Asterisks indicate the p-value after multiple hypothesis correction. (G) Relative stool bile acid profile measured by HPLC/MS. TCA, taurocholic acid and tauromuricholic acid; CA, cholate; LCA, lithocholic acid; UDCA, Ursodeoxycholic acid; DCA, deoxycholate. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05; NS, not significant. Scale bar is 200 μm. Results are representative of 3 independent experiments.
We then hypothesized that a select group of microbes within the Anaero-Biota play a protective role against campylobacteriosis. To investigate this possibility, we performed 16S rDNA sequencing using fecal samples of mice pre-colonized with Anaero- or Aero-Biota and infected with C. jejuni. PCoA analysis revealed distinct separation between Anaero- or Aero-Biota -reconstituted Il10-/- mice, and also between pre- and post- C. jejuni infection (Figure 4E). We further used t-test to compare Aero- and Anaero-Biota mice, and we found that the eight genera of Bifidobacterium, Clostridium XI, Butyricicoccus, Lactobacillus, Roseburia, Hydrogenoanaerobacterium, Coprobacillus, and Oscillibacter were significantly increased in the protected Anaero-Biota-colonized Il10-/- mice compared to susceptible Aero-Biota-colonized Il10-/- mice (Figure 4F, Supplementary Table 2), while two genera of Enterococcus and Clostridium sensu stricto were increased in susceptible Aero-Biota mice. Importantly, Campylobacter (red) relative abundance was not significantly different between Anaero- or Aero-Biota-colonized Il10-/- mice (Figure 4F), providing a culture-independent validation that C. jejuni colonization resistance is not the main mechanism by which Anaero-Biota protects against C. jejuni-induced colitis. Importantly, five (green color and Supplementary Table 2) of the eleven genera, significantly different between Aero- and Anaero-Biota are typically associated with an anti-inflammatory response27-30 and generate a range of metabolites including bile acid-derivatives and short chain fatty acids31-34. Limited or conflicted information is currently available on the role of the other six bacterial genera.
Beyond their role in digestion, bile acids participate in numerous physiological processes through their ability to activate various signaling pathways such as the farnesoid X receptor, the vitamin D receptor, and the pregnane X receptor.35 In addition, bile acids play an important anti-inflammatory role in the intestine.36 Because the genera Clostridium XI, Bifidobacterium, and Lactobacillus biotransform bile acids from conjugated (e.g. taurocholic acid, TCA) into primary (e.g. cholic acid, CA) and then to secondary forms (e.g. deoxycholic acid, DCA), we measured the five bile acid profile in the stool of GF, Aero-Biota or Anaero-Biota pre-colonized, C. jejuni-infected mice using HPLC/MS. In accordance with previous reports,37 bile acids of GF mice mainly consist of the conjugated bile acid TCA. Interestingly, DCA and UDCA, but not the primary bile acid (CA), were depleted in Aero-Biota colonized and C. jejuni-infected mice (Figure 4G, Supplementary Table 3), suggesting a potential protective role of these secondary bile acids in biota-mediated protection.
Microbial metabolite of secondary bile acid DCA protects against C. jejuni-induced colitis
We previously reported that rapamycin-treated Il10-/- mice do not develop campylobacteriosis, and observed that CONV-Biota inhibited C. jejuni-induced mTOR signaling in colonic tissue (Figure 2H and Supple Figure 4). Therefore, we test whether secondary bile acids, metabolites of the Anaero-Biota derived from CONV-Biota (Figure 4G), could block the important inflammatory signaling pathway. We showed that C. jejuni-induced mTOR signaling pathway in primary splenocytes isolated from Il10-/- mice 9, and we then used this cell system to test effect of CA and DCA on this pathway. Notably, secondary bile acid DCA but not primary bile acid CA inhibited C. jejuni-induced mTOR downstream target phosphor-p70S6K (Figure 5A). To functionally assess the potentially protective effect of secondary bile acids against C. jejuni-induced colitis, DCA, UDCA and LCA were gavaged to GF Il10-/- mice infected with C. jejuni. We also gavaged DCA to mice previously infected with C. jejuni for 5 days to evaluate treatment effect. Remarkably, both DCA prevention and treatment approaches reduced C. jejuni induced colitis (Figure 5 B and C) while C. jejuni luminal colonization level in DCA prevention mice was not significantly different from infection control mice (Figure 5D). Interestingly, Il10-/-mice exposed to UDCA and LCA (prevention) still developed colitis (Figure 6A and B). In addition, DCA strongly attenuated C. jejuni-induced proinflammatory gene expression of Il1β, Cxcl2 and Il17a mRNA in colonic tissue, while UDCA exacerbate the mRNA accumulation (Figure 6C). To further assess the protective role against campylobacteriosis, SPF Il10-/- mice were treated with clindamycin, an antibiotic especially efficient against anaerobic bacteria,19 including bile acid producing bacteria. We found that clindamycin-treated Il10-/- mice were susceptible to C. jejuni-induced colitis, whereas mice exposed to nalidixic acid, an antibiotic predominantly targeting gram negative bacteria,38 remained resistant to infection (Figure 7A and B). Using HPLC/MS analysis, we found that clindamycin treatment depleted all secondary bile acids, especially DCA, while nalidixic acid only reduced CA but failed to decrease DCA levels compared to SPF mice (Figure 7C, Supplementary Table 3).
Figure 5. The microbial metabolite deoxycholate inhibits mTOR activity and prevents and treats C. jejuni-induced colitis.
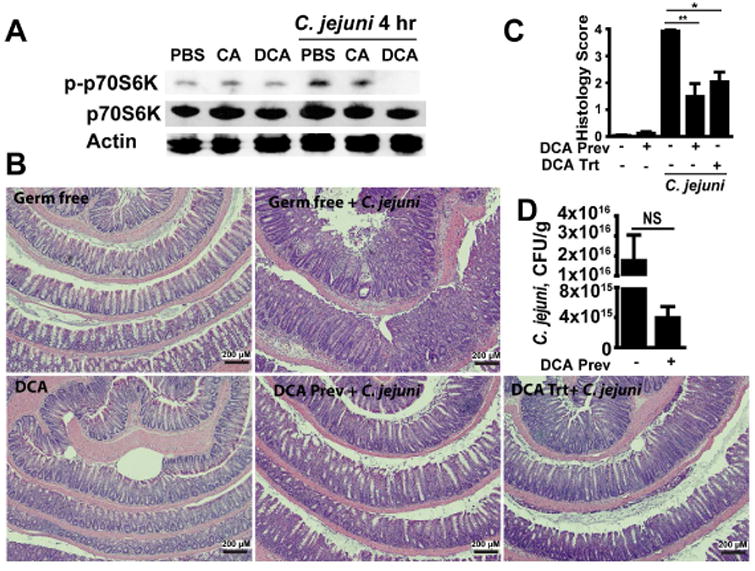
(A) Splenocytes isolated from Il10-/- mice were infected with C. jejuni (multiplicity of infection 50) and cultured in the presence of CA and DCA. Total and phosphorylated p70S6K (T389) and Actin was measured by Western Blot. (B) Cohorts of 4-7 GF Il10-/- mice infected with a single dose of 109 CFU C. jejuni/mouse were gavaged with the secondary bile acid DCA daily (DCA prevention (Prev)). For DCA treatment (DCA trt), mice infected with C. jejuni were gavaged with DCA on days 5-12 post-infection. H&E staining showing representative day 12 intestinal histology of C. jejuni-induced colitis in Il10-/- mice. (C) Quantification of histological intestinal damage score. (D) C. jejuni colonic luminal colonization level using culture. **, P < 0.01; *, P < 0.05; NS, not significant. Scale bar is 200 μm. Results are representative of 3 independent experiments.
Figure 6. Microbial metabolites of the secondary bile acids LCA and UDCA minimally impact C. jejuni-induced colitis.
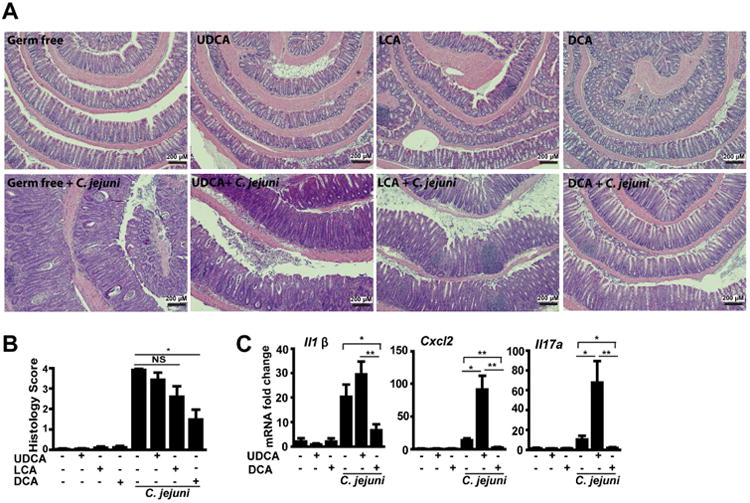
Cohorts of 4-7 GF Il10-/- mice infected with a single dose of 109 CFU C. jejuni/mouse were gavaged with the secondary bile acids UDCA, LCA or DCA daily. The mice were then euthanized 12-days after infection. (A) H&E staining showing representative intestinal histology of C. jejuni-induced colitis in Il10-/- mice. (B) Quantification of histological intestinal damage score. (C) Colonic Il1β, Cxcl2, and Il17a mRNA qPCR fold change relative to GF and normalized to Gapdh. **, P<0.01; *, P<0.05; NS, not significant. Scale bar is 200 μm. Results are representative of 3 independent experiments.
Figure 7. Targeted depletion of secondary bile acid-metabolizing microbiota promotes Il10-/- mouse susceptibility to C. jejuni.
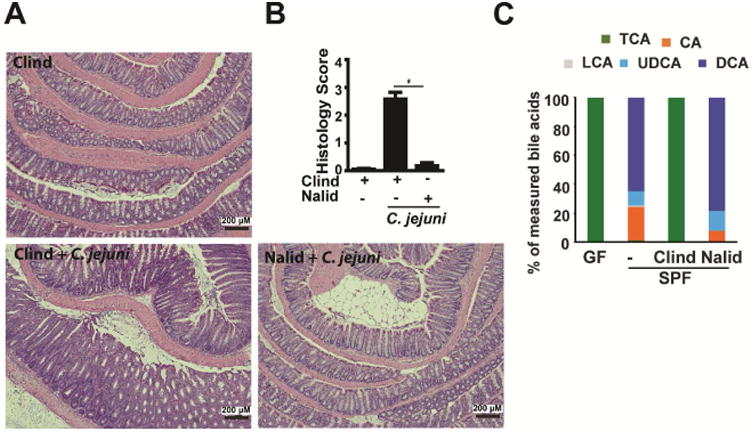
Cohorts of 4 SPF Il10-/- mice gavaged with the antibiotics clindamycin (Clind) or nalidixic acid (Nalid) for 7 days prior to infection with a single dose of 109 CFU C. jejuni/mouse. (A) H&E staining showing representative intestinal histology of C. jejuni-induced colitis in Il10-/- mice 21 days post-infection. (B) Quantification of histological intestinal damage score. (C) Relative stool bile acid profile measured by HPLC/MS (see Supplementary Table 4 for values). *, P<0.05. Scale bar is 200 μm. Results are representative of 3 independent experiments.
Discussion
Despite the prevalent role of Campylobacter jejuni in bacterial-derived diarrheal illness in developed countries, a paucity of information is available regarding host interaction with this important enteropathogen. Moreover, microbiota and cellular events implicated in host resistance/susceptibility to C. jejuni infection remain elusive.39, 40 Here we report that the commensal intestinal microbiota prevents C. jejuni-induced intestinal inflammation in ex-GF Il10-/- mice, an effect independent of pathogen luminal colonization level. Molecular and cellular analysis showed that the microbiota reduced C. jejuni-induced epithelial and lamina propria mTOR activation, inflammatory cytokine expression, neutrophil infiltration and C. jejuni invasion into colon tissue. Culture and fecal transplantation experiments identify anaerobic commensal microbiota as main protective contributors against C. jejuni infection. 16S rDNA sequencing in conjunction with HPLC/MS analysis identified DCA produced by a specific group of anaerobic bacteria as the mechanism protecting the host against C. jejuni infection. Direct supplementation of DCA to ex-GF mice protected against C. jejuni-induced colitis while targeted depletion of secondary bile acid-producers with the antibiotic clindamycin promoted campylobacteriosis in SPF Il10-/- mice. These findings pinpoint a novel mechanism by which biota-derived metabolites (e.g DCA) control resistance to C. jejuni infection by blocking activation of mTOR signaling.
A striking observation from our study is that microbiota-derived protection against C. jejuni pathogenesis operates independent of luminal colonization exclusion. Clearly, the presence of a conventionalized microbiota prevents C. jejuni invasion into intestinal mucosal tissues and MLN, but this response is not accompanied by a reduced level of luminal C. jejuni. This is in contrast with microbiota protection against C. difficile,19 Citrobacter rodentium41 and Salmonella typhimurium42 infections. In these models, colonization resistance is the main mechanism since the microbiota prevents pathogenic bacterial growth and reduced luminal colonization. Microbiota could control susceptibility to enteropathogen infection by targeting various host-mediated signaling pathways. Indeed, gut pathogens exploit various host signaling events to circumvent microbiota inhibitory effects. For example, S. typhimurium induces host-driven production of a new electron acceptor tetrathionate and uses the respiration to outgrow gut microbiota.43 Similarly, Escherichia coli utilizes nitrate generated during the inflammatory response to expand its niche in the gut lumen.44 These enteric pathogens “feed” on inflammation to gain a competitive growth advantage over an established commensal microbial community to break colonization resistance.43-46 Unlike these strategies, C. jejuni exploit mTOR signaling to again access and invade mucosal tissues and MLN, a process antagonized by microbiota through decreased mTOR activation and downstream targets p-p70S6K and p-S6 in both intestinal epithelial and lamina propria cells. It is not clear why C. jejuni targets or what the mechanism of mTOR signaling activation is. mTOR mediates innate signaling important in autophagy and bacterial clearance47 and targeting this pathway is expected to provide C. jejuni a survival advantage. We have previously shown that blocking mTOR signaling with rapamycin prevented C. jejuni-induced colitis and tissue invasion, although pathogen luminal load was not reduced.9 Our observation that the microbial metabolite DCA attenuated the C. jejuni-induced mTOR downstream target p-p70S6K in colonic tissues and immune cells clearly highlight the importance of mTOR in enteric infection. Interestingly, bacterial product-induced mTOR signaling is dysregulated in Il10-/- mice and lead to colitis, a phenomenon due to decreased expression of the mTOR inhibitor DNA-damage-inducible transcript 4 protein (DDIT4).48 Further investigation will be necessary to identify whether microbiota-derived DCA induced expression of an mTOR inhibitor or if other mechanisms are at play.
Microbial-derived metabolism is a relatively new concept for host-resistance/susceptibility to enteropathogen infection. For example, microbial-derived tryptophan catabolism leading to generation of aryl hydrocarbon receptor (AhR) ligand has emerged as an important part of host-microbe interaction in infectious diseases.49 Furthermore, bacterial-derived bile acid metabolites have been recently recognized as important modulators of C. difficile infection susceptibility.50 For example, anaerobic bacterium C. scindens transforms primary bile acids to secondary ones, which prevents C. difficile germination and growth,19 despite the bile acids' association with various chronic diseases. 20-22 In vitro, secondary bile acids LCA and DCA but not primary bile acid CA inhibit C. difficile vegetable growth and toxin production.19, 51 However, whether secondary bile acids prevent or treat CDI in human or animal models is still unknown. In our study, we directly supplemented secondary bile acids to C. jejuni-infected mice and observed that only the secondary bile acid DCA prevents and treats C. jejuni-induced colitis. Unlike C. difficile, C. jejuni growth is not impaired in medium containing DCA at concentrations as high as 0.2%, which is the upper limit of physiological range.52 Thus, the beneficial impact of DCA on campylobacteriosis is not mediated by impaired C. jejuni growth.
Taken together, our data reveal that the microbial metabolite secondary bile acid DCA attenuates campylobacteriosis through inhibition of C. jejuni-induced host inflammatory mTOR signaling. Our findings highlight the complex mechanism by which the microbiota controls host susceptibility to specific enteropathogen infection, and point to novel therapeutics approaches by targeting those microbial metabolites.
Supplementary Material
Supplemental Figure 1. Antibiotic cocktail depletes microbiota. Cohorts of 5 conventionally-derived Il10-/- mice were treated for 7 day with antibiotic. Stool samples were collected, serially diluted, and cultured on Brain Heart Infusion (BHI) agar for 48 hours under aerobic or anaerobic condition. Stool DNA was isolated and bacteria were estimated using real time PCR. (A) Live aerobic and anaerobic bacterial count on BHI. (B) Stool bacteria were estimated using PCR.
Supplemental Figure 2. C.jejuni-induced colitis in GF mice at 5- and 12-day post infection. Cohorts of 5-6 GF Il10-/- mice were infected with a single dose of 109 CFU C. jejuni/mouse and were euthanized 5 or 12 days post-infection. (A) Representative intestinal histology images. (B) Quantification of histological intestinal damage score. (C) C. jejuni was quantified using culture in stool and tissues of liver and MLN. (D) Colonic Il6, Cxcl1, and Cxcr2 mRNA qPCR fold change relative to GF and normalized to Gapdh at 5 and 12 days post-infection. All graphs depict mean ± SEM. *, P<0.05; NS, not significant. Scale bar is 200 μm. Results are representative of 3 independent experiments.
Supplemental Figure 3. GF Mice conventionalized for 14 days resist against C. jejuni-induced colitis. Cohorts of 5-6 GF IIl10-/- mice were transferred to SPF housing (conventionalization) for 3 or 14 days and were infected with a single dose of 109 CFU C. jejuni/mouse. The mice were euthanized 12 days post-infection. (A) Representative intestinal histology images. (B) Quantification of histological intestinal damage score. All graphs depict mean ± SEM. **, P<.01. Scale bar is 200 μm. Results are representative of 2 independent experiments.
Supplemental Figure 4. CONV-Biota attenuates C. jejuni-induced p-S6 positive cells in colon. Cohorts of 5-6 GF Il10-/- mice were orally gavaged a single dose of CONV-Biota for 14 days and were infected with C. jejuni as in Figure 2. The p-S6 positive cells (brown dots) in epithelial and lamina propria immune cells were detected using immunohistochemistry of anti-S6 antibody (S235/236).
Supplementary table 1. Statistical test results for genera in CONV-Biota
Supplementary table 2. Model results for all genera in Aero- and Anaero-Biota
Supplementary table 3. Bile acid profiles
Supplementary table 4. Bile acid statistical tests
Supplementary table 5. Aaero and Anaero Sequencing information
Acknowledgments
The authors would like to thank Dr. Ye Yang for anaerobic microbiota culture.
Grant Support: This research was supported by National Institutes of Health grants R01 DK47700 and from the University of Florida, Department of Medicine Gatorade Fund to C. Jobin; Arkansas Biosciences Institute grant to X. Sun and UF Health Cancer Center fund to R.Z. Gharaibeh. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- CONV-Biota
conventionalized microbiota
- Aero-Biota
aerobic CONV-Biota
- Anaero-Biota
anaerobic CONV-Biota
- DCA
Deoxycholic acid
- FISH
Fluorescence in situ hybridization
- IHC
immunohistochemistry
- mTOR
mammalian target of rapamycin
Footnotes
Transcript Profiling: Study sequence data are deposited in the National Center for Biotechnology Information Sequence Read Archive under access number PRJNA351834.
Author Contribution: X. S. and C. J. designed the experiments and wrote the manuscript with input from co-authors. X.S. performed animal and cell experiments and most analysis. K. W., R.Z.G. and A. F. performed microbiota inference modelling and analysis. J. G. performed 16S rDNA library and multiparallel sequencing. P. T. and S. B. performed quantification of bile acid species. Z. H performed immunohistochemistry staining. D.A contributed essential reagents.
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaakoush NO, Castano-Rodriguez N, Mitchell HM, et al. Global Epidemiology of Campylobacter Infection. Clin Microbiol Rev. 2015;28:687–720. doi: 10.1128/CMR.00006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaser MJ. Epidemiologic and clinical features of Campylobacter jejuni infections. J Infect Dis. 1997;176(Suppl 2):S103–5. doi: 10.1086/513780. [DOI] [PubMed] [Google Scholar]
- 3.van Spreeuwel JP, Duursma GC, Meijer CJ, et al. Campylobacter colitis: histological immunohistochemical and ultrastructural findings. Gut. 1985;26:945–51. doi: 10.1136/gut.26.9.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lippert E, Karrasch T, Sun X, et al. Gnotobiotic IL-10; NF-kappaB mice develop rapid and severe colitis following Campylobacter jejuni infection. PLoS One. 2009;4:e7413. doi: 10.1371/journal.pone.0007413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun X, Liu B, Sartor RB, et al. Phosphatidylinositol 3-kinase-gamma signaling promotes Campylobacter jejuni-induced colitis through neutrophil recruitment in mice. J Immunol. 2013;190:357–65. doi: 10.4049/jimmunol.1201825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janes MR, Fruman DA. Immune Regulation by Rapamycin: Moving Beyond T Cells. Science Signaling. 2009;2:pe25–pe25. doi: 10.1126/scisignal.267pe25. [DOI] [PubMed] [Google Scholar]
- 7.Weichhart T, Saemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Annals of the Rheumatic Diseases. 2008;67:iii70–iii74. doi: 10.1136/ard.2008.098459. [DOI] [PubMed] [Google Scholar]
- 8.Gomez-Cambronero J, Horn J, Paul CC, et al. Granulocyte-macrophage colony-stimulating factor is a chemoattractant cytokine for human neutrophils: involvement of the ribosomal p70 S6 kinase signaling pathway. J Immunol. 2003;171:6846–55. doi: 10.4049/jimmunol.171.12.6846. [DOI] [PubMed] [Google Scholar]
- 9.Sun X, Threadgill D, Jobin C. Campylobacter jejuni induces colitis through activation of mammalian target of rapamycin signaling. Gastroenterology. 2012;142:86–95 e5. doi: 10.1053/j.gastro.2011.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stahl M, Ries J, Vermeulen J, et al. A novel mouse model of Campylobacter jejuni gastroenteritis reveals key pro-inflammatory and tissue protective roles for Toll-like receptor signaling during infection. PLoS Pathog. 2014;10:e1004264. doi: 10.1371/journal.ppat.1004264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun X, Jobin C. Nucleotide-binding oligomerization domain-containing protein 2 controls host response to Campylobacter jejuni in Il10-/- mice. J Infect Dis. 2014;210:1145–54. doi: 10.1093/infdis/jiu148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deshmukh HS, Liu Y, Menkiti OR, et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med. 2014 doi: 10.1038/nm.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khosravi A, Yanez A, Price JG, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15:374–81. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukuda S, Toh H, Hase K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–7. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 15.Ubeda C, Djukovic A, Isaac S. Roles of the intestinal microbiota in pathogen protection. Clin Transl Immunology. 2017;6:e128. doi: 10.1038/cti.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–70. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Silverman MS, Davis I, Pillai DR. Success of self-administered home fecal transplantation for chronic Clostridium difficile infection. Clin Gastroenterol Hepatol. 2010;8:471–3. doi: 10.1016/j.cgh.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Buffie CG, Bucci V, Stein RR, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–8. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma H, Patti ME. Bile acids, obesity, and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 2014;28:573–83. doi: 10.1016/j.bpg.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernstein C, Holubec H, Bhattacharyya AK, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011;85:863–71. doi: 10.1007/s00204-011-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blackler RW, De Palma G, Manko A, et al. Deciphering the pathogenesis of NSAID enteropathy using proton pump inhibitors and a hydrogen sulfide-releasing NSAID. Am J Physiol Gastrointest Liver Physiol. 2015;308:G994–1003. doi: 10.1152/ajpgi.00066.2015. [DOI] [PubMed] [Google Scholar]
- 23.Uronis JM, Arthur JC, Keku T, et al. Gut microbial diversity is reduced by the probiotic VSL#3 and correlates with decreased TNBS-induced colitis. Inflamm Bowel Dis. 2011;17:289–97. doi: 10.1002/ibd.21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korlath JA, Osterholm MT, Judy LA, et al. A point-source outbreak of campylobacteriosis associated with consumption of raw milk. J Infect Dis. 1985;152:592–6. doi: 10.1093/infdis/152.3.592. [DOI] [PubMed] [Google Scholar]
- 25.Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scherer M, Gnewuch C, Schmitz G, et al. Rapid quantification of bile acids and their conjugates in serum by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3920–5. doi: 10.1016/j.jchromb.2009.09.038. [DOI] [PubMed] [Google Scholar]
- 27.Underwood MA, Arriola J, Gerber CW, et al. Bifidobacterium longum subsp. infantis in experimental necrotizing enterocolitis: alterations in inflammation, innate immune response, and the microbiota. Pediatr Res. 2014;76:326–33. doi: 10.1038/pr.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sgouras DN, Panayotopoulou EG, Martinez-Gonzalez B, et al. Lactobacillus johnsonii La1 attenuates Helicobacter pylori-associated gastritis and reduces levels of proinflammatory chemokines in C57BL/6 mice. Clin Diagn Lab Immunol. 2005;12:1378–86. doi: 10.1128/CDLI.12.12.1378-1386.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eeckhaut V, Machiels K, Perrier C, et al. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut. 2013;62:1745–52. doi: 10.1136/gutjnl-2012-303611. [DOI] [PubMed] [Google Scholar]
- 30.Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–83. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 31.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–59. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Kitahara M, Takamine F, Imamura T, et al. Clostridium hiranonis sp. nov. a human intestinal bacterium with bile acid 7alpha-dehydroxylating activity. Int J Syst Evol Microbiol. 2001;51:39–44. doi: 10.1099/00207713-51-1-39. [DOI] [PubMed] [Google Scholar]
- 33.Eeckhaut V, Van Immerseel F, Teirlynck E, et al. Butyricicoccus pullicaecorum gen. nov., sp. nov. an anaerobic, butyrate-producing bacterium isolated from the caecal content of a broiler chicken. Int J Syst Evol Microbiol. 2008;58:2799–802. doi: 10.1099/ijs.0.65730-0. [DOI] [PubMed] [Google Scholar]
- 34.Duncan SH, Aminov RI, Scott KP, et al. Proposal of Roseburia faecis sp. nov., Roseburia hominis sp. nov. and Roseburia inulinivorans sp. nov. based on isolates from human faeces. Int J Syst Evol Microbiol. 2006;56:2437–41. doi: 10.1099/ijs.0.64098-0. [DOI] [PubMed] [Google Scholar]
- 35.Copple BL, Li T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res. 2016;104:9–21. doi: 10.1016/j.phrs.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duboc H, Rajca S, Rainteau D, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut. 2013;62:531–9. doi: 10.1136/gutjnl-2012-302578. [DOI] [PubMed] [Google Scholar]
- 37.Swann JR, Want EJ, Geier FM, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4523–30. doi: 10.1073/pnas.1006734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crumplin GC, Smith JT. Nalidixic acid: an antibacterial paradox. Antimicrob Agents Chemother. 1975;8:251–61. doi: 10.1128/aac.8.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luethy PM, Huynh S, Ribardo DA, et al. Microbiota-Derived Short-Chain Fatty Acids Modulate Expression of Campylobacter jejuni Determinants Required for Commensalism and Virulence. MBio. 2017;8 doi: 10.1128/mBio.00407-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kampmann C, Dicksved J, Engstrand L, et al. Composition of human faecal microbiota in resistance to Campylobacter infection. Clin Microbiol Infect. 2016;22:61 e1–61 e8. doi: 10.1016/j.cmi.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Willing BP, Vacharaksa A, Croxen M, et al. Altering host resistance to infections through microbial transplantation. PLoS One. 2011;6:e26988. doi: 10.1371/journal.pone.0026988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bohnhoff M, Drake BL, Miller CP. The effect of an antibiotic on the susceptibility of the mouse's intestinal tract to Salmonella infection. Antibiot Annu. 1955;3:453–5. [PubMed] [Google Scholar]
- 43.Winter SE, Thiennimitr P, Winter MG, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–9. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winter SE, Winter MG, Xavier MN, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339:708–11. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiennimitr P, Winter SE, Winter MG, et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 2011;108:17480–5. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lupp C, Robertson ML, Wickham ME, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–29. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 47.Abdel-Nour M, Tsalikis J, Kleinman D, et al. The emerging role of mTOR signalling in antibacterial immunity. Immunol Cell Biol. 2014;92:346–53. doi: 10.1038/icb.2014.3. [DOI] [PubMed] [Google Scholar]
- 48.Ip WKE, Hoshi N, Shouval DS, et al. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513–519. doi: 10.1126/science.aal3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romani L, Zelante T, De Luca A, et al. Microbiota control of a tryptophan-AhR pathway in disease tolerance to fungi. Eur J Immunol. 2014;44:3192–200. doi: 10.1002/eji.201344406. [DOI] [PubMed] [Google Scholar]
- 50.Abt MC, McKenney PT, Pamer EG. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol. 2016;14:609–20. doi: 10.1038/nrmicro.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–12. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lertpiriyapong K, Gamazon ER, Feng Y, et al. Campylobacter jejuni type VI secretion system: roles in adaptation to deoxycholic acid, host cell adherence, invasion, and in vivo colonization. PLoS One. 2012;7:e42842. doi: 10.1371/journal.pone.0042842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Antibiotic cocktail depletes microbiota. Cohorts of 5 conventionally-derived Il10-/- mice were treated for 7 day with antibiotic. Stool samples were collected, serially diluted, and cultured on Brain Heart Infusion (BHI) agar for 48 hours under aerobic or anaerobic condition. Stool DNA was isolated and bacteria were estimated using real time PCR. (A) Live aerobic and anaerobic bacterial count on BHI. (B) Stool bacteria were estimated using PCR.
Supplemental Figure 2. C.jejuni-induced colitis in GF mice at 5- and 12-day post infection. Cohorts of 5-6 GF Il10-/- mice were infected with a single dose of 109 CFU C. jejuni/mouse and were euthanized 5 or 12 days post-infection. (A) Representative intestinal histology images. (B) Quantification of histological intestinal damage score. (C) C. jejuni was quantified using culture in stool and tissues of liver and MLN. (D) Colonic Il6, Cxcl1, and Cxcr2 mRNA qPCR fold change relative to GF and normalized to Gapdh at 5 and 12 days post-infection. All graphs depict mean ± SEM. *, P<0.05; NS, not significant. Scale bar is 200 μm. Results are representative of 3 independent experiments.
Supplemental Figure 3. GF Mice conventionalized for 14 days resist against C. jejuni-induced colitis. Cohorts of 5-6 GF IIl10-/- mice were transferred to SPF housing (conventionalization) for 3 or 14 days and were infected with a single dose of 109 CFU C. jejuni/mouse. The mice were euthanized 12 days post-infection. (A) Representative intestinal histology images. (B) Quantification of histological intestinal damage score. All graphs depict mean ± SEM. **, P<.01. Scale bar is 200 μm. Results are representative of 2 independent experiments.
Supplemental Figure 4. CONV-Biota attenuates C. jejuni-induced p-S6 positive cells in colon. Cohorts of 5-6 GF Il10-/- mice were orally gavaged a single dose of CONV-Biota for 14 days and were infected with C. jejuni as in Figure 2. The p-S6 positive cells (brown dots) in epithelial and lamina propria immune cells were detected using immunohistochemistry of anti-S6 antibody (S235/236).
Supplementary table 1. Statistical test results for genera in CONV-Biota
Supplementary table 2. Model results for all genera in Aero- and Anaero-Biota
Supplementary table 3. Bile acid profiles
Supplementary table 4. Bile acid statistical tests
Supplementary table 5. Aaero and Anaero Sequencing information