

Abstract
Macrophages are present in all vertebrate tissues and have emerged as multifarious cells with complex roles in development, tissue homeostasis and disease. Macrophages are a major constituent of the tumor microenvironment, where they either promote or inhibit tumorigenesis and metastasis, depending on their state. Successful preclinical strategies to target macrophages for anti-cancer therapy are now being evaluated in the clinic and provide proof-of-concept that this strategy may enhance current therapies; however, clinical success has been limited. This review discusses the promise of targeting macrophages for anti-cancer therapy, yet highlights how much is unknown regarding their ontogeny, regulation and tissue specific diversity. Further work might identify subsets of macrophages within different tissues, which could reveal novel therapeutic opportunities for anti-cancer therapy.
Keywords: Tumor associated macrophages, Tumor microenvironment, Novel anti-cancer therapy, CSF-1R
Tumor macrophages in the driver’s seat
The immune system is composed of both adaptive and innate immune cells, which are essential to coordinate effective immune responses to protect the host from invading pathogens, bacteria, malignant cells and to maintain homeostasis. Innate immune cells such as macrophages and dendritic cells (DCs) are professional antigen presenting cells (APCs) and are the first line of defense for the host, where they engulf and present foreign material in major histocompatibility complex (MHC) molecules classes I and II to T cells. T cell activation is complex, requiring activation signals from antigen presented by APCs as well as appropriate co-stimulatory molecules and cytokine signaling [1]. Upon T cell activation T cells proliferate and acquire effector function, which includes the production of effector molecules such as perforin and granzymes, which mediate cytotoxicity. Macrophages and T cells carefully regulate each other, as errors in this communication can lead to immunodeficiency, damage to host tissue in the form of autoimmunity, cancer and failure to destroy invading pathogens.
A variety of non-malignant cells including cells of both the innate and adaptive immune system make up the complex TME and generally support tumor growth and metastasis. Thus, targeting tumor-promoting immune cells has been of great clinical interest. Indeed, targeting the body’s own immune system, referred to as immunotherapy, has profoundly changed the way we think about and treat cancer and has shown great promise for the treatment of a number of human cancers (Box 1). Tumor associated macrophages (TAMs) have long been recognized as central regulators of the complex tumor microenvironment where while they general exhibit pro-tumor functions by promoting tumor survival, proliferation, angiogenesis, and dissemination [10–16], they can also be potent effector cells and recruit cytotoxic T lymphocytes (CTLs) to activate anti-tumor adaptive immune responses. This review will focus on the need to better characterize TAMs with the goal of being able to identify novel therapeutic strategies, but should not undermine the complexity of the TME and need for comprehensive profiling of the TME as a whole. Elucidating the intricacy of TAMs is essential, but will reveal only one piece of the puzzle.
Box 1. The Advent of Immunotherapy.
Immunotherapy dates back to at least 1890 when William Coley first observed that patients with inoperable sarcoma who had suffered a Streptococcus pyogenes infection obtained complete remission [2]. Coley went on to treat thousands of patients with mixtures, called Coley’s Toxins, of killed S. pyogenes and Serratia marcescens, achieving a complete remission in 10% of treated patients. This can impressively be compared to 5-year survival rates for metastatic sarcoma of 20% [3]. While the mechanisms of Coley’s toxins were not completely known at the time, it is now known that macrophages and dendritic cells acted as APCs to present foreign bacterial antigen to naïve T cells, initiating T cell priming and activation, leading to an anti-tumor immune response [4]. The advent of radiotherapy and use of chemotherapy limited the enthusiasm for Coley’s toxins, especially given its the high mortality rate. The history of immunotherapy has eloquently been reviewed [4, 5]. Currently there are over 2,004 immunomodulatory agents (940 in clinical stage and 1064 in preclinical stage) against 303 targets, from 864 companies being tested [6] highlighting the optimism of immunotherapy. T cell intrinsic immunotherapy such as anti-PD-1, anti-CTLA-4, and anti-PD-L1 has revolutionized cancer therapy – however only a subset of patients achieve durable clinical responses [7–9]. Resistance mechanisms are poorly understood and current biomarkers are lacking. Imperative to understanding resistance and developing biomarkers of response is to better understand the immune profile of tumors – including fully characterizing immune cells within the TME.
A significant pharmaceutical effort has been placed on targeting TAMs for anti-cancer therapy with pre-clinical and clinical outcomes holding great promise and has been reviewed recently [5, 17]. However, as our knowledge of macrophage biology continues to expand, the complex phenotypic and functional properties of macrophages are becoming apparent including the diversity of macrophage ontogeny, phenotype and tissue specific transcriptional regulation. Further understanding of the extent of which these factors influence macrophages in tissue during cancer will reveal new opportunities for cancer immunotherapy.
Macrophage ontogeny
The long and winding roads
Macrophages constitute 5–20% of the cells in every tissue of the body [18–21] and contribute to homeostatic tissue behaviors, disease and the tumor microenvironment [22]. There are various types of macrophages in tissues, which arise from at least 3 distinct sources, the embryonic yolk sac (YS), the fetal liver (FL) and the bone marrow (BM). TRM are initially derived from both the YS and FL and reach their tissues during embryonic development, persist into adulthood, and self-maintain through proliferation independent of hematopoietic stem cells (HSCs; Figure 1) [23–25]. In mice, the mononuclear phagocytic system (MPS) starts to develop at embryonic day 8 from the primitive ectoderm of the YS and gives rise to macrophages that do not have a monocytic progenitor. This primitive system is followed by definitive hematopoiesis in the FL, which is initially seeded by hematopoietic progenitors from the YS and subsequently from the hematogenic endothelium of the aorta-gonadal-mesonephros region of the embryo. Embryonic macrophages have an essential role in clearing areas of programmed cell death during development [26]. As postnatal formation of BM hematopoiesis begins, FL hematopoiesis declines and the HSC in the BM and spleen become the definitive source of circulating monocytes (Figure 1) [27]. YS-derived progenitors can be identified as lymphocyte antigen 6 complex (Ly6C)negative whereas HSC-derived monocytes can be identified by Ly6Chigh or Ly6Cnegative/low [28–30]. In contrast to YS-derived progenitors, bone-marrow derived macrophages (BDM) have a short life and give rise in the tissue only under inflammatory conditions (Table 1) [31].
Figure 1. The road of macrophage ontogeny in tissue.

Macrophages in tissues arise from at least 3 distinct sources. During primitive hematopoiesis macrophages arise from the YS independent of HSC precursors and populate the brain, constituting the major brain macrophage lineage, microglia. Additionally, the fetal liver is populated with YS-derived macrophages and become the major source of tissue resident macrophages (TRM) in the lung, spleen, liver, pancreas and kidney. Bone-marrow derived macrophages (BDM) constitute yet a third source of macrophages, which can populate all major organs upon homeostatic adaptations, infection, disease or cancer.
Table 1. Macrophages in tissue arise from at least 3 distinct origins.
Elegant fate mapping and genetic studies have revealed progenitors for tissue specific macrophages along with unique markers and transcriptional regulation [1–5].
| Tissue Resident Macrophages (TRM) | Bone-Marrow Derived Macrophages (BDM) | ||
|---|---|---|---|
| Progenitor | Early EMP | Late EMP | HSC |
| Source | Yolk sac | Yolk Sac to Fetal Liver | Bone marrow |
| Tissue | Brain (microglia) | Lung (alveolar) Liver (Kupffer) Spleen Skin (Langerhans) |
Intestinal tract, adipose tissue |
| Markers | Ly6CnegativeF480high | F480high | Ly6Chigh/low/negF480low CD11bhigh F4/80low |
| Longevity | Long | Long | Short |
| Proliferation | Upon insult/infection | Upon insult/infection | minimal |
| Transcriptional regulation dependency | Runx1 present, but not dependent PU.1 |
PU.1 Runx1 c-myb Notch 1 |
PU.1 Runx1 c-myb Notch 1 |
| Expression | CSF-1R | CSF-1R | Flt3 |
Lineage specific markers and transcriptional regulators that distinguish TRM and BDM have been identified (Table 1). Elegant genetic experiments using colony stimulating factor 1 receptor (CSF-1R) Csf1rMer-iCre-Mer;RosaLSL-YFP mice treated with OH-tamoxifen and progesterone at E8.5, in order to pulse-label Csf1r-expressing YS macrophages, provide evidence that CSF-1R can be a robust marker of YS-derived TRM [25]. This study revealed YFP labelled F480bright macrophages in all tissues examined: liver, skin, brain, pancreas, lung, spleen, and kidney; whereas YFP expression was not detected in the typical progeny of adult HSCs, such as blood leukocytes, or in tissue expressing the cluster of differentiation (CD) CD11bhigh F4/80low macrophages [25]. Further studies have shown that YS-derived macrophages are functionally dependent on CSF-1R, as CSF1R-deficient mice lack YS-derived microglia but retain monocytes [32] and selectively inhibiting the CSF-1R results in elimination of more than 99% of all microglia brain-wide [33]. To further examine the contribution of primitive myeloid precursors, mice that express the tamoxifen-inducible MER-Cre-MER recombinase gene under the control of an endogenous promoter of the runt-related transcription factor 1 (Runx1) locus were used for lineage tracing studies. Runx1+ progenitors migrate from the YS to the brain between E8.5 and E9.5 and serve as the cells of origin for microglia [32]. Treatment of pregnant Runx1-MER-Cre-MER mice (Runx1Cre/wt) mice crossed with the Cre-reporter mouse strain Rosa26R26R-eYFP/R26R-eYFP (enhanced yellow fluorescent protein) at E8.5–9.5 with 4-hydroxytamoxifen (4′OHT) further confirms that microglia arise from primitive myeloid progenitors early in embryonic development by YS precursors rather than BDM [32]. Alternatively, BDM but not YS-derived macrophages have a selective requirement for the gene c-myb in their development [25, 34]. Mice lacking c-Myb maintained tissue resident macrophages [35], providing evidence of the third precursor, the FL-derived TRM [24]. The cellular ontogeny and selective transcription factor dependence adds previously unrecognized complexity to tissue resident macrophages of different tissues. However, there is still little known regarding the individual contributions of TRMs and BDMs in disease and cancer, and additionally, how targeting one of these populations over another might affect cancer outcomes and/or response to therapy.
In adult tissues, it was previously thought that YS-derived TRM are terminally differentiated, non-dividing cells based on the original MPS model [36, 37]. However, emerging data indicates that under different conditions TRM are capable of proliferation and self-renewal. TRM in the brain (microglia) and of the skin (Langerhans cells) can proliferation and expand during development, infection and following recovery from acute inflammation [38–40]. To maintain homeostatic programs microglia do not require an ongoing input from circulating monocytes and can divide as needed to maintain appropriate numbers at their location [40–42]. In the pleural and peritoneal cavities after infection with the nematode Litomosoides sigmodontis or Brugia malayi, respectively, TRM are capable of proliferation and self-renewal, which was dependent on IL-4 [40]. Interestingly, IL-4 can induce local macrophage proliferation in the peritoneal cavity, pleural cavity, and liver without the requirement for infection, indicating that IL-4-mediated local proliferation can maintain tissue-macrophage homeostasis independent of bone marrow progenitors [40]. In other tissues, macrophages deriving from different developmental origins coexist, such as macrophages of the dermis [42, 43], intestine [44, 45], and a subpopulation of the heart [22], which require BM-derived precursors to maintain their pools. During homeostatic adaptations, macrophages of different phenotypes can also be recruited from the monocytes reservoirs of blood, spleen and bone marrow [27], but also from resident progenitors or through local proliferation [25, 40, 46, 47]. Which prompts the question, Are YS-derived TRMs and their BDMs replacements under pathological conditions functionally equivalent?
Diversity of tissue resident macrophages
Arriving at the destination
TRM have multiple functions in homeostasis, host protection and tissue injury, which are maintained at a fine balance as deregulation can lead to inflammation and disease as outlined in Table 2. Studies of gene-expression and enhancer activity of TRM have revealed distinct tissue-specific transcriptional and epigenetic programs highlighting their specialized tissue specific functions [48–51]. Transcriptional profiling of murine tissue macrophages isolated from the peritoneum, lung, splenic red pulp and the brain reveal there is high transcriptional diversity with minimal overlap. Only a few macrophage mRNA transcripts had uniform expression across the 4 tissue specific macrophage populations; for example, MerTK, which is involved in the phagocytosis of apoptotic cells, as well as mRNA encoding CD14, CD64, and Toll-like receptors (TLR)-4, TLR7, TLR8 and TLR13 [49]. However, hundreds of mRNA transcripts had a selective difference in expression of at least two-fold (higher or lower) expression in only one macrophage population, each with different predictive functions (metabolism, signaling, and interferon responsiveness) [49]. Notably, expression of gene families that were previously associated with macrophage function (those encoding chemokine receptors, TLRs, C-type lectin and efferocytic receptors) were considerably different among TRM [49]. In addition to the cellular ontogeny of TRM, the transcriptional diversity further sheds light on their complexity. Moving forward, given that this analysis was performed on cells recovered from uninfected mice, one would speculate that macrophages recovered from tumors would greatly amplify the transcriptional diversity both within tissues and between tissues. In fact, there are likely to be differences even within tumor types, depending on a multitude of factors including genetic drivers of disease, host-specific considerations including lifestyle, age, menopause status, as well as immune-related basis for disease. In fact, it is likely that while some macrophage populations may be similar between disease or among tumor type, it is also possible that revealing the diversity of macrophages on a personalized level will reveal great opportunity for anti-cancer therapy.
Table 2.
Tissue macrophages in homeostasis and their tissue specific functions.
| Source | Function | Consequences in disease |
|---|---|---|
| Circulating monocytes |
|
|
| Brain (microglia) |
|
|
| Bone (osteoclasts) |
|
|
| Adipose tissue |
|
|
| Heart |
|
|
| Liver (Kupffer) |
|
|
| Lung (alveolar) |
|
|
| Spleen |
|
|
| Testis |
|
|
Tissue macrophages play fundamental roles in regulating tissue homeostasis, disease, infection and cancer. The function of tissue macrophages in each tissue is specific for each tissue type. When macrophages are dysfunctional there are consequences in disease (column 3).
Diversity and cellular ontogeny of tumor macrophages
Do tumor macrophages follow the same road map as TRM or BDM?
Studies of the cellular ontogeny, diversity and tissue specificity of macrophages have revealed tissue specific disparities, which provokes important questions regarding whether TAMs may also be highly diverse between tissues and/or have broad cellular ontogeny. Recent evidence has emerged that in some cancers, BDM are recruited to the tumor and promote tumorigenesis whereas in other tissues, TRM proliferate and promote tumor growth and metastasis (Figure 2), as discussed below. In a pre-clinical mouse model of breast cancer, the loss of resident macrophages and a concomitant increase in BDM during tumorigenesis has been reported [52]. Similarly, in the Lewis lung carcinoma (LLC) model of non-small cell lung cancer and the genetic KrasLSL-G12D/+p53fl/fl model, the majority of TAMs are derived from BDM [53, 54]. On the other hand, in the brain, YS-derived, tissue resident microglia are highly prevalent in mouse brain tumor models [55] and significant proportions of YS-derived pancreas-resident macrophages expanded through in situ proliferation during tumor progression in murine pancreatic ductal adenocarcinoma (PDAC) models [56]. Using several different eloquent fate mapping techniques, where only BDM are labeled in one setting and only TRM are labelled in another, it was recently shown that both brain tumors (primary and metastatic) and PDAC are infiltrated with both TRM and BDM, however in both cases the TRM were the tumor promoting cell [55, 57]. During progression of pancreatic tumors, BDM played more potent roles in antigen presentation, whereas TRM exhibited a pro-fibrotic transcriptional profile, indicative of their role in producing and remodeling molecules in the extra cellular matrix [56]. The murine pancreatic TRM displayed an increase in CXCR4, a decrease of MHCII expression and a pro-fibrotic gene signature, which was validated in human pancreas TAMs, suggesting embryonic derived TAMs may play a role in human pancreatic cancer progression [56]. Analysis of TAM transcriptomes by RNA sequencing revealed that brain TAMs, regardless of if they originated from TRM or BDM, were transcriptionally distinct from normal microglia and blood monocytes. This is consistent with the inherent plasticity of the macrophage lineage, in which TAMs can undergo significant in situ programing in the TME [58]. Similar to the PDAC BDM TAMs, the BDMs in brain tumors had higher expression of gene transcripts involved with antigen presentation and T cell co-stimulation, compared to TRM-derived TAMs. The BDM and TRM-derived TAMs in brain tumors could be distinguished using the integrin ITGA4 (CD49d), which was expressed in the BDM but not TRM-derived TAMs.
Figure 2. The cellular ontogeny of TAMs.

TRM are derived from yolk-sac and fetal liver progenitors, which can proliferate in some tumors and have been shown to promote tumorigenesis in brain and pancreatic cancer. Hematopoietic stem cells of the bone marrow give rise to monocytes which can populate tumors and have been shown to promote tumorigenesis in breast and lung cancer.
Selectively targeting TAMs derived from either TRM or BDM may be a promising strategy for anti-cancer therapy and this will likely be different for each tissue. Indeed, through selective depletion of pancreas and glioblastoma-TRM and not BDM, there is clear evidence that the pancreas-TRM are more potent drivers of PDAC progression than their BDM counterparts [55, 57]. This is in contrast to the murine breast cancer models where depletion of HSC-derived BDM inhibits tumor growth and metastasis [52]. These reports highlight that macrophages may be programmed differently within the tumor microenvironment of different tissues and creates opportunity to fine tune therapy directed at modulating macrophages for anti-cancer therapy.
Lost on the highway: TAM characterization has suffered from lack of understanding of functional markers
Unlike T cells, which have clearly defined markers of activation and exhaustion, macrophages modulate expression of several markers simultaneously in response to their environment, which can change with further insult by local cytokines, hypoxia, necrosis and/or metabolites. Macrophages are currently identified clinically on the basis of expression of CD68 or CD163. Currently, the intracellular glycoprotein CD68 is widely used in clinical studies as a TAM marker, but it has recently also been shown to detect other cell types such as granulocytes, dendritic cells, fibroblasts, endothelial cells and some lymphoid subsets [59–63] and does not identify the phenotype or functional status of the macrophage. The scavenger receptor CD163 has also been used to identify some TAM populations. Cell surface expression of CD68 and CD163 have been used to show that a high density of TAMs correlates with a worse clinical outcome in most solid cancers such as breast, thyroid, head and neck, liver, bladder, kidney, pancreatic, ovarian, oral, endometrial and lung cancer as well as Hodgkin lymphoma [10, 64, 65]. By contrast, high TAM density has been associated with survival in colon [66], gastric [67] and endometrial [68] cancer. In some cancer types, CD68 could only be used to correlated to poor survival when combined with additional markers CD206, CD163 and CD204 [64, 69, 70]. The ambiguity of true macrophage markers has limited our ability to identify and characterize macrophages in human tumors and has limited our understanding of macrophages in disease progression.
Macrophages have the ability to rapidly change phenotype based on the environment, especially within tumors, which has made it difficult to use one or even two markers to functionally characterize macrophage within tumors. Recently, there have been several efforts to more precisely characterize macrophages in human tumors by employing the use of multiple markers. The use of multiple markers by either mass cytometry or multi-plex immunofluorescence further supports that TAM density correlates with more aggressive subtypes of cancer as well as worse clinical outcome [71, 72]. In addition, the diversity of TAMs has been further exposed. Using mass cytometry, 17 distinct TAM subsets were identified in renal cell carcinoma, which included blood monocytes, BM-derived macrophages and tissue resident macrophages [73]. Four populations of macrophages that correlated with grade II or higher disease expressed the highest expression of HLA-DR, CD68, and CD64 with no expression of CD11b or CD36 suggesting a mature macrophage phenotype. Interestingly these populations expressed combinations of previously classified “pro”-(CD163, CD204, and CD206) and “anti”-tumor (CD169, CD38) markers. While those markers have been broadly used in the past to distinguish macrophage phenotype, here they define distinct macrophage subsets, providing evidence that tumor macrophages can express both “pro”- and “anti”-tumor markers [73]. Further evidence for the diversity of TAM subsets comes from stage I early adenocarcinomas where 4 distinct macrophage and 2 monocyte populations were identified [74]. TAMs displayed a distinct transcriptional signature to that of tissue resident macrophages from matched normal lung tissue and showed an increase in TREM2, CD81, MARCO and APOE, PPARγ, CD64, CD14, and CD11c; and lower CD68 and CD206 expression. Using the TCGA database patients with this gene signature had a significant survival disadvantage [74]. Similar analysis in other cancer types is warranted and will likely provide further evidence that TAMs are more heterogenous and complex than previously thought. The complexity of TAMs prompts the questions: Which TAM subsets play a role in promoting tumorigenesis and which subtypes should be targeted for anti-cancer therapy?
Efforts to characterize the functional phenotype of TAM subsets will likely reveal novel therapeutic targets for anti-cancer therapy. To date, the difficulty in characterizing TAM phenotype and diversity has likely stemmed from bulk analysis methods which we and others have performed on total macrophage populations and/or have relied on only a handful of preselected markers [71, 73–76]. New, unbiased large-scale efforts for in-depth characterization of macrophages in human tumors should be of tremendous importance and interest. Preselected panels of antibodies used to characterize TAMs have already expanded our view and highlighted the diversity of TAMs in different tissues, however relevant phenotypic and functional biology may be lost due to limitations in selected antibodies. Therefore, approaches such as single cell RNA-sequencing may address TAM complexity by providing an unbiased classification of cells based on transcriptional profiles [77–79] and may be informative to guide future antibody panel selection; but is likely to be specific for each tumor type and potentially for individual patients even with the same tumor type. It will be critical to extrapolate findings from global transcriptomic and elegant fate-mapping studies to characterize TAM populations to guide future strategies that aim to optimize TAM modulation for anti-cancer therapy. In addition, a priority should be put on phenotypic characterization of TAM subsets, which is likely to be revealed utilizing unbiased characterization techniques. Macrophage phenotype classification has largely relied on the M1–M2 (anti- and pro-tumor) paradigm, which has largely skewed our perception of macrophage phenotype in vivo (table 3). The M1–M2 classification defines macrophages stimulated ex vivo without environmental influence: M0 (unstimulated); M1 (IFNγ+LPS), M2a (IL4), M2b (IFNγ+complexed Ig), M2c (dexamethasone) [80, 81]. Additional cytokines and stimuli have been reported to skew monocytes ex vivo towards either “M1” or “M2” macrophages and has been reviewed [82]. However, in vivo macrophages do not so neatly divide into the M1 and M2 classification schemes. Indeed, more than 60% of the upregulated genes in TAMs from brain tumors did not overlap with the M1, M2a, M2b, M2c ex vivo macrophage phenotypes, indicating ex vivo stimulation of monocytes may not be a true reflection of macrophages that are stimulated in the tumor microenvironment [81]. While TAMs generally fall much closer to that of M2-polarized macrophages the M1 and M2 characterization should not be used to characterize macrophages in vivo, there should be an effort to move away from the ambiguity of the M1–M2 characterization of TAMs. It will take enormous efforts to identify subsets of macrophages within tumors and coordinate these findings between patients with similar tumors as well as between tumor types, some of these efforts are described below. For now, characterizing TAMs based on functional, transcriptional or epigenetic status should lead to less ambiguous TAM characterization and direct new studies aimed at understanding the biology and characterization of TAMs.
Table 3. General markers of pro- and anti-tumor macrophages.
However, it will be important to use several markers from each category to distinguish the phenotype of TAMs. RNI – Reactive nitrogen intermediates.
| Pro-tumor | Anti-tumor | General |
|---|---|---|
| CD163, CD204, CD206, PD-L1, Arginase-I, YM1, FIZZI, MGL2, VEGF, Osteopontin, MMPs, CCR2 [1–3] | CD38, CD40, CD64, CD80, CD86, HLA-DR (CD74), CD169 | CD14, CD68, CD11c, CD11b, HLA-DR, CD14, |
| Secreted factors: IL-1RA, IL-10, CCL17, CCL18, CCL22, CXCL12, TGF-β, low IL-12, RNI, VEGFA | Secreted factors: CCL2, CCL5, CXCL9, CXCL10, CXCL16, IL-1β, IL-2, Il-6, IL-8, high IL-12, IL-23, IFN, ROS, NO, TNFα, iNOS, [1, 4] | IL-4α, |
| Transcription factors: IRF4, pSTAT3 [5], pSTAT 6 [6] | Transcription factors: IRF3, IRF5, pSTAT 1, pSTAT5 | AP-1, Kruppel-like factor 4, PPARγ |
| NF-κB (p50–p50) | NF-κB (p50–p65) | |
| miRNA: miR146-a | miRNA: miR127, miR155, miR223 | |
| HIF-2α | HIF-1α | |
| CSF-1R dependent | CSF-1R independent | |
| Down-regulation of H ferritin and haem oxygenase | Iron sequestration [7] |
Targeting TAMs for anti-cancer therapy
Tumor macrophages have taken a bad detour
Macrophages can represent up to 50% of leukocytes in the tumor microenvironment and generally promote tumorigenesis and metastasis (Box 2). In vitro and in vivo studies have revealed that macrophages can mediate chemotherapy resistance by providing survival factors and/or activating anti-apoptotic programs in malignant cancer cells [94, 97, 98]. Cytotoxic therapy has been shown to induce CSF1-dependent macrophage recruitment in a mouse model of breast cancer and inhibiting macrophage recruitment in combination with chemotherapy induces a reduction in primary and metastatic tumors [94]. In vitro co-culture studies with macrophages derived from bone marrow and mammary carcinoma cell lines revealed that macrophages contribute to chemotherapy resistance to paclitaxel, doxorubicin and etoposide [99], and to gemcitabine in murine PDAC cells [100]. In the PDAC cell lines, the resistance was dependent on activation of signal transducer and activator of transcription 3 (STAT3), which has been shown to promote cancer cell proliferation and survival, and resistance to carboplatin in vivo and synergize with IL-6 to enhance tumor cell growth [101]. Other mechanisms of how macrophages contribute to therapeutic resistance in cancer has been recently reviewed [10]. Therefore, taken together, targeting TAMs for anti-cancer therapy has great rationale. Both pre-clinical and clinical data have importantly established proof-of-principle that targeting TAMs can be employed as an anti-cancer strategy. Current strategies to target TAMs can be broadly characterized in two main categories: 1) Inhibiting pro-tumor macrophages or their suppressive effects and 2) Activating TAMs to an anti-tumor phenotype. These strategies are reviewed briefly below (Figure 3).
Box 2. Macrophages Supporting Tumors.
Originally, it was thought that the body recruits macrophages to tumors to defend itself from cancer, but there is increasing evidence that the tumor recruits them for its own support in a manner analogous to a wound attracting macrophages to assist in healing. Clinical correlative data suggests that increased abundance of TAMs is associated with poor prognosis in over 80% of human tumors [83]. High tumor density of TAMs has been significantly associated with increased vascular density, resistance to chemotherapy, and a worse clinical outcome in the majority of tumors including colorectal, breast, ovarian, non-small cell lung cancer, malignant melanoma, Hodgkin’s lymphoma, and multiple myeloma [15, 65, 84–92].
Macrophages represent a heterogeneous population of cells. Classically activated macrophages are potent effector cells that kill microorganisms and tumor cells, produce copious amounts of pro-inflammatory cytokines, and activate cytotoxic T lymphocytes. These macrophages are the so called “M1” macrophages. Conversely, macrophages can be activated in a manner that promotes tissue remodeling and repair and preferentially attract T cell subsets devoid of cytotoxic functions such as regulatory T cells (Tregs) and T helper type 2 (Th2) cells [86, 93]. The alternatively activated macrophages are the ones referred to as “M2” macrophages. When assessing the function of TAMs, they show pro-tumor functions promoting tumor survival, proliferation, angiogenesis, dissemination, and chemoresistance [11, 12, 15, 16, 86, 93–96].
Figure 3.
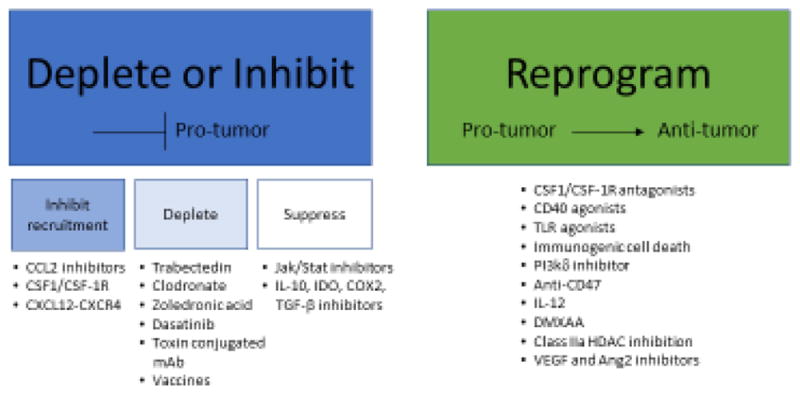
Current strategies to target tumor associated macrophages for anti-cancer therapy.
Inhibition of pro-tumor TAMs
Stop! Inhibiting monocyte and macrophage recruitment to tumor
On the basis that bone marrow derived monocytes are recruited to the tumor by the CCL2-CCLR2 axis or colony stimulating factor-1 (CSF-1)-CSF-1R receptor signaling, inhibitors against these ligands and receptors have been developed [102]. The chemokine (C-C motif) ligand 2 (CCL2; MCP-1) is secreted by a variety of cells including monocytic, endothelial, epithelial, fibroblasts and cancer cells. CCL2 and its receptor CCR2 are essential for recruitment of BDM monocytes into peripheral organs and solid tumors, resulting in their development into TAMs [103, 104]. CCL2 is elevated in the blood of some cancer patients as well as in a variety of tumor types, where increased expression correlates to poor clinical outcome [105]. Pre-clinical models have shown that CCL2 inhibition reduces metastasis [104], however cessation of therapy can accelerate breast cancer metastasis by promoting angiogenesis [106]. Further work has shown that combination therapy of CCL2 neutralization with irradiation, chemotherapy or immunotherapy reduces myeloid cells and results in improved anti-tumor effects in pre-clinical models [107]. CSF-1R inhibition is also currently being explored as CSF-1R-mediated signaling is crucial for the proliferation, differentiation and survival of the mononuclear phagocyte system and macrophages in particular. CSF-1R belongs to the type III protein tyrosine kinase receptor family and binding of its ligand CSF-1 or IL-34 induces homodimerization of the receptor and subsequent activation of receptor signaling. Similar to CCL2, high expression of CSF-1 has been shown to correlate with increased TAM density and poor progression in breast and ovarian cancer [83, 108].
Pre-clinical models have demonstrated that therapy targeting CSF-1R can enhance both chemotherapy and immunotherapy as well as prevent metastasis [109, 110]. Based on this success, a variety of strategies have been developed to inhibit the CSF-1/CSF-1R signaling axis, including their use as both monotherapy and combination therapy strategies in the clinic, which has recently been reviewed [111]. Among the small molecules, pexidartinib (PLX3397), an oral tyrosine kinase inhibitor of CSF-1R, cKIT, mutant fms-like tyrosine kinase 3 (FLT3) and platelet-derived growth factor receptor-β (PDGFR-β) has the broadest clinical development, as it is being tested in a variety of malignancies [111]. Several other small molecules are being tested clinically including ARRY-382, PLX7486, BLZ945 and JNJ-40346527. Another strategy involves mAb therapy targeting either CSF-1R (emactuzumab, AMG820, IMC-CS4 and cabiralizumab) or its ligand CSF-1 (MCS110 and PD-0360324). Mechanistically, in addition to blocking the recruitment of monocytes and macrophages to the tumor, blockade of CSF-1-CSF-1R has been shown to induce macrophage apoptosis [112].
The most promising clinical data targeting the CSF-1/CSF-1R axis has been observed in diffuse-type tenosynovial giant cell tumor (dt-GCT), a disease characterized by overexpression of CSF-1 due to a translocation involving chromosome 1p13 where the CSF-1 gene is located. These tumors are highly infiltrated with CSF-1R+ macrophages and targeting CSF-1R with either small molecules or mAb has shown promising overall response rates [113, 114]. More dismal clinical responses have been noted in brain cancer. While murine gliomas regress in response to inhibiting CSF-1R [115], clinical trials targeting CSF-1R have so far failed to increase overall survival [116], albeit, it should be noted that these patients had recurrent glioblastoma, which is a high bar to set to see a decrease in overall survival. In a Phase II study, orally administered PLX3397 in 38 patients with recurrent human glioblastoma did not show significant improvement in a 6-month progression free survival (PFS) compared to historical control data [116] and other clinical trials have not shown significant clinical benefit yet [111].
Several mechanisms have been observed that may account for the limited clinical efficacy of targeting CSF-1 or CSF-1R. While CSF-1R blockade reduces TAMs, as expected in a variety of pre-clinical mouse models, an increase of tumor-infiltrating myeloid derived suppressor cells (MDSC) has been observed [117]. Additionally, CSF-1R inhibition is thought to preferentially deplete the CSF-1R-dependent “pro”-tumor, while sparing the GM-CSF regulated “anti”-tumor TAMs. In line with this, it was recently shown that GM-CSF protects CSF-1R expressing macrophages from anti-CSF-1R antibody-induced apoptosis [102].
Get off the road: Depletion of TAMs
Depletion of macrophages and monocytes has been explored as a possible therapeutic strategy to target TAMs for cancer therapy. Several compounds have been shown to induce apoptosis of macrophages including trabectedin, Clodronate and Zoledronic acid as reviewed below. Trabectedin is a tetrahydroisoquinolone alkaloid and has been used as an anti-neoplastic agent. It’s mechanism of action on cancer cells is complex, as it binds to DNA covalently, blocks active transcription, and also interferes with the DNA repair efficiency, making homologous recombination deficient cancer cells particularly sensitive to this therapy. Trabectedin can also lead to DNA double strand breaks and cell cycle blockade resulting in cancer cell death [118, 119]. Interestingly, Trabectedin is highly cytotoxic to mononuclear phagocytes, including TAMs by engaging monocyte specific TRAIL receptors 1 and 2 and mediating a caspase 8 dependent apoptosis [120]. Patients treated with Trabectedin have reduced CD163+ cells in tumor biopsies [120]. The bisphosphonate clodronate encapsulated in liposomes is similarly efficient for the depletion of macrophages in vivo and has been shown to mediate macrophage-dependent reduction of angiogenesis, tumor burden and metastasis in pre-clinical models [121].
Other mechanisms for eliminating macrophages include Dasatinib, a tyrosine kinase inhibitor that targets MMP-9 expressing macrophages, which is currently used to treat CML [122]. In addition, toxin-conjugated mAb have also been shown to directly kill macrophages. An anti-scavenger receptor A (CD204) antibody conjugated to the saporin toxin reduced the number of vascular leukocytes and inhibited tumorigenesis in a pre-clinical mouse model of ovarian cancer [123]. Another strategy is to activate T cells to selectively eliminate TAMs. TAMs express abundant amounts of legumain, an asparaginyl endopeptidase that contributes to the degradation of the ECM and to angiogenesis. Immunization against legumain has been shown to lead to CD8+ dependent elimination of TAMs [124].
Slow down! Suppressing the of pro-tumor activity of tumor macrophages
Inhibiting the suppressive actions of TAMs has also been tested. Tyrosine kinase inhibitors sunitinib and sorafenib inhibit STAT3 in macrophages and thereby inhibit IL-10 and restore IL-12 secretion [125]. Similarly, fenretinide (4-HPR) inhibited the phosphorylation of STAT6 and skewed macrophage polarization [126]. Inhibiting IDO, TGF-b and IL-10 have also been employed to target suppressive effects of TAMs and other immune cells within the tumor microenvironment, this has recently been reviewed [127].
It is becoming clear that TAMs from different cancer types appear to respond differently to CSF-1R inhibition, perhaps a reflection of tissue specific TAM ontogeny or programming. For example, a small molecule inhibitor of CSF-1R, BLZ945, induced tumor regression in a mouse model of glioblastoma multiforme, but not through depletion of tumor macrophages [115]. Instead, brain microglia were depleted in the normal brain but not within the tumor region due to the presence to tumor derived survival factors. TAMs were instead re-educated by CSF-1R inhibition to a more anti-tumor-like phenotype [115], while the same compound depleted macrophages in preclinical models of breast cancer [128]. In both cases BLZ945 enhanced the anti-tumor response, but through different macrophage mechanisms; one through macrophage activation and the other through macrophage depletion. This highlights that TAMS may be programmed differently in response to the tissue specific environment and prompts further investigation of TAM molecular and epigenetic regulation between tissues.
Activation of anti-tumor TAMs
Correcting the course: Converting TAMs to anti-tumor macrophages
Macrophages are professional APC and have an essential role in activating T cells through cross-presentation of antigen, binding of co-stimulatory molecules and secretion of cytokines, all of which are required to activate and/or promote differentiation of T cells. In the context of cancer therapy, macrophages have been shown to be required for efficacy of chemotherapy [129], PD-1/PD-L1 [130, 131] and CTLA-4 [131] immunotherapy and anti-tumor monoclonal antibody therapy [132], which undermines the rationale for depleting TAMs during cancer therapy. Therefore, the strategy of re-orienting rather than depleting macrophages for cancer therapy has great scientific rationale and potential. There are several strategies that aim to activate TAMs. These include targeting both the tumor cell (anti-CD47) and the TAM (anti-CSF-1R, CD40 antagonism, TLR agonists, PI3kγ inhibition and class IIa HDAC inhibition), as reviewed below. CD47 is a “don’t eat me” signal overexpressed by cancer cells [133]. Blocking CD47 activates phagocytosis of cancer cells by macrophages [134]. While macrophages become highly phagocytic against tumor cells with blocked CD47 expression, the TAMs do not necessarily become anti-tumor macrophages and may retain their suppressive activities including secretion of pro-inflammatory cytokines. It remains to be determined if activating highly suppressive TAMs to phagocytose cancer cells is the best strategy, as these TAMs likely will continue to induce tumor suppression through secretion of cytokines that dampens a protect immune response. In line with this, initial data from Phase I clinical trials have not been as promising as the pre-clinical data [135]. Potentially, combinations of anti-CD47 therapy with therapy that converts TAMs to an anti-tumor phenotype may hold the most promise.
Other strategies to activate TAMs for anti-cancer therapy include targeting CD40, a member of the tumor necrosis factor receptor (TNFR) family, that is expressed by APCs including B cells, macrophages, endothelial cells, platelets and tumor cells [136]. When CD40 is activated on APCs, the APCs release pro-inflammatory cytokines such as TNFα and IL-12p70 and increase expression of CD80, CD86 and MHCII, which may help support anti-tumor T-cell activity [137]. An agonist monoclonal antibody against CD40 has demonstrated efficacy in mouse models of pancreatic ductal adenocarcinoma (PDAC) [138] and patients with PDAC [139] in combination with chemotherapy. Additionally, combination of anti-CSF-1R therapy with agonistic CD40 therapy reveals enhanced anti-tumor activity in preclinical syngeneic animal models, where reducing suppressive TAMs through anti-CSF-1R while activating APCs resulted in activated CTLs and reduced tumor burden [140].
PI3Kγ has recently been shown to selectively drive immunosuppressive transcriptional programing in macrophages that inhibit adaptive immune response and promotes myeloid cell invasion into syngeneic subcutaneous LLC, orthotopic pancreatic carcinoma and melanoma tumors [141]. PI3kγ is one of the four class I PI3K p110 catalytic isoforms and is overexpressed in some tumor types. In mouse tumor models IPI-549, a PI3Kγ-selective inhibitor [142], reprogramed TAMs, which reduced pro-tumor like macrophages while increasing anti-tumor like macrophages and CD8+ T cell responses [143] and enhanced anti-PD-1 therapy [144]. A Phase 1/1b First-In-Human, Dose-Escalation Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics is ongoing, and results are pending (NCT02637531).
Hypoxia in the TME is a critical factor that polarizes TAMs to a pro-tumor phenotype [145]. Several eloquent studies have shown that reducing hypoxia by inducing vascular normalization through selective or dual vascular endothelial growth factor (VEGF) and angiopoietin-2 (Ang2) inhibitors have been shown induce anti-tumor TAM phenotypes and reduction of tumor burden in mouse models of GBM and breast cancer [146, 147].
Class IIa HDAC inhibition has emerged as the newest target for converting TAMs to an anti-tumor phenotype. Class IIa HDACs have suffered from incomplete understanding of function and are not efficient deacetylases, likely due to the limited tools to study their function, including lack of selective inhibitors. TMP195 was identified through a high-throughput screen as a selective, first in class, class IIa HDAC inhibitor [148]. Unlike other HDAC inhibitors, TMP195 had no cytotoxic activity on tumor cells or immune cells. Instead, TMP195 selectively altered the transcription profile of macrophages but not T-cells, and sensitized macrophages to Th1 signaling while protecting them from Th2 signaling [75, 148]. In pre-clinical models of breast cancer, class IIa HDAC inhibition resulted in macrophage-mediated reduction in primary tumor burden and pulmonary metastasis. TMP195-activated macrophages became highly phagocytic and displayed an IFNγ gene signature [75]. Class IIa HDAC inhibition would not have previously been suspected to induce “anti”-tumor macrophage responses. This supports the rationale for the use of unbiased novel drug exploration to identify small molecules, regardless of their mechanism of action or class, to modulate TAMs for anti-cancer therapy.
Concluding remarks
The journey has only just begun. Future destination: targeting TAMs for successful clinical outcome
Several open questions remain to be answered (see Outstanding Questions Box). The appreciation of the extent of heterogeneity of TAMs comes with new challenges and opportunities. Macrophages are no longer considered end products of a hard-wired differentiation program from monocytic precursors. Instead, TAMs have emerged as highly dynamic with both inter-and intra-tissue heterogeneity. Future work will uncover subsets of TAMs which will help define their involvement in tumorigenesis and will lead to the identification of specific subsets of tumor macrophages that can be targeted for effective therapies, while sparing macrophages with anti-tumor functions as well as macrophages in other tissue types. Macrophages may hold more potential for therapeutic targeting than previously thought and great emphasis should be placed on understanding molecular regulation of TAMs, their cellular ontogeny, and persistence and function between different tissues. However, other more innovative approaches should also be explored. For example, macrophages are capable of facilitating regeneration of complex tissue in the fleshy part of the ear, including cartilage, hair follicles, and sweat glands in the African spiny mouse [149, 150] and are involved in the regeneration of axolotl limbs [151]. Future understanding of macrophages, at the stem cell level, may reveal novel insight to program macrophages for anti-cancer therapy. There is no doubt that further creative, precise investigation of macrophage and TAM biology will greatly enhance our ability to target macrophages for anti-cancer therapy. However, we should not loose site of the TME as a whole. While TAMs can play a fundamental role in regulating the immune environment of a tumor, including T cell activation, other immune cells of both innate and adaptive lineages should also be investigated. Like cars in a traffic circle, immune cells in a tumor must adapt and communicate for an effective immune response. Therefore, comprehensive understanding of all immune cells within the tumor, should translate to better clinical outcome.
Outstanding Questions Box.
-
How does the cellular ontogeny of TAMs influence tumor progression and influence cancer therapy?
Are the BDM that are recruited to the tumor identical to TRM?
-
Do BDM and TRM have similar molecular and epigenetic mechanisms that regulate their phenotype?
Is the phenotype pre-programmed?
How important is the tumor microenvironment (TME)?
Do tissue macrophages have immune memory? Do TAMs?
-
How should the distinct TAM subsets and states be characterized?
Are TAM states stable or are they in transient states of activation?
How are TAMs different from TRM?
How are TAMs different between tissue types?
-
Which TAM subsets promote and/or inhibit tumorigenesis?
What subsets of TAMs should be targeted for anti-cancer therapy?
Can the proliferation of protective macrophages be enhanced?
-
Which current macrophage targeting strategies will be most efficacious for cancer therapy?
Is this true for all tumor types?
Is this true for all stages of tumorigenesis?
Clinician’s Corner Box.
Much appropriate attention has been paid recently to exploiting the adaptive arm of the immune system to kill cancer cells. However, the innate arm offers a relatively untapped opportunity to kill cancer cells, either by eliminating tumor supportive TAMs, or by converting them to pro-inflammatory anti-tumor macrophages.
TAMs are a major population within most tumors
A high density of TAMs generally correlates with worse clinical outcome
-
TAMs have been broadly targeted for anti-cancer therapy through two strategies, which have shown some promise in the clinic
Inhibiting or depleting TAMs
Reprograming TAMs to an anti-tumor phenotype
Previously unrecognized diversity of TAMs has recently emerged, in which several distinct TAM subsets have been identified in human tumors, and the subsets appear to be different between tumor type
Further work is warranted to reveal tissue and TAM subset specific vulnerabilities to guide novel anti-cancer strategies aimed at targeting TAMs for anti-cancer therapy
Trends Box.
-
Macrophages in tissues arise from distinct sources
Tissue resident macrophages (TRM) derive from yolk sac and fetal liver progenitors
Bone-marrow derived macrophages (BDM) arise from HSC
Tissue macrophages have distinct transcriptional profiles between tissues
Tumor associated macrophages (TAMs) populate tumors through local proliferation of TRM or recruitment from BDM
Involvement of TAMs is tumor tissue specific; where TRM or BDM differentially promote tumorigenesis depending on the tissue type
Targeting TAMs for anti-cancer therapy has shown signs of pre-clinical and clinical success using either blunt targeting strategies (CSF-1R inhibitors) or more recently developed novel strategies such as PI3Kγ and class IIa HDAC inhibitors
Acknowledgments
The author apologizes to the authors whose work could not be cited due to space constraints. The author thanks A. Letai, J. Castrillon and A. Mehta for insightful discussions and critical feedback. This work was supported by the National Institutes of Health grant P50CA168504 and the Friends of Dana-Farber, Dancing for a Cure.
Glossary
- 4-hydroxytamoxifen
(4′OHT) A selective estrogen receptor modulator (SERM).
- Antigen presenting cell
(APC) A cell that presents antigen in a major histocompatibility complex at its surface; a process referred to as antigen presentation. T cells can recognize this complex through their T cell receptor.
- Bone-marrow derived macrophage
(BDM) A macrophage derived from hematopoietic stem cells in the bone marrow. BDM progenitors are recruited to tissue by cytokines or chemokines. Once in the tissue the progenitor becomes a mature macrophage.
- Bone marrow
(BM) A semi-solid tissue found within the spongy or cancellous portion of the bones. It is composed of HSC, marrow adipose tissue and stromal cells.
- Chemokine (C-C motif) ligand 2
(CCL2, MCP-1) A cytokine involved in the recruitment of monocytes and macrophages to tissue and tumors. CCL2 is secreted by cells undergo stress responses, malignant cells as well as stromal cells.
- Colony stimulating factor 1
(CSF-1) A cytokine required for the survival, proliferation and differentiation of some mature macrophage types.
- Colony stimulating factor 1 receptor
(CSF-1R) The receptor for CSF-1, which is expressed by some mature macrophage types.
- Coley’s toxins
A mixture of killed bacteria consisting of bacteria of specie Streptococcus pyogenes and Serratia marcescens, names after Dr. William Coley, a surgical oncologist from the Hospital for Special Surgery who developed the mixture in the late 19th century as a treatment for inoperable cancer.
- Dendritic cell
(DC) A cell of the myeloid lineage that functions as a professional APC.
- Diffuse-type tenosynovial giant cell tumor
(Dt-GCT) Alternatively known as pigmented villonodular synovitis (PVNS) is an orphan disease caused by the translocation involving chromosome 1p13 where the CSF-1 gene is located, causing CSF-1 overexpression and recruitment of CSF-1R+ macrophages.
- Erythromyeloid progenitors
(EMP) Cells from the yolk sac that are the primary source of fetal and adult tissue resident macrophages.
- Fetal liver
(FL) A major site of development of the human immune system. The FL is seed with EMP from the embryonic yolk sac and contributes to seeding fetal and adult tissue residence macrophages. T- and B-cell precursors are also present in the fetal liver.
- Hematopoietic stem cells
(HSC) Stem cells that give rise to blood cells through the process of hematopoiesis. They are derived from the mesoderm and located in the bone marrow.
- Lewis lung carcinoma
(LLC) A murine model of lung cancer. The tumor was originally discovered by Dr. Margaret R. Lewis of the Wistar Institute in 1951 when it spontaneously arose as a carcinoma of the lung in a C57BL mouse.
- Lymphocyte antigen 6 complex
(Ly6C) A GPI-anchored glycoprotein expressed on a range of hematopoietic cell lineages.
- M1 Macrophage
A functional state of macrophage polarization, also referred to as classically activated. The term M1 describes an ex vivo stimulated macrophage that shows properties similar to macrophages found in vivo that can protect the host from bacteria and malignant cancer cells.
- M2 Macrophage
A functional state of macrophage polarization, also referred to as alternatively activated. The term M2 describes an ex vivo stimulated macrophage that shows properties similar to macrophages found in vivo that are involved in wound repair and tumor progression.
- Myeloid derived suppressor cells
(MDSC) A heterogenous group of immature immune cells from the myeloid lineage. These cells derived from HSC-progenitors and in cancer are thought to contribute to angiogenesis, tumor-cell invasion and metastasis.
- Mononuclear phagocytic system
(MPS) a part of the immune system that consists of the phagocytic cells.
- Pancreatic ductal adenocarcinoma
(PDAC) A type of exocrine pancreatic cancer.
- Progression free survival
(PFS) A measurement of the length of time during and after treatment of cancer, or other diseases, that a patient lives with the disease but it does not get worse.
- Tumor associated macrophage
(TAM) A macrophage found within tumors.
- Tumor microenvironment
(TME) The cellular environment of the tumor, including immune cells, blood vessels, fibroblasts, cytokines and chemokines.
- Toll-like receptor
(TLR) A class of proteins that play a role in innate immune sensing. They are single, membrane-spanning, non-catalytic receptors expressed on innate immune cells such as macrophages and dendritic cells that can recognize structurally conserved molecules on foreign particles.
- Tumor necrosis factor receptor
(TNFR) A protein that is part of a superfamily of cytokine receptors. These receptors can bind tumor necrosis factors (TNFs) via an extracellular cysteine-rich domain.
- Tissue resident macrophage
(TRM) A population of immune cells that perform tissue specific functions related to homeostasis such as clearance of cellular debris and iron processing, they play roles in immunity through surveillance, responding to infection and the resolution of inflammation. This heterogenous population of cells can arise from either self-renewing, yolk sac derived progenitors or from HSCs from the bone marrow.
- Vascular endothelial growth factor
(VEGF) A signal protein produced by cells that stimulates the formation of blood vessels.
- Yolk sac
(YS) A membranous sac attached to the embryo. It is formed by cells of the hypoblast adjacent to the embryonic disk. It is important for early embryonic blood supply. Immune cells such as macrophages migrate from the YS to developing organs prior to HSC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith-Garvin JE, et al. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coley WB. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus) Proc R Soc Med. 1910;3(Surg Sect):1–48. [PMC free article] [PubMed] [Google Scholar]
- 3.Steen S, Stephenson G. Current treatment of soft tissue sarcoma. Proc (Bayl Univ Med Cent) 2008;21(4):392–6. doi: 10.1080/08998280.2008.11928435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D’Errico G, et al. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Transl Med. 2017;6(1):3. doi: 10.1186/s40169-016-0130-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engblom C, et al. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16(7):447–62. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 6.Tang J, et al. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol. 2018;29(1):84–91. doi: 10.1093/annonc/mdx755. [DOI] [PubMed] [Google Scholar]
- 7.Sharma P, Allison JP. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell. 2015;161(2):205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyi C, Postow MA. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett. 2014;588(2):368–76. doi: 10.1016/j.febslet.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 9.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 10.Ruffell B, Coussens LM. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell. 2015;27(4):462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solinas G, et al. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86(5):1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 12.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mantovani A, et al. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13(7):265–70. doi: 10.1016/0167-5699(92)90008-U. [DOI] [PubMed] [Google Scholar]
- 14.Allavena P, et al. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 16.Erreni M, et al. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron. 2011;4(2):141–54. doi: 10.1007/s12307-010-0052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang L, Zhang Y. Tumor-associated macrophages, potential targets for cancer treatment. Biomark Res. 2017:5. doi: 10.1186/s40364-017-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hume DA, Gordon S. Mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Identification of resident macrophages in renal medullary and cortical interstitium and the juxtaglomerular complex. J Exp Med. 1983;157(5):1704–9. doi: 10.1084/jem.157.5.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hume DA, et al. The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Relationship between macrophages, Langerhans cells, reticular cells, and dendritic cells in lymphoid and hematopoietic organs. J Exp Med. 1983;158(5):1522–36. doi: 10.1084/jem.158.5.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hume DA, et al. Immunohistochemical localization of a macrophage-specific antigen in developing mouse retina: phagocytosis of dying neurons and differentiation of microglial cells to form a regular array in the plexiform layers. J Cell Biol. 1983;97(1):253–7. doi: 10.1083/jcb.97.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasmono RT, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101(3):1155–63. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- 22.Epelman S, et al. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ginhoux F, Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity. 2016;44(3):439–449. doi: 10.1016/j.immuni.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 24.Gomez Perdiguero E, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 26.Hopkinson-Woolley J, et al. Macrophage recruitment during limb development and wound healing in the embryonic and foetal mouse. J Cell Sci. 1994;107(Pt 5):1159–67. doi: 10.1242/jcs.107.5.1159. [DOI] [PubMed] [Google Scholar]
- 27.Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–61. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung K, et al. Ly6Clo monocytes drive immunosuppression and confer resistance to anti-VEGFR2 cancer therapy. J Clin Invest. 2017;127(8):3039–3051. doi: 10.1172/JCI93182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–6. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung K, et al. Targeting CXCR4-dependent immunosuppressive Ly6C(low) monocytes improves antiangiogenic therapy in colorectal cancer. Proc Natl Acad Sci U S A. 2017;114(39):10455–10460. doi: 10.1073/pnas.1710754114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serbina NV, et al. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–52. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elmore MRP, et al. CSF1 receptor signaling is necessary for microglia viability, which unmasks a cell that rapidly repopulates the microglia-depleted adult brain. Neuron. 2014;82(2):380–97. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mucenski ML, et al. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. 1991;65(4):677–89. doi: 10.1016/0092-8674(91)90099-k. [DOI] [PubMed] [Google Scholar]
- 36.Daems WT, de Bakker JM. Do resident macrophages proliferate? Immunobiology. 1982;161(3–4):204–11. doi: 10.1016/S0171-2985(82)80075-2. [DOI] [PubMed] [Google Scholar]
- 37.Naito M, et al. Development, differentiation, and phenotypic heterogeneity of murine tissue macrophages. J Leukoc Biol. 1996;59(2):133–8. doi: 10.1002/jlb.59.2.133. [DOI] [PubMed] [Google Scholar]
- 38.Davies LC, et al. A quantifiable proliferative burst of tissue macrophages restores homeostatic macrophage populations after acute inflammation. Eur J Immunol. 2011;41(8):2155–64. doi: 10.1002/eji.201141817. [DOI] [PubMed] [Google Scholar]
- 39.Mildner A, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10(12):1544–53. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 40.Jenkins SJ, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332(6035):1284–8. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hashimoto D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakubzick C, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39(3):599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamoutounour S, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. 2013;39(5):925–38. doi: 10.1016/j.immuni.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 44.Bain CC, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929–937. doi: 10.1038/ni.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zigmond E, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37(6):1076–90. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 46.Kovtunovych G, et al. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood. 2010;116(26):6054–62. doi: 10.1182/blood-2010-03-272138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haldar M, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell. 2014;156(6):1223–34. doi: 10.1016/j.cell.2014.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haldar M, Murphy KM. Origin, development, and homeostasis of tissue-resident macrophages. Immunol Rev. 2014;262(1):25–35. doi: 10.1111/imr.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–28. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gosselin D, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159(6):1327–40. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lavin Y, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–26. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franklin RA, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laoui D, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74(1):24–30. doi: 10.1158/0008-5472.CAN-13-1196. [DOI] [PubMed] [Google Scholar]
- 54.Cortez-Retamozo V, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109(7):2491–6. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowman RL, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016;17(9):2445–2459. doi: 10.1016/j.celrep.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu Y, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 2017;47(3):597. doi: 10.1016/j.immuni.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu Y, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 2017;47(2):323–338. e6. doi: 10.1016/j.immuni.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Palma M. Origins of Brain Tumor Macrophages. Cancer Cell. 2016;30(6):832–833. doi: 10.1016/j.ccell.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 59.Gottfried E, et al. Expression of CD68 in non-myeloid cell types. Scand J Immunol. 2008;67(5):453–63. doi: 10.1111/j.1365-3083.2008.02091.x. [DOI] [PubMed] [Google Scholar]
- 60.Hameed A, et al. Immunohistochemical expression of CD68 antigen in human peripheral blood T cells. Hum Pathol. 1994;25(9):872–6. doi: 10.1016/0046-8177(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 61.Pulford KA, et al. Distribution of the CD68 macrophage/myeloid associated antigen. Int Immunol. 1990;2(10):973–80. doi: 10.1093/intimm/2.10.973. [DOI] [PubMed] [Google Scholar]
- 62.Kaiserling E, et al. Aberrant expression of macrophage-associated antigens (CD68 and Ki-M1P) by Schwann cells in reactive and neoplastic neural tissue. Light- and electron-microscopic findings. Mod Pathol. 1993;6(4):463–8. [PubMed] [Google Scholar]
- 63.Ruffell B, et al. Leukocyte composition of human breast cancer. Proc Natl Acad Sci U S A. 2012;109(8):2796–801. doi: 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang QW, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7(12):e50946. doi: 10.1371/journal.pone.0050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Steidl C, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362(10):875–85. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forssell J, et al. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13(5):1472–9. doi: 10.1158/1078-0432.CCR-06-2073. [DOI] [PubMed] [Google Scholar]
- 67.Ohno S, et al. The degree of macrophage infiltration into the cancer cell nest is a significant predictor of survival in gastric cancer patients. Anticancer Res. 2003;23(6D):5015–22. [PubMed] [Google Scholar]
- 68.Ohno S, et al. Correlation of histological localization of tumor-associated macrophages with clinicopathological features in endometrial cancer. Anticancer Res. 2004;24(5C):3335–42. [PubMed] [Google Scholar]
- 69.Colvin EK. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front Oncol. 2014;4:137. doi: 10.3389/fonc.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu L, et al. Prognostic value of diametrically polarized tumor-associated macrophages in renal cell carcinoma. Ann Surg Oncol. 2014;21(9):3142–50. doi: 10.1245/s10434-014-3601-1. [DOI] [PubMed] [Google Scholar]
- 71.Tsujikawa T, et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017;19(1):203–217. doi: 10.1016/j.celrep.2017.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gil Del Alcazar CR, et al. Immune Escape in Breast Cancer During In Situ to Invasive Carcinoma Transition. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-17-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chevrier S, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169(4):736–749. e18. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lavin Y, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell. 2017;169(4):750–765. e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guerriero JL, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543(7645):428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Broz M, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen Presenting Cells, Critical for T cell Immunity. Cancer cell. 2014;26(5):638–652. doi: 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muller S, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18(1):234. doi: 10.1186/s13059-017-1362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang Q, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2017;32(1):42–56. e6. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chung W, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081. doi: 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Szulzewsky F, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10(2):e0116644. doi: 10.1371/journal.pone.0116644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hambardzumyan D, et al. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–7. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol. 2014:5. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bingle L, et al. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196(3):254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 84.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–70. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campbell MJ, et al. Proliferating macrophages associated with high grade, hormone receptor negative breast cancer and poor clinical outcome. Breast Cancer Res Treat. 2011;128(3):703–11. doi: 10.1007/s10549-010-1154-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 87.Balkwill F, et al. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 88.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 89.Zheng Y, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood. 2009;114(17):3625–8. doi: 10.1182/blood-2009-05-220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brocker EB, et al. Infiltration of primary and metastatic melanomas with macrophages of the 25F9-positive phenotype. Cancer Immunol Immunother. 1987;25(2):81–6. doi: 10.1007/BF00199945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Torisu H, et al. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int J Cancer. 2000;85(2):182–8. [PubMed] [Google Scholar]
- 92.Bailey C, et al. Chemokine expression is associated with the accumulation of tumour associated macrophages (TAMs) and progression in human colorectal cancer. Clin Exp Metastasis. 2007;24(2):121–30. doi: 10.1007/s10585-007-9060-3. [DOI] [PubMed] [Google Scholar]
- 93.Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 94.DeNardo DG, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discovery. 2011;1(1):54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mantovani A, et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 96.Sica A, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 2008;18(5):349–55. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 97.Castells M, et al. Ovarian ascites-derived Hospicells promote angiogenesis via activation of macrophages. Cancer Lett. 2012;326(1):59–68. doi: 10.1016/j.canlet.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 98.Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat. 2012;15(1–2):39–49. doi: 10.1016/j.drup.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shree T, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25(23):2465–79. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mitchem JB, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–41. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jinushi M, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108(30):12425–30. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ries CH, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–59. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 103.Kitamura T, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212(7):1043–59. doi: 10.1084/jem.20141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang J, et al. Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst. 2010;102(8):522–8. doi: 10.1093/jnci/djq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bonapace L, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–3. doi: 10.1038/nature13862. [DOI] [PubMed] [Google Scholar]
- 107.Moisan F, et al. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol Oncol. 2014;8(7):1231–9. doi: 10.1016/j.molonc.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scholl SM, et al. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J Natl Cancer Inst. 1994;86(2):120–6. doi: 10.1093/jnci/86.2.120. [DOI] [PubMed] [Google Scholar]
- 109.Linde N, et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun. 2018;9(1):21. doi: 10.1038/s41467-017-02481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhu Y, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cannarile MA, et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5(1):53. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.MacDonald KP, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116(19):3955–63. doi: 10.1182/blood-2010-02-266296. [DOI] [PubMed] [Google Scholar]
- 113.Cassier PA, et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015;16(8):949–56. doi: 10.1016/S1470-2045(15)00132-1. [DOI] [PubMed] [Google Scholar]
- 114.Tap WD, et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N Engl J Med. 2015;373(5):428–37. doi: 10.1056/NEJMoa1411366. [DOI] [PubMed] [Google Scholar]
- 115.Pyonteck SM, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–72. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Butowski N, et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 2016;18(4):557–64. doi: 10.1093/neuonc/nov245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kumar V, et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell. 2017;32(5):654–668. e5. doi: 10.1016/j.ccell.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.D’Incalci M, et al. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer. 2014;111(4):646–50. doi: 10.1038/bjc.2014.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Herrero AB, et al. Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res. 2006;66(16):8155–62. doi: 10.1158/0008-5472.CAN-06-0179. [DOI] [PubMed] [Google Scholar]
- 120.Germano G, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 121.Zeisberger SM, et al. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95(3):272–81. doi: 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liang W, et al. Antitumor activity of targeting SRC kinases in endothelial and myeloid cell compartments of the tumor microenvironment. Clin Cancer Res. 2010;16(3):924–35. doi: 10.1158/1078-0432.CCR-09-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bak SP, et al. Scavenger receptor-A-targeted leukocyte depletion inhibits peritoneal ovarian tumor progression. Cancer Res. 2007;67(10):4783–9. doi: 10.1158/0008-5472.CAN-06-4410. [DOI] [PubMed] [Google Scholar]
- 124.Smahel M, et al. Enhancement of DNA vaccine potency against legumain. J Immunother. 2014;37(5):293–303. doi: 10.1097/CJI.0000000000000040. [DOI] [PubMed] [Google Scholar]
- 125.Edwards JP, Emens LA. The multikinase inhibitor sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE(2) in murine macrophages. Int Immunopharmacol. 2010;10(10):1220–8. doi: 10.1016/j.intimp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dong R, et al. The involvement of M2 macrophage polarization inhibition in fenretinide-mediated chemopreventive effects on colon cancer. Cancer Lett. 2017;388:43–53. doi: 10.1016/j.canlet.2016.11.029. [DOI] [PubMed] [Google Scholar]
- 127.Sharma P, et al. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Strachan DC, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. OncoImmunology. 2013;2(12):e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guerriero JL, et al. DNA Alkylating Therapy Induces Tumor Regression through an HMGB1-Mediated Activation of Innate Immunity. J Immunol. 2011;186(6):3517–26. doi: 10.4049/jimmunol.1003267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dahan R, et al. FcgammaRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell. 2015;28(3):285–95. doi: 10.1016/j.ccell.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 131.Simpson TR, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gul N, et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014;124(2):812–23. doi: 10.1172/JCI66776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–85. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chao MP, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142(5):699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sikic BI, et al. A first-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. Journal of Clinical Oncology. 2016;34(15_suppl):3019–3019. doi: 10.1200/JCO.18.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Elgueta R. Molecular mechanism and function of CD40/CD40L engagement in the immune system. 2009;229(1) doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Quezada SA, et al. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. 2004;22:307–28. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 138.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Beatty GL, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–95. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wiehagen KR, et al. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol Res. 2017;5(12):1109–1121. doi: 10.1158/2326-6066.CIR-17-0258. [DOI] [PubMed] [Google Scholar]
- 141.Schmid MC, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19(6):715–27. doi: 10.1016/j.ccr.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Evans CA, et al. Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-gamma Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate. ACS Med Chem Lett. 2016;7(9):862–7. doi: 10.1021/acsmedchemlett.6b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kaneda MM, et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016;6(8):870–85. doi: 10.1158/2159-8290.CD-15-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kaneda MM, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Raggi F, et al. Regulation of Human Macrophage M1–M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front Immunol. 2017;8:1097. doi: 10.3389/fimmu.2017.01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kloepper J, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci U S A. 2016;113(16):4476–81. doi: 10.1073/pnas.1525360113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Peterson TE, et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci U S A. 2016;113(16):4470–5. doi: 10.1073/pnas.1525349113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lobera M, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9(5):319–25. doi: 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- 149.Gawriluk TR, et al. Comparative analysis of ear-hole closure identifies epimorphic regeneration as a discrete trait in mammals. Nat Commun. 2016;7:11164. doi: 10.1038/ncomms11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Simkin J, et al. Macrophages are necessary for epimorphic regeneration in African spiny mice. Elife. 2017:6. doi: 10.7554/eLife.24623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Godwin JW, et al. Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A. 2013;110(23):9415–20. doi: 10.1073/pnas.1300290110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 155.Mucenski ML, et al. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. 1991;65:677–689. doi: 10.1016/0092-8674(91)90099-k. [DOI] [PubMed] [Google Scholar]
- 156.Gomez Perdiguero E, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chen S, et al. CNS Macrophages Control Neurovascular Development via CD95L. Cell reports. 2017;19:1378–1393. doi: 10.1016/j.celrep.2017.04.056. [DOI] [PubMed] [Google Scholar]
- 158.Gosselin D, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017:356. doi: 10.1126/science.aal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Boyle WJ, et al. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 160.Chawla A, et al. Macrophage-mediated inflammation in metabolic disease. Nature reviews Immunology. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sergin I, et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nature communications. 2017;8:15750. doi: 10.1038/ncomms15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hulsmans M, et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell. 2017;169:510–522. e520. doi: 10.1016/j.cell.2017.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mossadegh-Keller N, et al. Developmental origin and maintenance of distinct testicular macrophage populations. The Journal of experimental medicine. 2017;214:2829–2841. doi: 10.1084/jem.20170829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Biswas SK, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 165.Ojalvo LS, et al. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J Immunol. 2010;184:702–712. doi: 10.4049/jimmunol.0902360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stout RD, Suttles J. Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunological reviews. 2005;205:60–71. doi: 10.1111/j.0105-2896.2005.00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu H, et al. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yan D, et al. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1alpha Activation. Cell reports. 2016;16:2914–2927. doi: 10.1016/j.celrep.2016.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Recalcati S, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. European journal of immunology. 2010;40:824–835. doi: 10.1002/eji.200939889. [DOI] [PubMed] [Google Scholar]