

Abstract
JAK2 constitutive activation/overexpression is common in classical Hodgkin lymphoma, and several cytokines stimulate Hodgkin lymphoma cells by recognizing JAK1-/JAK2-bound receptors. JAK blockade may thus be therapeutically beneficial in Hodgkin lymphoma. In this phase II study we assessed the safety and efficacy of ruxolitinib, an oral JAK1/2 inhibitor, in patients with relapsed/refractory Hodgkin lymphoma. The primary objective was overall response rate according to the International Harmonization Project 2007 criteria. Thirty-three patients with advanced disease (median number of prior lines of treatment: 5; refractory: 82%) were included; nine (27.3%) received at least six cycles of ruxolitinib and six (18.2%) received more than six cycles. The overall response rate after six cycles was 9.4% (3/32 patients). All three responders had partial responses; another 11 patients had transient stable disease. Best overall response rate was 18.8% (6/32 patients). Rapid alleviation of B-symptoms was common. The median duration of response was 7.7 months, median progression-free survival 3.5 months (95% CI: 1.9–4.6), and the median overall survival 27.1 months (95% CI: 14.4–27.1). Forty adverse events were reported in 14/33 patients (42.4%). One event led to treatment discontinuation, while 87.5% of patients recovered without sequelae. Twenty-five adverse events were grade 3 or higher. These events were mostly anemia (n=11), all considered related to ruxolitinib. Other main causes of grade 3 or higher adverse events included lymphopenia and infections. Of note, no cases of grade 4 neutropenia or thrombocytopenia were observed. Ruxolitinib shows signs of activity, albeit short-lived, beyond a simple anti-inflammatory effect. Its limited toxicity suggests that it has the potential to be combined with other therapeutic modalities. ClinicalTrials.gov: NCT01877005
Introduction
Hodgkin lymphoma (HL) is regarded as a curable malignancy in most cases, yet treatment failure still occurs in about 10% of patients with early-stage disease.1 In advanced-stage disease, up to 10% of cases do not reach complete remission and are thus considered to have primary refractory HL,2 while 20–30% of primary responders eventually relapse following first-line treatment.3
For most patients with relapsed or refractory HL (R/R HL), the standard of care consists of high-dose salvage chemotherapy followed by autologous stem-cell transplantation (SCT). For patients who develop R/R HL within 1 year of autologous SCT, the prognosis proves extremely poor, since they have a median survival of 1.2 years.4 For patients in whom all classical approaches have failed, new strategies, including checkpoint inhibitors targeting PD-1 and antibody-drug conjugates targeting CD30, have become part of the therapeutic armamentarium against R/R HL.5–8 However, patients with multiple relapses or those who develop refractory disease remain in medical need, especially those in whom treatment with brentuximab-vedotin (BV) and PD-1 blockers fails.
Classical HL is characterized by the presence of Hodgkin and Reed-Sternberg (HRS) cells and their variants.9 HRS cells were demonstrated to shape their environment by secreting immunosuppressive cytokines and chemokines.10 With this in mind, the Janus kinase (JAK) – signal transducer and activator of transcription (STAT) pathway appears to be a relevant cytokine-induced signal transduction pathway that has been shown to transfer signals directly from cell surface cytokine receptors to the cell nucleus. Given that enhanced JAK-mediated signaling has been demonstrated in a significant number of HL patients,11 this signaling pathway has become a focus for developing novel therapeutic agents for the disease. Van Roosbroeck et al. reported the translocation of JAK2 in several cases of HL,12 and JAK inhibition was shown to decrease the proliferation of cell lines. Although such translocations are relatively rare, 9p24.1 genomic amplification including the JAK2 locus appears common in HL, along with increased protein expression and activity, resulting in the constitutive activation of STAT6, an essential messenger of tumor cell growth.13–15 In corollary, JAK 1/2 inhibition may be suitable to target the constitutive activation caused by either JAK2 translocation or JAK2 amplification and to modify the reactive microenvironment which contributes to HL growth via aberrant cytokine production.16
Ruxolitinib is the first potent, selective, and oral inhibitor of JAK1/2 being developed for clinical use.17 Its major effects include inhibition of proliferation, induction of apoptosis, and reduction in cytokine plasma levels, all mediated by the drug’s ability to inhibit JAK-induced phosphorylation of STAT.18 Used in the treatment of myelofibrosis, ruxolitinib had durable efficacy in reducing splenomegaly and alleviating constitutional symptoms, the patients gained weight and their general physical condition improved.19 The dose-limiting toxicity was thrombocytopenia, which was fairly well managed by dose reductions or brief interruptions of treatment. In the present phase II study, we sought to investigate the safety and efficacy of ruxolitinib in patients with R/R HL. Exploratory biomarker analyses pertaining to plasma cytokine profiles and aberrations of JAK2 were also carried out.
Methods
Patients’ eligibility
Patients aged 18 years or older with a diagnosis of R/R HL for whom no treatment with proven efficacy was available were eligible to enter the trial after having receiving at least one prior therapy provided that they had measurable nodal disease at baseline (≥1 cm in the longest transverse diameter, clearly measurable in at least two perpendicular dimensions) on computed tomography or magnetic resonance imaging, as well as an Eastern Cooperative Oncology Group performance score of ≤3. Additional inclusion criteria were an absolute neutrophil count ≥1.0 × 109/L, platelet count ≥75 × 109/L, serum creatinine ≤1.5 × upper limit of normal, serum bilirubin ≤1.5 × upper limit of normal, and ALT and AST levels ≤2.5 or ≤5.0 × upper limit of normal in the event the transaminase increase was due to HL-related liver disease. Pregnant or lactating patients were not allowed to enter the trial, and men and women of childbearing potential had to agree to employ an adequate contraceptive method during the study treatment. Patients were permitted to have received an undefined number of prior lines of therapy, and a previous allogeneic SCT was likewise allowed provided that patients had not received any immunosuppressive therapy within the 90 days prior to starting the screening procedures. Patients were required to have a life expectancy of ≥3 months.
Study design and treatment
This multicenter, open-label, phase II study (HIJAK, NCT01877005) was conducted at ten LYSA centers in France and Belgium, with patients recruited from July 2013 through December 2014. Its primary efficacy endpoint was overall response rate (ORR), defined as the proportion of patients with a complete response or partial response at 6 months of treatment by investigator assessment based on the revised 2007 International Harmonization Project response criteria for malignant lymphoma.20 Secondary objectives included relief of B symptoms, best ORR (occurring at any time during study), duration of response, progression-free survival, overall survival, as well as the incidence and severity of adverse events.
The study was carried out in line with the ethical principles of the Helsinki Declaration and in compliance with the International Conference on Harmonization Guideline for Good Clinical Practice. The protocol was approved by the institutional review board of each study site and written informed consent was obtained from all patients.
The starting dose of ruxolitinib was 20 mg given twice daily during six 28-day cycles for the induction period if the platelet count was >200 × 109/L. The ruxolitinib dose was decreased to 15 mg twice daily in patients with platelet counts between 75 × 109/L and 200 × 109/L. Patients who achieved at least stable disease at the end of cycle 6 and who had, in the investigator’s opinion, a clinical benefit were eligible to continue ruxolitinib (15 mg or 20 mg), which was defined as “maintenance” therapy. Treatment could be continued for up to 2 years or until progressive disease, intolerability, or as long as the investigator thought that there was clinical benefit.
Administration of the study drug could be stopped for any grade ≥3 non-hematologic toxicity, with the exception of deep venous thrombosis and alopecia. Following event resolution to grade ≤1, ruxolitinib could be resumed, with a 5 mg dose reduction and a maximum delay of 4 weeks. Mandatory dose decreases or interruptions for hematologic toxicity as well as the rules for permanent discontinuation are detailed in the Online Supplementary Appendix. Growth factors were allowed as per American Society of Clinical Oncology guidelines and infectious prophylaxis as per the guidelines of Heine et al.21
Study assessments
Baseline assessments comprised documentation of disease- related symptoms, physical examination, laboratory tests, and imaging studies of the neck, chest, abdomen, and pelvis, using computed tomography or magnetic resonance imaging. Biopsy prior to inclusion was recommended, but not mandatory. Tumors were measured at baseline, at the end of every two cycles of ruxolitinib, and following the six-cycle induction, as well as during maintenance therapy. Given the exploratory nature of the study, there was no centralized review of computed tomography response. However, positron emission tomographic images of the responders were all centrally reviewed by a nuclear physician (ASC) to confirm partial or complete metabolic response based on the Deauville five-point scale. The evaluable study population for efficacy was restricted to patients who had received at least 28 days of the study drug.
Safety was monitored for up to 1 month after treatment. Adverse events were summarized by means of the Medical Dictionary for Regulatory Activities, and graded using the National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI-CTCAE), version 3.0. Laboratory abnormalities were assessed according to NCI-CTCAE version 4.0. Only grade 3 or 4 toxicities and grade 2 infections were to be reported. All patients were included in the toxicity analysis.
Exploratory biomarker analysis
Blood samples (5 mL) were taken at baseline prior to drug administration and on day 1 of cycle 2 for the measurement of 27 cytokines related to the immune system using bead-based immunoassays. JAK2 gains, amplifications, and gene rearrangements were also investigated using fluorescent in situ hybridization with two tri-color sets of probes associating JAK2/9p24 break-apart probes with a control centromeric probe (CEP9/9q21): the already prepared probes from Empire genomics on the one hand, and the association of the JAK2 B/A probe from Kreatech with the CEP9 probe from Vysis on the other hand. The CD274/PDL1 and PDCD1LG2/PDL2 loci at 9p24 were studied with home-made prepared bacterial artificial chromosome probes purchased from the Chori BACPAC Resources Center (Oakland, CA, USA). Extraction, labeling and hybridization were performed on paraffin-embedded tissue, as previously reported.22
Statistical methods
The sample size for this phase II study was calculated using an exact single-stage phase II design.23 A two-stage design with interim analysis for activity or toxicity was not planned given the very advanced stage of the patients, the relative paucity of alternative options, and the potential toxicity of ruxolitinib that was expected to be in the low range, based on myelofibrosis data. The treatment was considered ineffective if the ORR was ≤15%, and effective if the ORR was ≥35%. Under the assumption of an alpha first-order risk error set at 5% and beta at 20% with a one-sided test, it was deemed necessary to include a total of 28 evaluable patients with a cut-off number of eight. If at least eight patients had a response, the hypothesis of an ORR ≤15% was rejected with both a target error rate and an actual error rate of 0.05. If seven or fewer patients had a response, the hypothesis of an ORR ≥35% was rejected with a target error rate of 0.2 and an actual error rate of 0.187. The ORR estimate and its 90% confidence intervals (CI) were calculated for all patients who completed at least one cycle of the study drug.
The Kaplan-Meier method was employed to estimate the median value and its 95% CI for time to response, duration of response, progression-free survival and overall survival. The safety analysis comprised all patients who received at least one dose of the study drug. All statistical analyses were performed using SAS software, version 9.2. P-values <0.05 were considered statistically significant. All available data were included in data listings and tabulations, with no imputations of values for missing data. An interim analysis was neither planned nor performed.
Results
Patients’ disposition and characteristics
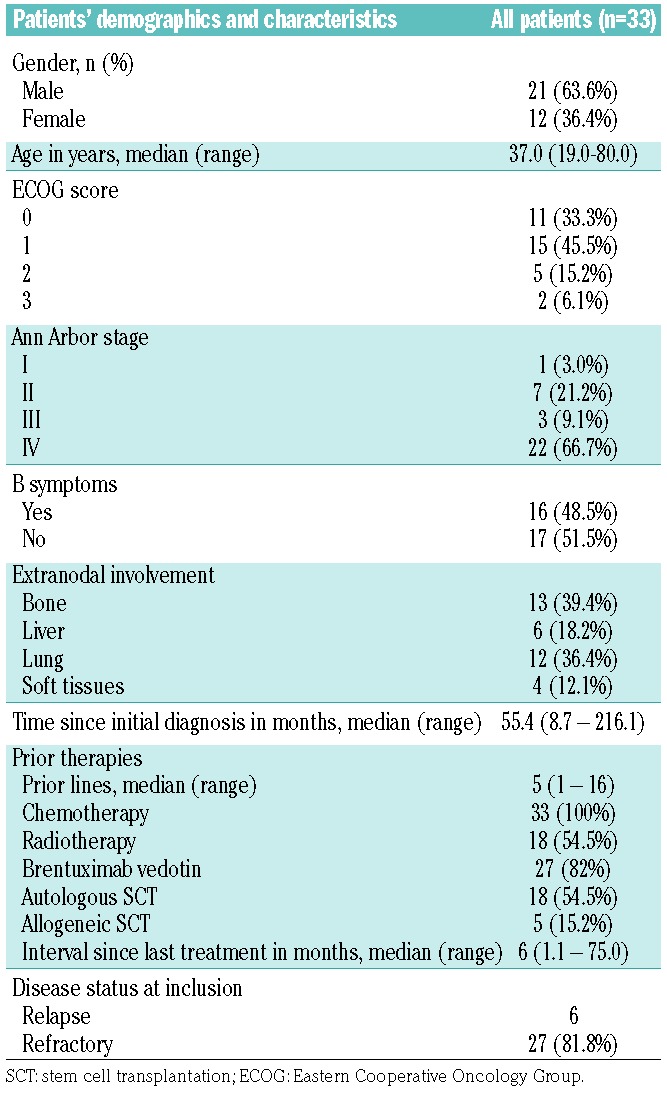
The patients’ characteristics are listed in Table 1. From July 2013 to December 2014, a total of 33 patients with R/R HL were recruited. Their median age was 37 years (range, 19–80). Most of the patients had advanced HL (stage III/IV) and had been heavily pretreated, with a median number of five prior regimens including autologous SCT (54%), allogeneic SCT(15%), and BV (82%). Of the 33 patients recruited, 27 (82%) had refractory HL and 22 had biopsy-confirmed relapse of HL. Among the six patients displaying a response, a biopsy was performed in five of them at relapse [8 days, 12 days, 6 weeks (n=2) and 14 months prior to inclusion in the study].
Table 1.
Patient’s demographics and characteristics.
Patients’ exposure to the study drug
The median number of ruxolitinib cycles administered was four (range, 1 to 12) (Table 2). Nine patients received all six of the planned cycles of ruxolitinib and six of these patients continued on maintenance therapy with the JAK inhibitor. The remainder discontinued ruxolitinib therapy, most because of progressive disease and in one case due to adverse events.
Table 2.
Treatment exposure and modifications.

Responses and outcomes
The patients’ disposition through the study is illustrated in Figure 1. Among the 33 HL patients included in the trial, one patient did not complete the first cycle of treatment because of progressive disease and was not, therefore, included in the efficacy analysis. At the end of the ruxolitinib induction period (6 months) three of 32 patients had a response, for an ORR of 9.4% (90% CI: 2.6–22.5%); the response in all three was partial. At some point during induction six of the 32 patients had a response, which was, in all six cases a partial response, for a best ORR of 18.8% (95% CI: 7.2–36.4%). A detailed analysis of the responders’ characteristics is provided in Table 3. Figures 2 and 3 illustrate metabolic evolution in two patients. Interestingly, UPN 611001, who had achieved a partial response after six cycles of treatment, eventually entered complete remission during the follow-up, beyond the six cycles. Achievement of complete metabolic response was confirmed by central review. At the time of writing, two patients (UPN 611001 and 881001) are still taking ruxolitinib. Figure 4 illustrates changes in target tumor measurements in individual patients. The best reduction, if any, at any time throughout treatment is shown.
Figure 1.

Patients’ disposition. *N months on maintenance therapy: 4, 6, 6, 21, 16, 22; PD: progressive disease.
Table 3.
Characteristics of responders (best response achieved during the 6-month ruxolitinib induction).

Figure 2.

Response after ruxolitinib. Illustrative patient (UPN 601004). (A) Positron emission tomography (PET)-computer tomography (CT) frontal view. (B) PET-CT sagittal view. Partial response with allievation of B symptoms and blood inflammation was achieved 2 months after starting ruxolitinib. At month 6, the patient had slowly progressive disease but refused to stop ruxolitinib. CRP: C- reactive protein.
Figure 3.

Response after ruxolitinib. Illustrative patient (UPN 601001). Comparison of frontal positron emission tomography (PET)-scan prior to inclusion and after 2 months of ruxolitinib. There was a rapid improvement of constitutional symptoms after a few days on ruxolitinib. PET after 2 months showed metabolic partial response with a total volume reduction of tumor lung lesions of 64%.
Figure 4.

Waterfall plot demonstrating percent change from baseline in target tumor dimensions (best response, n=27). Of note, among the 32 patients evaluable for disease response, five had no end-of-treatment SPD measurements by computer tomography (CT) scan as planned by protocol because there were obvious signs of disease progression. *Persisting positive positron emission tomography scan, considered as partial response.
In addition, during the 6-month induction, transient stable disease was recorded in 11 patients, albeit of limited duration. Overall, the disease control rate (including stable disease with complete and partial responses) was 53.1% (17/32 patients) (95% CI: 34.7–70.9%) with a median duration of 1.9 months.
The alleviating effect on systemic symptoms, such as pruritus, fever, and sweating, was noteworthy, starting within the first month of drug administration and commonly lasting. The impact was most remarkable on the control of pruritus, which affected 35.5% of patients prior to initiating therapy but only 6.6% after the first cycle of ruxolitinib. Sweating, which was present in 32.2% of the patients at inclusion, was reduced, with 20% still having this symptom after one cycle of treatment. Fever was abolished in 3/4 patients with this symptom at inclusion.
The median follow-up was 17.5 months. As illustrated in Figure 5, the median progression-free survival was 3.5 months (95% CI: 1.9–4.6). The median duration of response was 7.7 months (95% CI: 1.8-NA) for the six patients who eventually a achieved response (data not shown). Overall, 30 patients had progressive disease, with 97% at the initial site and/or 60% at new sites. Following ruxolitinib discontinuation, 25 (83.3%) patients were given further treatments, consisting of chemotherapy in 19, and immunotherapy in nine, the latter comprising rituximab in four, BV in three, and nivolumab in two. Transplantation was eventually carried out in five patients, and was allogeneic in four cases and autologous in the remaining one. Among the 25 patients prescribed further therapy, the observed complete and partial response rates were 10% and 15%, respectively. Overall, 12 patients died on account of lymphoma progression (83.3%), toxicity of other treatments (8.3%), or other reasons (8.3%). The median overall survival was 27.1 months (95% CI: 14.4–27.1).
Figure 5.

Kaplan-Meier estimate of progression-free survival in 32 evaluable patients with Hodgkin lymphoma receiving ruxolitinib.
Safety
All patients enrolled in the study received at least one dose of study medication and were, therefore, included in the safety analysis. Overall, 40 adverse events were observed in 14/33 patients (42.4%). In six patients, the adverse events were related to the ruxolitinib therapy. In eight of them, the events were of grade ≥3 (Table 4A). Among the 40 adverse events recorded, 30 (75%) occurred during induction and 18 (45%) were related to ruxolitinib. No drug-related deaths were recorded. One adverse event resulted in permanent drug discontinuation, while 87.5% of the adverse events resolved without sequelae. The characteristics and grade of the adverse events, listed by system organ class and preferred terms, are displayed in Table 4B. Twenty-five (62.5%) were of grade ≥3. These were mostly anemia (n=11), all considered related to ruxolitinib. Other main causes of grade ≥3 adverse events included lymphopenia (n=4), infections (n=3) and miscellaneous causes. Of note, no cases of grade 4 neutropenia or thrombocytopenia were observed.
Table 4.
Treatment-emergent adverse events.
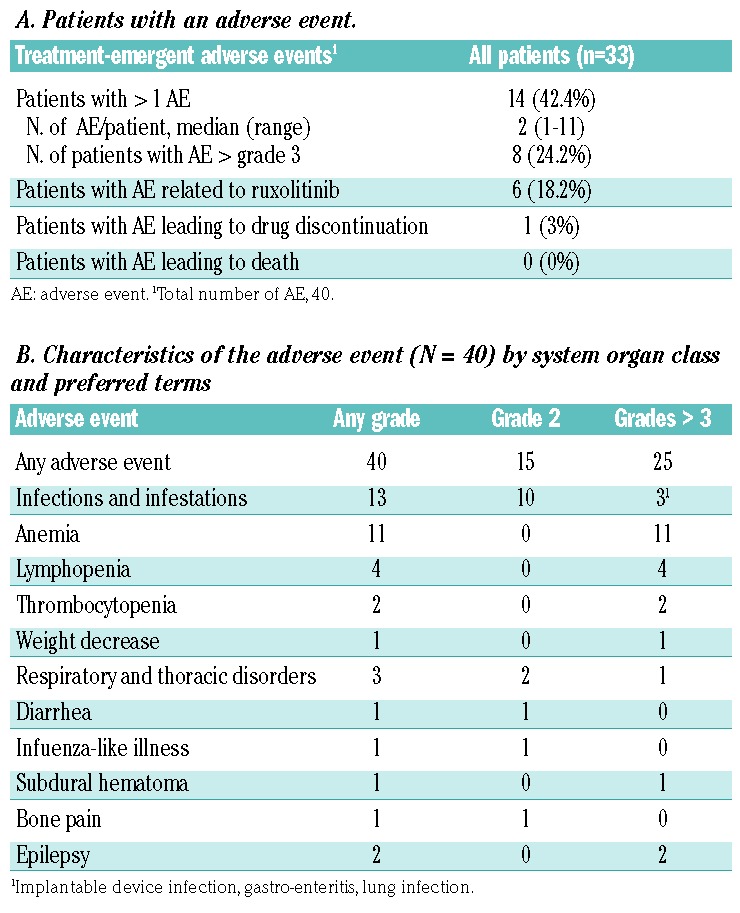
Eight serious adverse events were reported in four patients, during induction (n=5), maintenance (n=1) or after the end of treatment (n=2). These serious adverse events consisted of infection in three patients (device- related sepsis, gastroenteritis, and lung infection). The other serious adverse events were anemia, diarrhea, subdural hematoma, bone pain, and pulmonary embolism. Two serious adverse events (anemia, lung infection) were deemed drug-related and six were considered grade ≥3: infection (n=3), anemia, subdural hematoma, and pulmonary embolism. Of the eight serious adverse events, six resolved without sequelae, while the device-related sepsis and pulmonary embolism, observed in the same patient, persisted until the patient died due to progressive disease and were thus not considered as the cause of death. Among the 33 patients, one second primary malignancy was observed (adenocarcinoma of the colon in an 80-year old male patient).
Biomarker analysis
Using bead-based immunoassays, plasma levels of 27 cytokines related to the immune system were measured at baseline and after the first cycle of treatment. At baseline, there was no difference in cytokine levels between responders and non-responders. In responders, the only cytokine that decreased significantly was CX-CL10 (P=0.01). In patients presenting with pruritus (n=11), the levels of platelet-derived growth factor-BB (PDGF-BB) (Online Supplementary Appendix), interleukin (IL)-5, IL-10, IL-12, IL-13, IL-17, eotaxin, fibroblast growth factor basic (FGF basic), macrophage inflammatory protein 1b (MIP1b), regulated on activation, normal T-cell expressed and secreted (RANTES), and vascular endothelial growth factor (VEGF) were significantly increased. In the latter patients, ruxolitinib treatment significantly decreased the levels of PDGF-BB, IL-10, IL-12, IL-13, IL-17, FGF basic and VEGF. Among the patients who could be analyzed for JAK2 amplification in HRS cells (n=12), polysomy (suggesting hyperdiploidy) was detected in all of them, and specific JAK2 amplification in only one. This latter patient achieved a partial response as determined by computed tomography criteria and also a positron emission tomography-determined response lasting 4 months. It is note- worthy that the PDL1 and PDL2 loci (which are in the vicinity of the JAK2 locus at 9p24), analyzed by fluorescent in situ hybridization with bacterial artificial chromosome probes, showed the same pattern of gains as for the JAK2 locus.
Discussion
JAK/STAT activation, driven by an aberrant network of cytokines and chemokines in the HL microenvironment, is critical for the proliferation and survival of neoplastic HRS cells.24,25 The JAK/STAT pathway also plays a role in immune evasion by HL cells via the secretion of chemokines leading to Th2 homing or via the regulation of PD-L1/L2 expression, which confers an immune privilege to HRS cells. Chromosome 9p24.1/PD-L1/PD-L2 alterations increase the abundance of the PD-1 ligands, PD-L1 and PD-L2, and their further induction through JAK/STAT signaling.26–28 This complex crosstalk between malignant HRS cells and the reactive microenvironment could be targeted to overcome chemoresistance. Based on this rationale, we explored JAK 1/2 inhibition in a phase II study of fixed dose ruxolitinib in patients with advanced HL patients before the onset of the era of PD-1 blockers. With an ORR of 9.4% at the end of the 6-month induction period, this study did not reach its primary efficacy goal. Nevertheless, when including transient responses seen before the 6-month evaluation, the ORR was 18.8% in some heavily pretreated patients, most of whom were refractory and had failed treatment with BV. These responses were sometimes durable (median=7.7 months). Some other patients had disease control, but with uncertain clinical benefit. A notable finding to be highlighted was the relief of B symptoms and pruritus, which was quick and long-lasting, resulting in a number of patients being reluctant to discontinue the compound, despite progressive disease. The latter effect should not be interpreted as a proven surrogate of anti-lymphoma activity.
These results tend to lend some support to the concept of JAK1/2 inhibition as a potential therapeutic means in HL. There are presently only scarce data available on the use of ruxolitinib in HL. In a preliminary report of an ongoing study, Kim et al. described rapid achievement of disease control (1 complete response, 5 partial responses, 1 stable disease) in 13 patients with advanced HL treated with ruxolitinib at a dose of 20 mg bid.29 Younes et al. reported changes in tumor measurements in HL patients treated in a phase I study with SB1518, an inhibitor of JAK2 and FLT-3.30 In vitro, AZD1480, an inhibitor of JAK1 and JAK2, could regulate proliferation in HL cell lines.27 The multikinase inhibitor lestaurtinib also inhibited growth and increased apoptosis of HL cell lines and HL cells from lymph nodes.31 Finally, a clinical grade JAK2 inhibitor, fedratinib, inhibited the proliferation of classical HL cell lines in a JAK2 copy number-dependent manner implying decreased phosphorylation of STAT and expression of downstream targets including PD-L1 showing immunomodulation by JAK inhibitors.32
If JAK2 is actually an appropriate target, questions arise as to why the study outcome was not more convincing. Could the drug’s limited activity be attributed to insufficient dosage? Given that we observed unambiguous cytokine profile changes and frequent improvements in B symptoms, it would seem that the dosage of 20 mg twice daily, a dosage at which target inhibition occurs in myelofibrosis,33 was appropriate. Another factor possibly influencing the outcome was our patients’ disease stage, represented by a high percentage of refractoriness. At this late stage, the genetic changes would be so complex that selective inhibition of JAK is insufficient in cells dependent on other signaling pathways to promote their survival, thus further curbing the study’s potential. It is known that genomic aberrations, such as chromosome breakpoints, are more numerous in later clinical stages of HL.34 Mechanisms of resistance to JAK/STAT inhibition have been reported such as a feedback loop of paradoxically activated extracellular signal-regulated kinases 1 and 2 (ERK1/2).27 Aberrations of the 9p24.1 amplicon, which contains the JAK2 gene, are more frequent in advanced disease.28 Surprisingly, in our patients, a low incidence of JAK2 amplification was seen, suggesting a low proportion of patients harboring the target of ruxolitinib, although this inference should be considered with caution since not all patients could be analyzed.
With respect to safety, ruxolitinib was by and large well-tolerated, with no drug-related mortality reported. The most prominent toxicities included drug-related anemia and manageable infectious events with no specific pattern. The relative lack of hematologic toxicity suggests that it could be feasible to combine ruxolitinib treatment with genotoxic compounds. For patients who discontinued ruxolitinib therapy, a switch to chemotherapy and/or immunotherapy was feasible, suggesting that the compound does not jeopardize further treatment.
The question now remains as to how this compound can best be utilized in the future. The exploratory nature of our study did not allow identification of the best candidates on the basis of clinical stage or biomarkers. The cytokine profile showed some changes in patients with pruritus, but these changes were not correlated with clinical response. Although JAK2 status was explored in a minority of patients, the only patient with JAK2 amplification achieved a response. It will be important to focus on biomarker results in ongoing studies of JAK inhibition in HL. Given ruxolitinib’s limited benefits as monotherapy, use in combination with other drugs may possibly enhance its therapeutic potential. Ruxolitinib, which has no overlapping toxicity with chemotherapy, has been combined with hypomethylating agents, lenalidomide, and even intensive chemotherapy.35–38 In vitro data have shown that ruxolitinib could restore the sensitivity of cisplatin-resistant cell lines with higher Jak2 expression.39 Interestingly, the combination of BV with ruxolitinib resulted in additive and synergistic killing in a xenograft mouse model of HL through a mechanism involving mitochondrial control of apoptosis.40 Another means to boost ruxolitinib’s potential would be to combine it with agents blocking other signaling pathways. Interestingly, the combination of ruxolitinib with a Bcl2/Bcl-xL inhibitor displayed dramatic synergy in an adult T-cell leukemia cell line via a mechanism implying BAX activation.41 Finally, the effect of combining chemical JAK blockade and an anti-PD1/L1 strategy should be analyzed in HL, keeping in mind, however, that a potential antagonism may be encountered due to these two drugs acting on the same target, given that PD1-L1 expression is dependent on JAK2 activity.
In conclusion, based on a strong biological rationale for clinical evaluation of JAK2 blockade in HL, we initiated a phase II study of ruxolitinib in R/R HL patients. The study failed to fulfill the efficacy criteria for further development of the drug as monotherapy. Nonetheless, in patients with very advanced disease ruxolitinib showed hints of activity that surpassed solely an anti-inflammatory effect. This may suggest that further improvements will come from a more complete inhibition of signaling pathways involved in HRS cell survival or from combination with chemotherapy, such as BV.
Supplementary Material
Acknowledgments
The authors would like to thank the patients, their families and their caregivers who made this study possible. We also thank all the study investigators and study staff at each of the clinical sites. For the LYSARC, we acknowledge the project manager and all members of the data monitoring committee. We express our thanks to Loïc Chartier and Sami Boussetta, biostatisticians at the LYSARC, who contributed to the statistical design and analysis of the study. Editorial assistance was provided by Cremer Consulting.
Footnotes
Preliminary results were presented at the 58th Annual Meeting of the American Society of Hematology, held on December 3 – 6, 2016, in San Diego, USA.
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/5/840
References
- 1.Meyer RM, Hoppe RT. Point/counterpoint: early-stage Hodgkin lymphoma and the role of radiation therapy. Hematology Am Soc Hematol Educ Program. 2012;2012:313–321. [DOI] [PubMed] [Google Scholar]
- 2.Horning SJ. Primary refractory Hodgkin’s disease. Ann Oncol. 1998; 9(Suppl 5): S97–101. [DOI] [PubMed] [Google Scholar]
- 3.Kuruvilla J, Keating A, Crump M. How I treat relapsed and refractory Hodgkin lymphoma. Blood. 2011;117(16):4208–4217. [DOI] [PubMed] [Google Scholar]
- 4.Arai S, Fanale M, DeVos S, et al. Defining a Hodgkin lymphoma population for novel therapeutics after relapse from autologous hematopoietic cell transplant. Leuk Lymphoma. 2013;54(11):2531–2533. [DOI] [PubMed] [Google Scholar]
- 5.Ansell SM. Hodgkin lymphoma: MOPP chemotherapy to PD-1 blockade and beyond. Am J Hematol. 2016;91(1):109–112. [DOI] [PubMed] [Google Scholar]
- 6.Armand P, Shipp MA, Ribrag V, et al. Programmed death-1 blockade with pembrolizumab in patients with classical hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol. 2016. June 27 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363(19):1812–1821. [DOI] [PubMed] [Google Scholar]
- 8.Moskowitz C. Novel agents and strategies in transplant-eligible patients with relapsed and refractory Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2016;2016(1):331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slovak ML, Bedell V, Hsu YH, et al. Molecular karyotypes of Hodgkin and Reed- Sternberg cells at disease onset reveal distinct copy number alterations in chemosensitive versus refractory Hodgkin lymphoma. Clin Cancer Res. 2011;17(10):3443–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aldinucci D, Gloghini A, Pinto A, De Filippi R, Carbone A. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumour growth and immune escape. J Pathol. 2010;221(3):248–263. [DOI] [PubMed] [Google Scholar]
- 11.Navarro A, Diaz T, Martinez A, et al. Regulation of JAK2 by miR-135a: prognostic impact in classic Hodgkin lymphoma. Blood. 2009;114(14):2945–2951. [DOI] [PubMed] [Google Scholar]
- 12.Van Roosbroeck K, Cox L, Tousseyn T, et al. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood. 2011;117(15):4056–4064. [DOI] [PubMed] [Google Scholar]
- 13.Meier C, Hoeller S, Bourgau C, et al. Recurrent numerical aberrations of JAK2 and deregulation of the JAK2-STAT cascade in lymphomas. Mod Pathol. 2009;22(3):476–487. [DOI] [PubMed] [Google Scholar]
- 14.Hartmann S, Martin-Subero JI, Gesk S, et al. Detection of genomic imbalances in microdissected Hodgkin and Reed- Sternberg cells of classical Hodgkin’s lymphoma by array-based comparative genomic hybridization. Haematologica. 2008;93(9): 1318–1326. [DOI] [PubMed] [Google Scholar]
- 15.Joos S, Granzow M, Holtgreve-Grez H, et al. Hodgkin’s lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int J Cancer. 2003;103(4):489–495. [DOI] [PubMed] [Google Scholar]
- 16.Aldinucci D, Celegato M, Casagrande N. Microenvironmental interactions in classical Hodgkin lymphoma and their role in promoting tumor growth, immune escape and drug resistance. Cancer Lett. 2016;380(1): 243–252. [DOI] [PubMed] [Google Scholar]
- 17.Assi R, Verstovsek S, Daver N. ‘JAK-ing’ up the treatment of primary myelofibrosis: building better combination strategies. Curr Opin Hematol. 2017;24(2):115–124. [DOI] [PubMed] [Google Scholar]
- 18.Vannucchi AM, Harrison CN. Emerging treatments for classical myeloproliferative neoplasms. Blood. 2017;129(6):693–703. [DOI] [PubMed] [Google Scholar]
- 19.Massaro F, Molica M, Breccia M. How ruxolitinib modified the outcome in myelofibrosis: focus on overall survival, allele burden reduction and fibrosis changes. Expert Rev Hematol. 2017;10(2):155–159. [DOI] [PubMed] [Google Scholar]
- 20.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. [DOI] [PubMed] [Google Scholar]
- 21.Heine A, Brossart P, Wolf D. Ruxolitinib is a potent immunosuppressive compound: is it time for anti-infective prophylaxis? Blood. 2013;122(23):3843–3844. [DOI] [PubMed] [Google Scholar]
- 22.Duhoux FP, Ameye G, Lambot V, et al. Refinement of 1p36 alterations not involving PRDM16 in myeloid and lymphoid malignancies. PLoS One. 2011;6(10):e26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.A’Hern RP. Sample size tables for exact single-stage phase II designs. Stat Med. 2001;20(6):859–866. [DOI] [PubMed] [Google Scholar]
- 24.Aldinucci D, Pinto A, Gloghini A, Carbone A. Chemokine receptors as therapeutic tools in Hodgkin lymphoma: CCR4 and beyond. Blood. 2010;115(3):746–747; author reply 748. [DOI] [PubMed] [Google Scholar]
- 25.Carbone A, Gloghini A, Castagna L, Santoro A, Carlo-Stella C. Primary refractory and early-relapsed Hodgkin’s lymphoma: strategies for therapeutic targeting based on the tumour microenvironment. J Pathol. 2015;237(1):4–13. [DOI] [PubMed] [Google Scholar]
- 26.Green MR, Monti S, Rodig SJ, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Derenzini E, Lemoine M, Buglio D, et al. The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood Cancer J. 2011;1(12):e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roemer MG, Advani RH, Ligon AH, et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol. 2016;34(23):2690–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SJ, Kang HJ, Dong-Yeop S, et al. The efficacy of JAK2 inhibitor in heavily pretreated classical Hodgkin lymphoma: a prospective pilot study of ruxolitinib in relapsed or refractory classical Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2016;128(22): 1820. [Google Scholar]
- 30.Younes A, Romaguera J, Fanale M, et al. Phase I study of a novel oral Janus kinase 2 inhibitor, SB1518, in patients with relapsed lymphoma: evidence of clinical and biologic activity in multiple lymphoma subtypes. J Clin Oncol. 2012;30(33):4161–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diaz T, Navarro A, Ferrer G, et al. Lestaurtinib inhibition of the Jak/STAT signaling pathway in Hodgkin lymphoma inhibits proliferation and induces apoptosis. PLoS One. 2011;6(4):e18856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res. 2014;20(10):2674–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–4053. [DOI] [PubMed] [Google Scholar]
- 34.Falzetti D, Crescenzi B, Matteuci C, et al. Genomic instability and recurrent breakpoints are main cytogenetic findings in Hodgkin’s disease. Haematologica. 1999;84(4):298–305. [PubMed] [Google Scholar]
- 35.Naqvi K, Daver N, Pemmaraju N, et al. Clinical use of ruxolitinib in an academic medical center in unselected patients with myeloproliferative neoplasms not on clinical study. Leuk Lymphoma. 2017;58(4): 866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daver N, Cortes J, Newberry K, et al. Ruxolitinib in combination with lenalidomide as therapy for patients with myelofibrosis. Haematologica. 2015;100(8):1058–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devillier R, Raffoux E, Rey J, et al. Combination therapy with ruxolitinib plus intensive treatment strategy is feasible in patients with blast-phase myeloproliferative neoplasms. Br J Haematol. 2016;172(4):628–630. [DOI] [PubMed] [Google Scholar]
- 38.Mayfield JR, Czuchlewski DR, Gale JM, et al. Integration of ruxolitinib into dose-intensified therapy targeted against a novel JAK2 F694L mutation in B-precursor acute lymphoblastic leukemia. Pediatr Blood Cancer. 2017;64(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu Y, Hong Y, Xu Y, Liu P, Guo DH, Chen Y. Inhibition of the JAK/STAT pathway with ruxolitinib overcomes cisplatin resistance in non-small-cell lung cancer NSCLC. Apoptosis. 2014;19(11):1627–1636. [DOI] [PubMed] [Google Scholar]
- 40.Ju W, Zhang M, Wilson KM, et al. Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or Navitoclax in a murine model of human Hodgkin’s lymphoma. Proc Natl Acad Sci USA. 2016;113(6):1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M, Mathews Griner LA, Ju W, et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2- dependent adult T-cell leukemia. Proc Natl Acad Sci USA. 2015;112(40):12480–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.