

Abstract
Lamins have important roles in nuclear structure and cell signaling. Several diseases are associated with mutations in the lamin A/C gene (LMNA in humans). Patients with familial partial lipodystrophy caused by LMNA mutations develop pancreatitis, but it is not clear how these mutations affect pancreatic function. We generated mice with inducible exocrine pancreas-specific disruption of Lmna and showed that LMNA is lost from most exocrine pancreas cells. LMNA-knockout pancreata develop endoplasmic reticulum stress with loss of acinar cell markers, increased autophagy, apoptosis, and cell proliferation, compared to CreERT2– mice (littermate controls). Disruption of Lmna led to a phenotype that resembled chronic pancreatitis, with increased Sirius-Red staining and ACTA in male LMNA-knockout mice compared to littermate males, but not in female mice. LMNA-knockout pancreata have reduced levels of RB and activation of E2F, based on increased expression of its target genes. Therefore, lamins maintain pancreatic homeostasis by regulating RB stability and E2F activity.
Keywords: Intermediate Filament, Nuclear Lamina, Mouse Model, Partial Lipodystrophy
Main text
Lamins are intermediate filaments proteins that comprise the major components of the nuclear lamina1, and are classified as A- or B-types which vary in cellular functions and expression patterns2. Lamin A and C proteins (LMNA) are A-type lamins; translated from splice variants encoded by the mouse Lmna gene (LMNA in humans)3. A broad spectrum of diseases termed laminopathies are caused by lamin mutations, particularly A-type lamins2,3. Despite the disease-relevance and critical functions of lamins, nothing is known about their role in the pancreas. Notably, patients with Dunnigan-type familial partial lipodystrophy (FPLD2) due to LMNA mutations4 are susceptible to pancreatitis that is typically attributed to hypertriglyceridemia5, but FPLD2-associated pancreatitis also occurs without hypertriglyceridemia (unpublished observations). This suggests that LMNA may play an important role in maintaining exocrine pancreas homeostasis.
Pancreas-specific conditional knockout (KO) mice were generated by crossing CreERT2+ mice with mice having floxed Lmna exons 10 and 11 (Fig.S1A). LMNA-staining demonstrated protein absence in most exocrine pancreas cells at 3, 4, 6 and 21d post-tamoxifen injection (Fig.1A; Fig.S1B). Histopathological analysis showed decreased eosin staining and smaller acini 4d but not 3d after Cre-induction (Fig.S1C). Immunoblotting of KO-pancreata (4d) showed decreased amylase and elevated Grp78 (markers of acinar injury), in addition to elevated chop in males (Fig.S1D).
Fig. 1. Exocrine pancreas-specific Lmna-disruption triggers ER stress-response and loss of acinar cell phenotype.
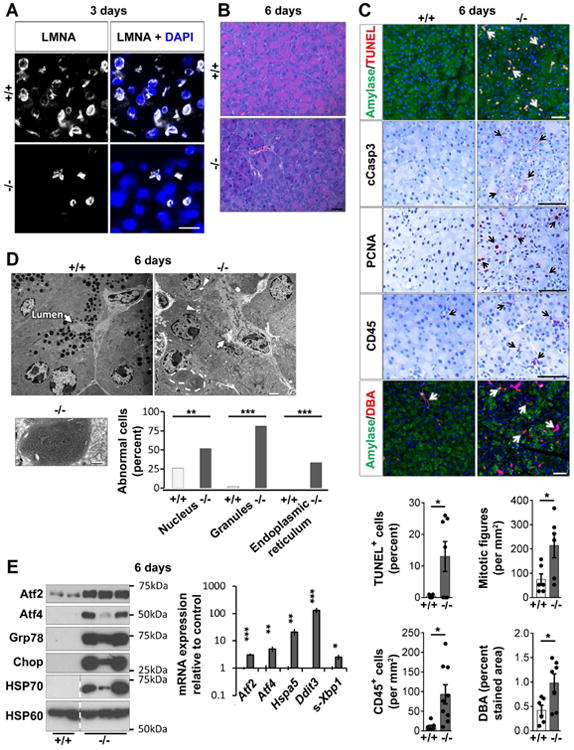
(A) Loss of LMNA in most pancreatic acinar cells 3d post-tamoxifen injection. Bar=20μm. (B) Representative H&E-stained sections 6d post-tamoxifen injection. Bar=20μm. (C) Pancreata (6d) were stained as indicated; arrows highlight positive staining. Bar=50μm. Histograms: quantification using tissues from 6-9 mice/group. (D) Transmission electron microscopy of 6d control and KO-pancreata. Arrows: acinus lumen, void of secretions in KO but not control acini; arrowheads: vacuoles with electron-dense material. Bar=2μm. Lower panel: Condensed and tightly-wound ER (high magnification; bar=0.8μm). Histogram: electron-micrographs quantification (3-4 mice/group). (E) Immunoblots of indicated ER stress-related proteins, HSP70, and the mitochondrial protein HSP60. Dashed lines mark non-adjacent wells of same gel. Histogram: RT-qPCR of the indicated transcripts (N=6 mice/group). *p≤0.05,**p≤0.01,***p≤0.001.
Pancreata harvested 6d post-tamoxifen injection had less eosin staining (Fig.1B), suggesting loss of acinar cell phenotype. RT-qPCR analysis showed decreased amylase transcription and increased expression of ductal cell markers (Fig.S1E), and several acinar cell markers were down-regulated (Fig.S1F). Staining for DBA (Dolichos biflorus agglutinin) confirmed ductal cell increase (Fig.1C). Recruitment of inflammatory cells was noted after staining for the pan-leukocyte marker CD45 (Fig.1C). Acinar cell apoptosis was observed and confirmed using TUNEL/amylase co-staining, and cleaved-caspase-3 (cCasp3) staining (Fig.1C). Mice treated with tamoxifen by oral gavage had similar histological and biochemical findings to intraperitoneally-injected mice (Fig S2A-D). KO-pancreata had significantly more apoptosis (Fig.S2E) than control, but comparable CD45+ cell infiltration (not shown).
Many KO-acinar cells expressed high levels of PCNA and mitotic figures indicating increased cell proliferation (Fig.1C). Electron microscopy showed 81% of KO-cells with fewer and smaller zymogen granules (vs 2% in littermates), and 52% with irregular-shaped nuclei vs 26% in littermates (Fig.1D; Fig.S3A,B). These observations are similar to previous findings in human fibroblasts with LMNA mutation6. Notably, 33% of acinar cells contained areas of tightly-coiled ER (Fig.1D; Fig.S3A). The ultrastructural alterations were accompanied by increased Atf2, Atf4, Grp78, chop, and HSP70 proteins, and increased spliced-Xbp1 mRNA (s-Xbp1), consistent with ER stress. HSP60 remained unchanged thereby indicating organelle-specific stress (Fig.1E). Elevated LC3-II:I ratio and the presence of electron-dense material-containing vacuoles (Fig.1D; Fig.S3A,C) in KO-pancreata support increased autophagy.
We used expression-profiling analysis of pancreata 6d post-tamoxifen injection to determine potential dysregulated signaling pathways. Three of the top four alterations (p53 signaling, cell cycle, basal transcription factors) are known RB-associated pathways7 that are regulated by LMNA8. Indeed, pancreatic RB levels decreased 4d after Cre-induction (Fig.2A). To determine if the E2-factor (E2F) family of transcription factors was activated in KO-pancreata, we tested the induction of E2F target genes9 and found them elevated (including RB-E2F family, canonical-E2F, pro-apoptotic-E2F and MAPK targets (Fig.2A)). RB activation is cell-autonomous since 47% of cells that stained positive for PCNA, a well-known E2F target, were LMNA-negative (Fig.2B).
Fig. 2. LMNA-KO mice develop a phenotype that resembles chronic pancreatitis associated with RB destabilization and E2F target-activation.
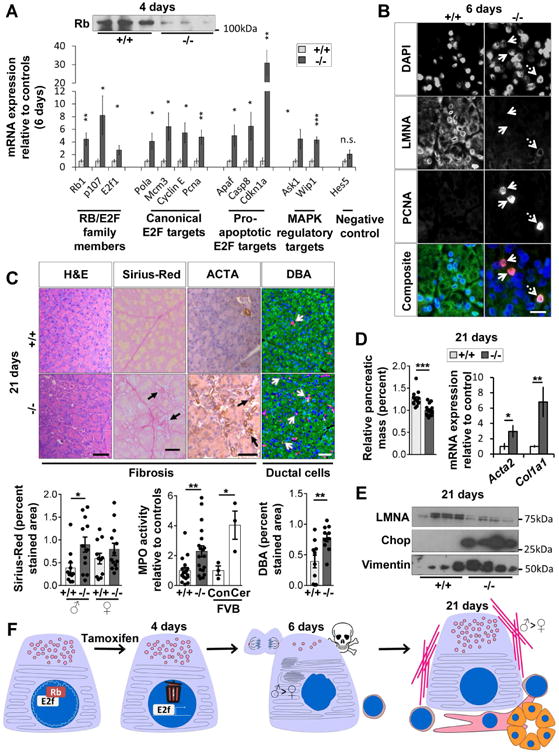
(A) RB protein is decreased in LMNA-KO pancreata (4d). E2F target-transcripts were quantified using RT-qPCR (6d). N=6 mice/group. (B) Immunofluorescence of pancreata (6d) from panel-A. Solid arrows: PCNA+/LMNA- cells. Dashed arrow: PCNA+/LMNA+ cells. Bar=20μm. (C) Pancreata (21d) were stained as indicated. Arrows highlight positive staining. Bar=50μm. Histograms: Quantification of Sirius-Red, MPO-activity and DBA-staining. Wild-type mice (FVB background), injected with saline (Con) or cerulein (Cer), were used as controls for the MPO assay. (D) Relative pancreatic mass (N=13-15 mice/group) and mRNA expression of fibrosis markers determined by RT-qPCR (N=8 mice/group). (E) Immunoblot of pancreata lysates (21d) from control/KO mice (4 pancreata/genotype). (F) Proposed model of LMNA role in pancreatic homeostasis. (*p≤0.05,**p≤0.01,***p≤0.001).
Pancreata harvested 21d post-tamoxifen injection revealed modest fibrosis, atrophy and ductal cell increase (Fig.2C,D), evidenced by Sirius-Red, alpha-smooth muscle actin (ACTA), and DBA-staining (Fig.2C). Increased myeloperoxidase (MPO) activity and Il1b, Tnf, and Ccl2 transcription (Fig.2C; Fig.S3D) suggested inflammation, although CD45 staining was comparable between groups (not shown). KO-pancreata had increased number of granules (Fig.S3E), but normal ER morphology and LC3 expression (not shown), suggesting partial recovery. Chop expression remained elevated (Fig2E). Fibrosis was supported by increased Acta2 and Col1a1 transcription, vimentin and ACTA proteins, increased hydroxyproline (Fig.2D,E; Fig.S4A-C), with Sirius-Red-staining and ACTA being more pronounced in male KO-pancreata (Fig.2C; Fig.S4A,B).
Collectively, we describe a role for LMNA in maintaining pancreatic acinar cell homeostasis through RB-E2F pathway regulation. LMNA-KO in the exocrine pancreas causes ER stress, loss of acinar cell markers, increased autophagy, apoptosis, and proliferation as a result of E2F activation by RB destabilization, which culminates in a phenotype that resembles chronic pancreatitis (Fig.S4D). The ER stress-response and fibrosis are more pronounced in KO males than females (Fig.2F). This male-preferential phenotype is similar to what is observed after hepatocyte-selective disruption of Lmna, which causes male (but not female) age-associated spontaneous mouse hepatosteatosis10.
RB is a nuclear lamina-associated transcription factor that is regulated through association with lamina-associated-polypeptide-2α and LMNA, while LMNA protects RB from proteasome-mediated degradation8. Pancreas-specific E2F1-E2F2 double-mutation generates a similar phenotype to what we observed, with increased transcription of E2F targets Pcna and Mcm311. Since we expect RB destabilization to upregulate E2F activity, our findings might appear contradictory to the E2F1-E2F2 double-deficient findings. However, several E2F and RB family members exist and play complex, interconnected, context and tissue-specific roles12. Therefore, we cannot exclude the possibility that additional signaling pathways, independent of RB or E2F, are dysregulated in LMNA-KO pancreata. The ER stress we observed is unlikely to have direct connection to RB or E2F, but it's not surprising that ER homeostasis is disrupted since the ER membrane is continuous with the nuclear membrane13. Further studies are needed to determine if patients with laminopathies have increased susceptibility to pancreatic disease. We posit that patients with FPLD2, and potentially other laminopathies, may have increased susceptibility to pancreatic dysfunction, independent of serum lipid levels (LMNA-KO mice used herein have normal serum triglycerides, not shown). RB regulation by LMNA is particularly relevant to FPLD2 since the RB-E2F pathway is important for lipogenesis in adipocytes, liver, and pancreas11,14,15.
A. Materials and Methods
Mouse experiments
Transgenic Cela1-CreERT2 and floxed Lmna mice were described previously1,2 and obtained from the laboratories of John Williams and Colin Stewart, respectively. All mice were in a C57BL/6 background. Briefly, CreERT2 expression is driven by the acinar cell-specific Cela1 promotor and exons 10 and 11 of the Lmna gene are flanked by loxP sites (Figure S1A). Tamoxifen (Sigma-Aldrich) was dissolved in corn oil (Dyets Inc.) at a concentration of 20mg/mL and incubated at 37°C overnight under constant shaking. Mice were weighed at day 0 and tamoxifen was administered on three consecutive days (days 0, 1, and 2) by intra-peritoneal (IP) injection at a dose of 75mg/kg as previously described1, unless noted otherwise. Tamoxifen administration results in recombination and disruption of Lmna in pancreatic acinar cells. Homozygous floxed, Cre-positive (Lmnafl/fl, Cre+) mice were used for experiments and Cre- littermates were used as controls. For all mice receiving tamoxifen, it was administered intraperitoneally or by oral gavage at the same time, frequency, and dose. Tissues were collected from euthanized mice between six and nine weeks of age at the indicated number of days after tamoxifen administration (Figures S1A).
Expression profiling and RT-qPCR analysis
Total RNA was isolated using a combination of Trizol (Invitrogen) and RNeasy mini-kit (Qiagen) isolation and quantified using NanoDrop (Thermo Fisher Scientific). For the microarray analysis, RNA quality and concentration were further assed using an RNA 6000 Nano BioAnalyzer (Agilent). Processing and analysis was determined using the Affymetrix protocol with Affymetrix Mouse Gene ST 2.1 array strips. Three male and one female KO and control sex-matched mice (6 days post-tamoxifen injection) were used for analysis. Criterion for dysregulated transcript expression includes an adjusted P-value of less than 0.05 and a relative expression of at least 2-fold above or below expression in controls. The Database for Annotation, Visualization and Integrated Discovery (DAVID)3,4 was used to analyze the list of transcripts that were significantly up-regulated in KO-pancreata using gene enrichment algorithms and the KEGG pathway database3. The gene expression omnibus series accession number is GSE97396. For qPCR analysis, cDNA was synthesized using the TaqMan Reverse Transcription Kit (Life Technologies). Eppendorf realplex2 (Eppendorf) and SYBR Green Supermix (Bio-Rad Laboratories, Inc.) were used for qPCR and quantification was performed as previously described5. Inflammation-related gene (IL-1β, IL-6, TNF-α, CCL2) expression level was assessed for all time points (3, 4, 6, and 21 days post-tamoxifen injection). The primers used for qPCR are listed in Table S1.
Histology and transmission electron microscopy
Samples for histology were fixed overnight in 10% buffered formalin and dehydrated in ethanol before processing and embedding in paraffin. Embedded tissues were then cut into 5 μm sections and stained using standard protocols with hematoxylin and eosin (H&E), PicroSirius Red stain, or the biotinylated lectin DBA (Dolichos biflorus agglutinin, Vector Laboratories). PicroSirius Red-stained areas were quantified using ImageJ7 software from 11-13 mice per sex per genotype (three fields of view per animal). DBA staining was quantified using Metamorph software (Molecular Devices) from 6-10 mice per group (at least three fields of view per animal). Samples were prepared and imaged by transmission electron microscopy as described previously5,6. Abnormalities were determined by the following criteria—for nuclei: acinar cell nuclei with irregular perimeter; for granules: acinar cells with few or small zymogen granules; for ER: acinar cells with condensed, swirling ER. Examples are shown in Figure S3A. Zymogen granule number and area were quantified using Metamorph software from five electron micrographs from 3-5 mice per genotype per time point.
TUNEL assay, immunofluorescence, and immunohistochemistry
Immunofluorescent staining and TUNEL assay were performed on 6-1 μm thick cryosections of tissue embedded in OCT. Samples were fixed in 100% ice-cold methanol or 4% neutral buffered paraformaldehyde, depending on antibody specifications. Sections were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) and washed with PBS. Sections were blocked with bovine serum albumin and goat serum before addition of the primary antibody in diluted blocking solution. Fluorophore-conjugated secondary antibodies (Invitrogen) were added after washing. Tissues were then washed and counterstained with DAPI (Life Technologies) before imaging. TUNEL staining was performed using the ApopTag Red In Situ Apoptosis Detection Kit (EMD Millipore). Sections were counterstained with anti-amylase antibody and DAPI before imaging. Immunohistochemistry was performed similar to described previously8. Briefly, paraffin sections were deparaffinized, rehydrated, and epitopes unmasked by boiling with Citra buffer (Biogenex). Tissues were then blocked with a solution of bovine serum albumin and goat serum before incubation with the indicated primary antibody. Vectashield DAB Peroxidase Substrate Kit was used to detect the primary antibody. Sections were counterstained with hematoxylin, mounted and imaged.
Tissue lysate preparation, immunoblot analysis and antibodies
Pancreata were dissected, homogenized, and protein fractionation was performed followed by concentration determination as described previously9 with the addition of the following phosphatase inhibitors: 1 μM (β-glycerophosphate disodium salt hydrate, 100 nM okadaicacid, and 5 mM sodium fluoride. Reducing Laemmli sample buffer was added to the samples before separation under denaturing conditions using sodium dodecyl sulfate polyacrylamide gel electrophoresis. Protein was then transferred to an Immobilon-P Membrane (EMD Millipore), followed by immunoblotting and visualization by chemiluminescence (Thermo Fisher Scientific). Equal loading was confirmed with Coomassie blue staining. Relative quantification of immunoblots band densitometry was performed using ImageJ7 software and was calculated relative to littermate mean value set to 1. The antibodies used included (company/source, target protein, clone/catalog number, application (WB, IF, IHC)): BD Biosciences, CD45, 30-F11, IHC; Cell Signaling Technology, Atf2, 20F1, WB; BiP/Grp78, PA5, WB; Casp7, 9492S, WB; cCasp3, ASP175.IHC; cCasp7, D198, WB; Chop, C50B12, WB; LMNA, 4C11, IF; PCNA, D3H8P, IF, IHC; PDI, C81H6, WB; LC3B, 2775, WB; Developmental Studies Hybridoma Bank, K19, Troma 3, WB; RB, 4.1C, WB; Neomarkers Inc, Hsp60, LK2, WB; Hsp70, W27, WB; Santa Cruz Biotechnology, Atf4, C-20, WB; LMNA, H-110, WB, IF; Lipase, A-3, WB; Sigma-Aldrich, Vimentin, 13.2, WB; ACTA, A5228, WB; Amylase, A8273, WB, IF; Thermo Scientific, ACTA, 710487, IHC. High background using multiple RB antibodies made RB determination difficult in some experimental replicates.
Myeloperoxidase (MPO) activity
MPO activity assay was performed as previously described10 for days 3, 4, 6 and 21 after tamoxifen injection. Briefly, pancreata were homogenized in 20 mM sodium phosphate buffer (pH 7.4), followed by centrifugation for 10 min (13,000g, 4°C). Pellets were suspended in 50 mM sodium phosphate buffer (pH 6) with 0.5% hexadecylmethylammonium bromide (Sigma-Aldrich) followed by four freeze/thaw cycles. Lysate was centrifuged and the supernatant containing MPO was collected. MPO-containing lysate was mixed with TMB ELISA substrate (Sigma-Aldrich) and reaction was stopped by addition of 2N H2SO4. MPO activity was normalized to DNA content and was calculated relative to littermate mean value set to 1.
Hydroxyproline assay
Pancreatic hydroxyproline content was assessed as previously described11. Briefly, pancreata were weighed, homogenized in distilled water, and incubated overnight at 110°C in 6 N HCl. The hydrolyzed homogenate was then filtered, evaporated by speed vacuum centrifugation and resuspended in distilled water. Samples (and serial dilutions of trans-4-hydroxy-L-proline (Sigma-Aldrich) to generate a standard curve) were mixed with 0.6% chloramine-T solution for 10 min at room temperature. Freshly prepared Ehrlich's solution (Sigma-Aldrich) was then added to the mixture, which was incubated for 45 min at 50°C, fo llowed by absorbance reading at 570 nm. Hydroxyproline amount was calculated based on a standard curve and values were normalized to pancreas wet weight.
Statistics
Unpaired, two tailed, student's T-tests were used for statistical analysis in GraphPad Prism 7 unless noted otherwise. Error bars represents standard error of measurement between biological replicates from at least two independent experiments. Positive cell counting, zymogen granule area and number, and stained surface area were performed by a blinded scorer after de-identifying and randomizing samples by shuffling files. P*≤0.05, P**≤0.01, P***≤0.001.
Study approval
Animal care guidelines were followed as approved by the University of Michigan Animal Care and Use Committee, as well as the recommendations in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health12.
Supplementary Material
Figure S1: Characterization of LMNA-KO pancreata. (A) Transgenic mice expressing CreERT2 driven by the Celal promotorwere bred with Lmnafl/fl mice. Tamoxifen was administered to promote recombination of the loxP sites and excision of exons 10 and 11 of thefloxed Lmna gene. (B) Representative LMNA immunostaining in control and KO-pancreata at 4-, 6- and 21 days- post-tamoxifen injection. White: LMNA; blue: DAPI. LMNA is efficiently deleted in -/- mice (Lmnafl/fl Cre+), but not in +/+ mice (Lmnafl/flCre-). Scale bar = 20 μm. (C) Representative hematoxylin and eosin stained sections for control and KO mice 3 and 4 days post-tamoxifen administration. No obvious phenotype was observed at the 3-day time point. Decreased eosin staining and smaller acini were noted 4 days after tamoxifen administration (insets). Scale bar= 50μm. (D) Immunoblotting of 4-day lysates from pancreata of control and KO mice (each lane corresponds to an animal), using supernatant soluble (amylase/Grp78) and pellet (chop) fractions. Protein loading was confirmed by Coomassie staining (lower 2 rows). The link between ER stress and lamins was recently reported in several contexts: (i) LMNA mislocalization to the ER triggers ER stress and cellular dysfunction13, (ii) an anti-viral drug causes an ER stress-like transcriptional response while impairing lamin A maturation14, and (iii) a LMNA mutation that causes familial dilated cardiomyopathy increases the unfolded protein response15. Although none of these studies report findings in the absence of LMNA, they all have altered lamins and ER stress response in common. (E) RT-qPCR was used to quantify changes in expression of acinar and ductal cell markers six days after tamoxifen injection. Decreased amylase transcription and increased transcription of keratins 8, 18, and 19 was observed. N = 6 mice/group. (F) Relative expression of several acinar cell markers was determined by microarray analysis and is shown with a heat map using CIMminer. Black indicates relative expression ∼1, red indicates expression >1, and blue indicates expression <1. *p≤0.05, **p≤0.01, ***p≤0.001.
Figure S2: Mice administered tamoxifen by oral gavage develop a similar phenotype to mice treated by the intraperitoneal route six days after administration. (A) Mice were administered tamoxifen (75mg/kg mouse weight) by oral gavage. Representative hematoxylin and eosin stained sections of KO and control pancreata. Decreased eosin staining, smaller acini and apoptotic bodies were noted. Scale bar = 20μm. (B) Immunoblots of the indicated proteins from pancreas lysates of mice treated with tamoxifen by oral gavage are similar to those treated by IP. The last lane represents a pancreas lysate from a mouse treated by IP for comparison. Similarities include increased Grp78 (S - supernatant), chop and presence of insoluble Grp78 in males (P - pellet). Apoptosis was assessed by cCasp7, which was elevated in KO mice. Coomassie stained gels confirm equal loading (bottom two rows). (C) Immunoblots of pancreatic amylase and lipase in oral gavage group showing exocrine pancreas enzymes are reduced in KO-pancreata. (D) DBA staining of pancreata isolated from the oral gavage group shows increased ductal cell staining in the KO-pancreata. (E) TUNEL staining in the oral gavage group shows increased apoptosis in KO-pancreata. *p≤0.05, **p≤0.01.
Figure S3: Characterization of ultrastructural abnormalities, inflammation and LC3 in KO-pancreata. (A) Ultrastructural abnormalities observed in KO-pancreata six days post-tamoxifen administration. Left panel: control acinar cells contained more round and regular-shaped nuclei, classified as “normal” (as quantified in Figure 1D). Middle panel: LMNA-/- pancreatic acinar cells often contained nuclei with irregular perimeters and apparent disruption of heterochromatin, classified as “abnormal” (as quantified in Figure 1D). Arrowheads indicate electron-dense material-containing vacuoles likely due to increased autophagy. Right panel: Swirling ER with lipid-like deposits in the center. Image was captured from LMNA-/- pancreata and represents “abnormal” ER (as quantified in Figure 1D). Scale bar= 2μm. (B) Granule quantification at six days post-tamoxifen injection. Note the decreased number of granules and granule area. (C) Immunoblots of LC3B from three independent experiments show increased expression of LC3-II in KO-pancreata (observed six days post-tamoxifen administration) compared to controls (left). Relative quantification of LC3B immunoblots shows increased LC3-II:LC3-I ratio in KO-pancreata (right). (D) RT-qPCR analysis of genes associated to pancreatitis 21 days post-tamoxifen injection. N = 5-8 mice/group. (E) Granule quantification at 21 days post-tamoxifen injection. Representative electron micrographs display the increased number of granules observed in KO cells at 21 days. *p≤0.05, ***p≤0.001.
Figure S4. Disruption of Lmna leads to alterations consistent with pancreatic fibrosis, preferentially in males. (A) Male KO-pancreata have higher expression of ACTA at 21 days after tamoxifen injection. Immunoblots for ACTA from two representative experiments show expression in KO males is consistently higher than in control males. (B) Relative quantification of ACTA immunoblots from four independent experiments (8-10 mice/genotype/sex). KO male pancreata have increased ACTA expression compared to male controls. (C) Quantification of pancreatic hydroxyproline (21d). KO-pancreata have increased hydroxyproline content compared to controls. *p≤0.05.
Table S1. Forward and reverse qPCR primers are listed 5′ to 3′ for the indicated target gene.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant R01 DK47918, and a Department of Veterans Affairs Merit Award (M.B.O.); and institutional NIH grant DK034933 to the University of Michigan. We thank Craig Johnson for assistance with the expression profiling analysis, and Stephen Lentz for assistance with immunofluorescence microscopy and quantification. Due to limit of 15 references, we apologize that we could not include all the citations we would have otherwise.
Footnotes
The authors have declared that no conflict of interest exists.
Author contributions: M.B.O. conceived of the project. J.S.E., J.B.C. and M.B.O. conceived of and designed the experiments. J.S.E., J.B.C. and R.A.D. performed the experiments. B.N. performed the electron microscopy and immunostaining. C.L.S. provided essential reagents and important input. J.S.E., J.B.C., M.B.O., J.A.W. and E.A.O. analyzed and interpreted the data. J.S.E., J.B.C. and M.B.O. wrote the paper. All authors reviewed and approved the manuscript.
Authors names in bold designate shared co-first authorship.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gruenbaum Y, et al. Annu Rev Biochem. 2015;84:131–64. doi: 10.1146/annurev-biochem-060614-034115. [DOI] [PubMed] [Google Scholar]
- 2.Worman HJ. J Pathol. 2012;226:316–25. doi: 10.1002/path.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davidson PM, et al. Trends Cell Biol. 2014;24:247–56. doi: 10.1016/j.tcb.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shackleton S, Lloyd DJ, et al. Nat Genet. 2000;24:153–6. doi: 10.1038/72807. [DOI] [PubMed] [Google Scholar]
- 5.Haque WA, et al. Diabet Med. 2002;19:1022–5. doi: 10.1046/j.1464-5491.2002.00796.x. [DOI] [PubMed] [Google Scholar]
- 6.Toth JI, et al. Proc Natl Acad Sci U S A. 2005;102:12873–8. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinberg RA. Cell. 1995;81:323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 8.Johnson BR, et al. Proc Natl Acad Sci U S A. 2004;101:9677–82. doi: 10.1073/pnas.0403250101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polager S, et al. Trends Cell Biol. 2008;18:528–35. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Kwan R, et al. Cell Mol Gastroenterol Hepatol. 2017;4:365–383. doi: 10.1016/j.jcmgh.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iglesias A, et al. J Clin Invest. 2004;113:1398–407. doi: 10.1172/JCI18879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong JL, et al. Nature. 2009;462:930–4. doi: 10.1038/nature08677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke B, et al. Nat Rev Mol Cell Biol. 2013;14:13–24. doi: 10.1038/nrm3488. [DOI] [PubMed] [Google Scholar]
- 14.Hansen JB, et al. Proc Natl Acad Sci U S A. 2004;101:4112–7. doi: 10.1073/pnas.0301964101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denechaud PD, et al. J Clin Invest. 2016;126:137–50. doi: 10.1172/JCI81542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Characterization of LMNA-KO pancreata. (A) Transgenic mice expressing CreERT2 driven by the Celal promotorwere bred with Lmnafl/fl mice. Tamoxifen was administered to promote recombination of the loxP sites and excision of exons 10 and 11 of thefloxed Lmna gene. (B) Representative LMNA immunostaining in control and KO-pancreata at 4-, 6- and 21 days- post-tamoxifen injection. White: LMNA; blue: DAPI. LMNA is efficiently deleted in -/- mice (Lmnafl/fl Cre+), but not in +/+ mice (Lmnafl/flCre-). Scale bar = 20 μm. (C) Representative hematoxylin and eosin stained sections for control and KO mice 3 and 4 days post-tamoxifen administration. No obvious phenotype was observed at the 3-day time point. Decreased eosin staining and smaller acini were noted 4 days after tamoxifen administration (insets). Scale bar= 50μm. (D) Immunoblotting of 4-day lysates from pancreata of control and KO mice (each lane corresponds to an animal), using supernatant soluble (amylase/Grp78) and pellet (chop) fractions. Protein loading was confirmed by Coomassie staining (lower 2 rows). The link between ER stress and lamins was recently reported in several contexts: (i) LMNA mislocalization to the ER triggers ER stress and cellular dysfunction13, (ii) an anti-viral drug causes an ER stress-like transcriptional response while impairing lamin A maturation14, and (iii) a LMNA mutation that causes familial dilated cardiomyopathy increases the unfolded protein response15. Although none of these studies report findings in the absence of LMNA, they all have altered lamins and ER stress response in common. (E) RT-qPCR was used to quantify changes in expression of acinar and ductal cell markers six days after tamoxifen injection. Decreased amylase transcription and increased transcription of keratins 8, 18, and 19 was observed. N = 6 mice/group. (F) Relative expression of several acinar cell markers was determined by microarray analysis and is shown with a heat map using CIMminer. Black indicates relative expression ∼1, red indicates expression >1, and blue indicates expression <1. *p≤0.05, **p≤0.01, ***p≤0.001.
Figure S2: Mice administered tamoxifen by oral gavage develop a similar phenotype to mice treated by the intraperitoneal route six days after administration. (A) Mice were administered tamoxifen (75mg/kg mouse weight) by oral gavage. Representative hematoxylin and eosin stained sections of KO and control pancreata. Decreased eosin staining, smaller acini and apoptotic bodies were noted. Scale bar = 20μm. (B) Immunoblots of the indicated proteins from pancreas lysates of mice treated with tamoxifen by oral gavage are similar to those treated by IP. The last lane represents a pancreas lysate from a mouse treated by IP for comparison. Similarities include increased Grp78 (S - supernatant), chop and presence of insoluble Grp78 in males (P - pellet). Apoptosis was assessed by cCasp7, which was elevated in KO mice. Coomassie stained gels confirm equal loading (bottom two rows). (C) Immunoblots of pancreatic amylase and lipase in oral gavage group showing exocrine pancreas enzymes are reduced in KO-pancreata. (D) DBA staining of pancreata isolated from the oral gavage group shows increased ductal cell staining in the KO-pancreata. (E) TUNEL staining in the oral gavage group shows increased apoptosis in KO-pancreata. *p≤0.05, **p≤0.01.
Figure S3: Characterization of ultrastructural abnormalities, inflammation and LC3 in KO-pancreata. (A) Ultrastructural abnormalities observed in KO-pancreata six days post-tamoxifen administration. Left panel: control acinar cells contained more round and regular-shaped nuclei, classified as “normal” (as quantified in Figure 1D). Middle panel: LMNA-/- pancreatic acinar cells often contained nuclei with irregular perimeters and apparent disruption of heterochromatin, classified as “abnormal” (as quantified in Figure 1D). Arrowheads indicate electron-dense material-containing vacuoles likely due to increased autophagy. Right panel: Swirling ER with lipid-like deposits in the center. Image was captured from LMNA-/- pancreata and represents “abnormal” ER (as quantified in Figure 1D). Scale bar= 2μm. (B) Granule quantification at six days post-tamoxifen injection. Note the decreased number of granules and granule area. (C) Immunoblots of LC3B from three independent experiments show increased expression of LC3-II in KO-pancreata (observed six days post-tamoxifen administration) compared to controls (left). Relative quantification of LC3B immunoblots shows increased LC3-II:LC3-I ratio in KO-pancreata (right). (D) RT-qPCR analysis of genes associated to pancreatitis 21 days post-tamoxifen injection. N = 5-8 mice/group. (E) Granule quantification at 21 days post-tamoxifen injection. Representative electron micrographs display the increased number of granules observed in KO cells at 21 days. *p≤0.05, ***p≤0.001.
Figure S4. Disruption of Lmna leads to alterations consistent with pancreatic fibrosis, preferentially in males. (A) Male KO-pancreata have higher expression of ACTA at 21 days after tamoxifen injection. Immunoblots for ACTA from two representative experiments show expression in KO males is consistently higher than in control males. (B) Relative quantification of ACTA immunoblots from four independent experiments (8-10 mice/genotype/sex). KO male pancreata have increased ACTA expression compared to male controls. (C) Quantification of pancreatic hydroxyproline (21d). KO-pancreata have increased hydroxyproline content compared to controls. *p≤0.05.
Table S1. Forward and reverse qPCR primers are listed 5′ to 3′ for the indicated target gene.