

Abstract
Mantle cell lymphoma patients have variable clinical courses, ranging from indolent cases that do not require immediate treatment to aggressive, rapidly progressing diseases. Thus, diagnostic tools capable of stratifying patients according to their risk of relapse and death are needed. This study included 83 samples from the Fondazione Italiana Linfomi MCL-0208 clinical trial. Through gene expression profiling and quantitative real-time PCR we analyzed 46 peripheral blood and 43 formalin-fixed paraffin-embedded lymph node samples. A prediction model to classify patients was developed. By analyzing the transcriptome of 27 peripheral blood samples, two subgroups characterized by a differential expression of genes from the B-cell receptor pathway (B-cell receptorlow and B-cell receptorhigh) were identified. The prediction model based on the quantitative real-time PCR values of six representative genes (AKT3, BCL2, BTK, CD79B, PIK3CD, and SYK), was used to classify the 83 cases (43 B-cell receptorlow and 40 B-cell receptorhigh). The B-cell receptorhigh signature associated with shorter progression-free survival (P=0.0074), selected the mantle cell lymphoma subgroup with the shortest progression-free survival and overall survival (P=0.0014 and P=0.029, respectively) in combination with high (>30%) Ki-67 staining, and was an independent predictor of short progression- free survival along with the Mantle Cell Lymphoma International Prognostic Index-combined score. Moreover, the clinical impact of the 6- gene signature related to the B-cell receptor pathway identified a mantle cell lymphoma subset with shorter progression-free survival intervals also in an external independent mantle cell lymphoma cohort homogenously treated with different schedules. In conclusion, this 6-gene signature associates with a poor clinical response in the context of the MCL- 0208 clinical trial. (clinicaltrials.gov identifier: 02354313).
Introduction
Mantle cell lymphoma (MCL) is a distinctive B-cell malignancy accounting for 5–10% of all lymphomas,1–3 whose molecular hallmark and initiating oncogenic event, the t(11;14)(q13;q32) translocation, leads to constitutive overexpression of the proto-oncogene cyclin D1 (CCND1).2,4
Once considered as uniformly characterized by a poor prognosis, MCL has been demonstrated to have unexpectedly variable clinical courses, ranging from indolent cases that do not require immediate treatment to aggressive, rapidly progressing disease.2,5–10 Even among patients requiring treatment, prognosis is highly heterogeneous, with patients experiencing prolonged remissions and others rapidly relapsing even after cytarabine-containing induction regimens followed by autologous transplantation. Thus, diagnostic tools capable of stratifying MCL patients in different risk classes are warranted in order to direct treatment strategies.11 For this reason, many attempts have been made to identify clinical, histological, and molecular markers that can stratify patients according to their risk of relapse and death.12–25
In addition to the clinical MCL prognostic score (MCL- International Prognostic Index, MIPI)12,14 capable of stratifying patients into risk groups with different overall survival (OS),14 the Ki-67 proliferation index has been proposed as one of the most powerful and independent predictors of survival in MCL even in the context of prospective trials and modern therapies,5,13,26 and for these reasons has been integrated into the so-called MIPI-combined (MIPI-c) score.13,26 Moreover, effective prognostic discrimination is achieved by post-treatment response monitoring by positron emission tomography (PET)-scan and minimal residual disease (MRD). Furthermore, a seminal study identified a specific signature associated with proliferation as the strongest predictor of OS in a large MCL series.20 In this context, a cohort of 20 proliferation-associated genes constructed on the basis of gene expression analysis was demonstrated to be superior to other molecular markers.20 Since approaches based on microarray technology have not yet been incorporated into routine clinical practice, a PCR-based surrogate method investigating expression of five genes has been proposed and applied to paraffin- embedded tissues.18
Recent evidence suggests that the B-cell receptor (BCR) pathway may contribute to the pathogenesis of several histological types of B-cell non-Hodgkin lymphomas, including MCL.27–30 The importance of BCR signaling pathway in B-cell malignancy pathogenesis has driven interest in the use of small-molecule inhibitors of BCR-associated kinases, potentially preventing the activation of one or more of the distal BCR signaling pathway proteins.28,31
In the present study, we developed a survival predictive model for younger patients with advanced MCL treated in the context of the Fondazione Italiana Linfomi (FIL) MCL- 0208 Phase III randomized clinical trial. This model is based upon the quantitative evaluation of six genes, mostly from the BCR pathway, selected from a gene expression profile (GEP) of peripheral blood (PB) MCL cells and was applied to formalin-fixed paraffin-embedded (FFPE) tissue specimens. Notably, the model predicts poor response in the context of the FIL-MCL-0208 trial.
Methods
Primary MCL cases
The study included 83 out of 300 samples of adult patients under 66 years of age with advanced stage MCL, enrolled in the FIL-MCL-0208 prospective, multicenter, Phase III randomized clinical trial (clinicaltrials.gov identifier: 02354313),32 divided as follows: i) a panel of 27 PB samples utilized for GEP upon positive sorting of the clonal CD5+/CD19+ MCL cells; ii) an additional panel of 19 PB samples utilized for quantitative real-time PCR (qRT-PCR) of the identified gene signature in the purified MCL cell component; iii) a panel of 43 lymph node (LN) samples utilized for qRT-PCR of the identified gene signature (in this LN panel 6 samples had a matched PB sample). The clinical and histopathological details of the 83 MCL cases used in this study are reported in Table 1. No significant differences were found between the 83 cases entering the study versus the 217 remaining cases enrolled in the clinical trial in terms of median age, MIPI score, Ki-67 index and PFS intervals (Online Supplementary Table S1 and Online Supplementary Figure S1). No differences in clinical and biological parameters were observed between PB and LN MCL samples (data not shown). All patients were treated according to the FIL-MCL-0208 clinical trial, as reported in Online Supplementary Figure S2.
Table 1.
Characteristics of 83 mantle cell lymphoma cases entering the study.
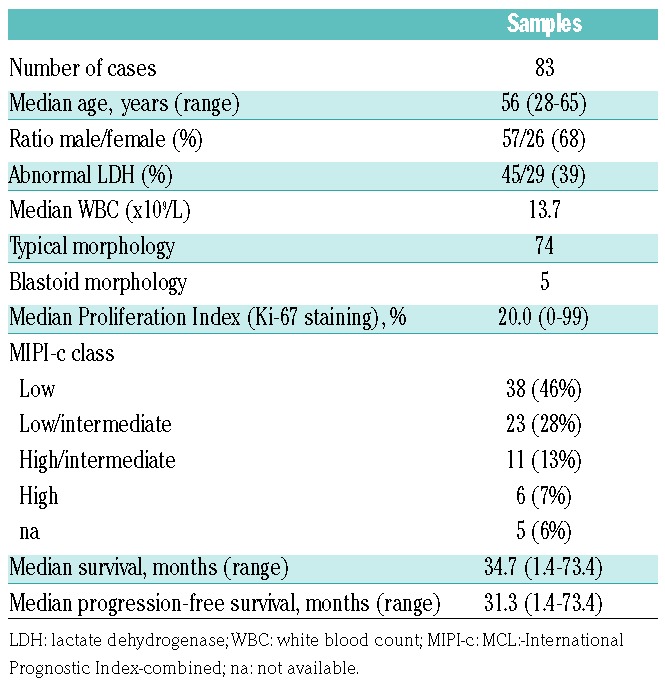
Mantle cell lymphoma diagnosis was prospectively confirmed by centralized histological review according to the World Health Organization (WHO) 2008 criteria.3,33 All patients provided informed consent in accordance with Institutional Review Board requirements (0016331-BZ 09/02/2010) and the Declaration of Helsinki, and protocol consent included use of MRD sample left- overs for the study.
All the procedures employed for RNA extraction, GEP and downstream analyses, qRT-PCR, analyses and qRT-PCR validations were carried out according to standard protocols, as reported previously.34–37 (See the Online Supplementary Appendix for details). Microarray data are available in Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE89447. Cases used for these procedures are reported in Online Supplementary Table S2.
Validation procedures
The 6-gene signature was tested in the MCL cohort described by Saba et al.,30 enrolled in another clinical trial (clinicaltrials.gov identifier: 00114738), by using the sum of the array gene expression values, as reported.30 Gene signatures related to MCL outcome were retrieved from previous papers,30,38 and imported in the GeneSpring GX and tested in the present cohort with GEP data available.
Statistical analysis
Overall survival was computed from trial registration to death as a result of any cause, censored at the latest follow up in patients who were still alive. Progression-free survival (PFS) was computed from trial registration to progression or death as a result of any cause, censored at the latest tumor assessment if no progression was observed. Clinical correlations, performed with the MedCalc v.9.5 software, were made using Kaplan-Meier plots and log-rank test. The Cox proportional model was chosen for multivariable analysis. Clinical outcome results were up-dated as of January 2017.32 Investigators are still blinded to the investigation arm as the primary study end point has still not been met.
Results
GEP identifies MCL patients with distinct expression of genes belonging to the BCR pathway
Global GEP was performed in purified MCL cells from 27 PB samples. An unsupervised analysis performed by principal component analysis (PCA) divided the cohort into two groups of 14 cases and 13 cases, respectively (Figure 1A). Consistently, a hierarchical clustering, which was run with all the GEP features, split MCL cases into two major groups perfectly resembling the PCA groups (Figure 1B).
Figure 1.

Gene Expression Profile (GEP) analysis of 27 mantle cell lymphoma samples. (A) Principal Component Analysis (PCA) scores represented in a 3D scatter plot. One point per array/sample is shown. Black line indicates separation between PCA classes. (B) Hierarchical clustering of 14 group-1 cases and 13 group-2 cases, using 50,739 probes. (C) Hierarchical clustering of 14 group1 cases and 13 group2 cases, using the 922 differentially expressed probes. Color codes for gene expression values refer to mean centered log-ratio values.
Supervised analysis according to the PCA classification defined a gene expression signature composed of 922 probes, 713 up-regulated and 209 down-regulated in group-2 versus group-1 samples (Figure 1C and Online Supplementary Table S3).
Pathway analysis revealed that “Antigen processing and presentation” and “B-cell receptor signaling pathway” were among the top ranked pathways enriched in the group-2 category (Online Supplementary Table S4). Similar results were obtained by GSEA which highlighted a constitutive overexpression of genes related to the BCR signaling pathways in the context of group-2 patients (Figure 2A and Online Supplementary Table S5). Therefore, here- after the two PCA groups were identified as BCRlow (group-1) and BCRhigh (group-2).
Figure 2.

6-gene signature and Decision Tree (DT) prediction model. (A) Gene Expression Profile data of BCRlow and BCRhigh MCL samples were tested using Gene set enrichment analysis (GSEA). Reported are the significant gene sets differentially expressed and related to the B-Cell Receptor (BCR) pathway. (B) Venn diagram derived by merging the differentially expressed probes and the genes belonging to the BCR related gene sets. In bold genes selected as the 6-gene signature. (C) Hierarchical clustering of 14 BCRlow cases and 13 BCRhigh cases, using the six gene values. Color codes for gene expression values refer to mean centered log-ratio values. (D) Hierarchical clustering of 8 BCRlow cases and 9 BCRhigh cases belonging to the training set of DT prediction model, using the six gene qRT-PCR values. (E) Hierarchical clustering of 6 BCRlow cases and 4 BCRhigh cases belonging to the validation set of DT prediction model, using the six gene qRT-PCR values. Bar under the heat-map refers to prediction generated by the DT prediction model. Color codes for gene expression values refer to mean centered log-ratio values.
A 6-gene signature identifies BCRlow and BCRhigh MCL samples
Having identified two different groups of MCL patients at diagnosis with a different expression of genes related to the BCR pathway, we overlapped the genes included in the gene sets related to the BCR pathway (115 probes) and the differentially expressed genes (922 probes) to create a reduced signature (Figure 2B). In this way, 18 probes corresponding to 15 genes, all over-expressed in BCRhigh cases were identified (Figure 2B). Among these genes, a subgroup of six genes (AKT3, BCL2, BTK, CD79B, PIK3CD, and SYK) was selected for further validations due to their direct involvement in the BCR pathway and/or the existence of drugs targeting the related proteins. A hierarchical cluster using only these six genes was able to discriminate patients belonging to the BCRlow or BCRhigh groups (Figure 2C).
Development of a qRT-PCR-based predictor for BCRlow and BCRhigh in MCL samples
By analyzing the expression levels of the selected six genes in the same 27 MCL PB samples by qRT-PCR approach, a strict correlation with GEP data was found (Online Supplementary Figure S3B). Moreover, the 27 MCL cases were randomly divided into a training set (17 cases; 8 BCRlow and 9 BCRhigh samples) and a validation set (10 cases; 6 BCRlow and 4 BCRhigh samples) to develop and test a decision tree (DT) model based on qRT-PCR data capable of categorizing patients into one of the two categories. The DT model based on qRT-PCR data correctly classified 16 of 17 cases belonging to the training set and 10 of 10 cases of the validation cohort, and allowed the classification of 19 additional PB samples screened with qRT-PCR (9 BCRlow and 10 BCRhigh (Figure 2D and E and Online Supplementary Table S2).
Association between BCR categories and biological and clinical parameters
Collectively, the 6-gene signature was re-evaluated by setting up a validated qRT-PCR approach (see Online Supplementary Appendix and Online Supplementary Table S6) in PB samples from 46 MCL cases, 23 were identified as BCRlow and 23 as BCRhigh. By correlating the BCR groups with the available biological parameters, no association was found between the 6-gene signature and IGHV gene status (P=0.93) (Online Supplementary Table S2 and Online Supplementary Figure S4A), Ki-67 expression, white blood cells, hemoglobin, lymphocytes, platelets, and neutrophil count (data not shown). The only significant difference was between the BCR classification and lactate dehydrogenase (LDH) levels; BCRhigh cases showed a higher level of LDH with respect to BCRlow MCL (416.6±191.6 vs. 292.2±127.4; P=0.023) (Online Supplementary Figure S4B).
Clinically, MCL patients classified as BCRhigh experienced shorter PFS with respect to BCRlow MCL cases (median PFS: 21.6 months vs. not reached; P=0.0375) (Figure 3).
Figure 3.

BCRhigh mantle cell lymphoma (MCL) group is associated with a worse clinical outcome. Kaplan-Meier curves obtained by comparing progression-free survival intervals of 23 BCRlow MCL cases with 23 BCRhigh MCL cases. The number of patients in each group is reported under relative categories; P refers to log- rank test.
Application of the 6-gene signature to LN samples from MCL patients
To evaluate the capability of the 6-gene signature to identify different subgroups also in the context of MCL LN cases, we tested our qRT-PCR approach in a series of 43 LN samples preserved as FFPE LN specimens. Thirty- five (81%) out of 43 samples were amplifiable for all six genes, and using a DT model based on qRT-PCR values from FFPE, 23 cases were classified as BCRlow and 20 classified as BCRhigh (Online Supplementary Table S2). Notably, for 6 out of 43 LN samples, a PB matched sample was available, and by comparing qRT-PCR results performed on PB samples and LN FFPE samples from these cases, a good concordance was overall observed, although FFPE samples generally amplified at higher Ct values (Online Supplementary Figure S5). Of note, 5 out of 6 these MCL cases were consistently classified. The misclassified case was considered as BCRlow according to GEP data. Also in the context of LN samples, no correlation was found between the different biological parameters and BCR groups (data not shown).
By merging the MCL cases analyzed either in PB or in LN, a total of 83 cases were collected, 43 BCRlow and 40 BCRhigh. BCRhigh patients had a shorter PFS with respect to BCRlow patients (median PFS: 42.1 months vs. not reached; P=0.0074) (Figure 4A). Since Ki-67 is a well-known prognosticator in MCL,26 we combined the BCR groups with the prognostic groups defined by Ki-67 score. Cases with high Ki-67 (≥30% of Ki-67 expressing cells) and classified in the BCRhigh group experienced the shortest PFS, while cases classified as BCRlow had similar longer PFS intervals irrespective of the high or low Ki-67 score (median PFS: 20.5 months vs. not reached for all the other combinations; P=0.0014) (Figure 4B). Consistently, multivariable analysis carried out by including the BCR signature and the MIPI-c categories selected the BCRhigh and the high risk MIPI-c category as independent predictors of PFS (Table 2). Regarding OS, while the BCR readout failed to identify groups with different OS intervals, possibly due to the low rate of events and short follow up (Figure 4C), the combination of high Ki-67 score and a BCRhigh 6-gene signature was able again to select the MCL subgroup with the shortest OS (46.7 vs. not reached; P=0.029) (Figure 4D).
Figure 4.

BCRhigh mantle cell lymphoma (MCL) group is associated with a worse clinical outcome (overall series). (A) Kaplan-Meier curves obtained by comparing progression-free survival (PFS) intervals of 43 BCRlow MCL cases with 40 BCRhigh MCL cases. (B) Kaplan-Meier curves obtained by comparing PFS intervals of 19 BCRlow and low Ki-67 MCL cases, with 20 BCRlow and high Ki-67 MCL cases, with 21 BCRhigh and Ki-67 low MCL cases, with 10 BCRhigh and Ki-67 high MCL cases. (C) Kaplan-Meier curves obtained by comparing overall survival (OS) intervals of 43 BCRlow MCL cases with 40 BCRhigh MCL cases. (D) Kaplan-Meier curves obtained by comparing OS intervals of 19 BCRlow and low Ki-67 MCL cases, with 20 BCRlow and high Ki-67 MCL cases, with 21 BCRhigh and Ki- 67 low MCL cases, with 10 BCRhigh and Ki-67 high MCL cases. The number of patients in each group is reported under relative categories; P refers to log-rank test.
Table 2.
Cox regression analysis on mantle cell lymphoma cases.

Validations of BCR signature
To verify whether the BCR signature maintained its prognostic impact in an independent set of patients, we used the gene expression data of MCL LN biopsies reported by Saba et al.30 Also in this different setting, a high expression of the 6-gene signature, as in the context of BCRhigh cases, identified an MCL patient subset with inferior PFS (P=0.049) (Online Supplementary Figure S6).
In another set of analyses, by taking advantage of our 27 MCL cases with GEP data available, we correlated our BCR signature with other MCL signatures with proven clinical impact.30,38 As reported in Online Supplementary Figure S7A, the BCR signature reported in Saba et al.30 divided MCL cases in two groups that corresponded exactly to our BCR definition (Online Supplementary Figure S7B).30 Similarly, the 17 genes of the proliferation signature reported by Scott et al.38 split our MCL cases in 3 different groups resembling the 3 different groups originally defined (Online Supplementary Figure S8A). In this context, the shortest PFS and OS intervals were observed in the third group characterized by a higher expression of genes related to proliferation and a BCRhigh phenotype in keeping with our findings (Online Supplementary Figure S8A-C).
Discussion
In this study, we demonstrated that a BCR-derived signature based on the differential expression of six genes correlated with shorter PFS intervals in the context of a Phase III prospective clinical trial (FIL-MCL-0208) for younger MCL patients receiving R-CHOP induction, followed by high-dose cytarabine and autologous stem cell transplantation (clinicaltrials.gov identifier: 023541313).32
Notably, the BCR-related 6-gene signature reported here was able to identify an MCL subset with shorter PFS intervals also in the context of an external independent MCL cohort homogenously treated with different schedules.30 On the other hand, when the signature described by Saba et al.30 and Scott et al.38 was applied to our MCL cases, the patient subsets with the worse prognosis turned out to be particularly enriched in BCRhigh cases, even though these signatures did not include any gene from our signature. Therefore, although composed of genes located upstream of the BCR machinery, our signature was able to identify cases with an active BCR pathway as defined by other signatures.30 In this regard, however, experiments with primary MCL cases and/or MCL cell lines combining BCR stimulation with the use of specific BCR inhibitors should be performed to investigate the contribution of the 6-gene signatures described here to the actual activation of the BCR pathway.
Again in agreement with this line of reasoning, BCRhigh samples presented a significant upregulation of PAX5 (see GEP data in Online Supplementary Table S3), a gene whose product is known to prevent plasma cell differentiation thus preserving the capacity to respond to antigen- induced activation and proliferation.39 Taken together these data corroborate recent findings of ongoing active BCR signaling in MCL cell in vivo,29,30 and further underline the role of antigen stimulation in the ontogeny of MCL, as suggested by the skewed IGVH gene repertoire found in MCL cells.40
In order to discriminate between BCRlow and BCRhigh MCL samples, we developed a DT model based on the expression of the selected six genes.28 This DT model was applied in an independent cohort of PB samples and then to a further series of FFPE LN samples, thus demonstrating that two MCL subsets with different expression levels of BCR-related genes could also be recognized in the LN compartment, mirroring PB. Taken together, by combining data from the PB and LN compartments, MCL cases classified as BCRhigh showed higher LDH levels and shorter PFS with respect to BCRlow patients, suggesting that activation of BCR signaling drives tumor proliferation and determines clinical outcome of MCL patients, which is in keeping with recent findings.30
By combining the predictive capacity of the 6-gene BCR signature with the Ki-67 index, we identified a particularly unfavorable category (BCRhigh and high Ki-67) with a substantially shorter PFS and OS than the other groups. Consistently, the BCRhigh signature turned out to be an independent prognosticator along with the high-risk MIPI-c category for short PFS by multivariate analysis. There is no indication that the validity of the model may be affected by the different recruitment site (PB vs. LN), or by different sample storage (frozen vs. FFPE) because the main clinical parameters were equally distributed between the different series (PB/frozen vs. LN/FFPE) (R Bomben et al., 2018, unpublished observation). In this regard, an important feature of this model/assay is its applicability to both PB and LN FFPE samples, having, therefore, the chance to combine results of qRT-PCR with Ki-67 staining in all the cases.
Our data underscore the increasing importance of BCR- related genes in the pathogenesis and development of MCL, further underlined by the clinical significance of drugs specifically targeting genes belonging to this pathway. In particular, therapeutic targeting of BTK41 can be rationally exploited in lymphoid malignancies that have been proved to be dependent on an antigen-dependent BCR-mediated active signaling. However, despite the relatively high response rate to single agent ibrutinib in relapsed/refractory MCL, it remained unclear as to why some patients showed clear responses, while others received little therapeutic benefit.31,42 The BCR-related signature described here may provide insights into molecular factors that explain the divergent responses of MCL patients to ibrutinib, although other causes of primary resistance might be related to gene mutations in the other pathways, e.g. NF-κB pathway and epigenetic modifiers, as recently reported.43,44
In conclusion, in the present study we developed a survival model for patients with MCL composed of six genes (AKT3, BTK, CD79B, PIK3CD, SYK, BCL2) whose expression can easily be investigated by qRT-PCR and also in FFPE specimens. The signature was associated with a poor clinical response in the context of a high-dose chemoimmunotherapy regimen, and might, therefore, be considered for validation and application in future clinical trials.
Supplementary Material
Acknowledgments
The authors would like to thank Progetto Giovani Ricercatori GR-2011-02347441, GR-2009-1475467, and GR-2011- 02351370, Ministero della Salute, Rome, Italy; Progetto Ricerca Finalizzata RF-2009-1469205, and RF-2010-2307262, Ministero della Salute, Rome, Italy; Associazione Italiana contro le Leucemie, linfomi e mielomi (AIL), Venezia Section, Pramaggiore Group, Italy; Associazione Italiana Ricerca Cancro (AIRC), Investigator Grant IG-2015 (17622); “5×1000 Intramural Program”, Centro di Riferimento Oncologico, Aviano, Italy; Provincia Autonoma di Bolzano/Bozen, Italy; A.O. S. Maurizio, Bolzano/Bozen, Italy; Progetto di Rilevante Interesse Nazionale (PRIN2009) 7.07.02.60 AE01, Ministero Italiano dell’Università e della Ricerca (MIUR), Roma, Italy; Fondi di Ricerca Locale, Università degli Studi di Torino, Italy; Fondazione Neoplasie Del Sangue (Fo.Ne.Sa), Torino, Italy; CRT 2015.1044, Fondazione CRT, Torino, Italy. We are grateful to all the Clinical Investigators, to the Pathologists, to Luigia Monitillo, Daniela Barbero, Marina Ruggeri, Paola Ghione, and Gian Maria Zaccaria.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/5/849
References
- 1.Dreyling M, Ferrero S, Hermine O. How to manage mantle cell lymphoma. Leukemia. 2014;28(11):2117–2130. [DOI] [PubMed] [Google Scholar]
- 2.Cheah CY, Seymour JF, Wang ML. Mantle Cell Lymphoma. J Clin Oncol. 2016; 34(11):1256–1269. [DOI] [PubMed] [Google Scholar]
- 3.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122(10):3416–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dreyling M, Ferrero S, Vogt N, Klapper W. New paradigms in mantle cell lymphoma: is it time to risk-stratify treatment based on the proliferative signature? Clin Cancer Res. 2014;20(20):5194–5206. [DOI] [PubMed] [Google Scholar]
- 6.Ghielmini M, Zucca E. How I treat mantle cell lymphoma. Blood. 2009;114(8):1469–1476. [DOI] [PubMed] [Google Scholar]
- 7.Herrmann A, Hoster E, Zwingers T, et al. Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol. 2009;27(4):511–518. [DOI] [PubMed] [Google Scholar]
- 8.Zucca E, Roggero E, Pinotti G, et al. Patterns of survival in mantle cell lymphoma. Ann Oncol. 1995;6(3):257–262. [DOI] [PubMed] [Google Scholar]
- 9.Barista I, Romaguera JE, Cabanillas F. Mantle-cell lymphoma. Lancet Oncol. 2001;2(3):141–148. [DOI] [PubMed] [Google Scholar]
- 10.Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol. 2009; 27(8):1209–1213. [DOI] [PubMed] [Google Scholar]
- 11.Dreyling M, Ferrero S. The role of targeted treatment in mantle cell lymphoma: is transplant dead or alive? Haematologica. 2016;101(2):104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geisler CH, Kolstad A, Laurell A, et al. The Mantle Cell Lymphoma International Prognostic Index (MIPI) is superior to the International Prognostic Index (IPI) in predicting survival following intensive first-line immunochemotherapy and autologous stem cell transplantation (ASCT). Blood. 2010;115(8):1530–1533. [DOI] [PubMed] [Google Scholar]
- 13.Hoster E, Rosenwald A, Berger F, et al. Prognostic Value of Ki-67 Index, Cytology, and Growth Pattern in Mantle-Cell Lymphoma: Results From Randomized Trials of the European Mantle Cell Lymphoma Network. J Clin Oncol. 2016; 34(12):1386–1396. [DOI] [PubMed] [Google Scholar]
- 14.Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111(2):558–565. [DOI] [PubMed] [Google Scholar]
- 15.Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250–18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123(19):2988–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katzenberger T, Petzoldt C, Holler S, et al. The Ki67 proliferation index is a quantitative indicator of clinical risk in mantle cell lymphoma. Blood. 2006;107(8):3407. [DOI] [PubMed] [Google Scholar]
- 18.Hartmann E, Fernandez V, Moreno V, et al. Five-gene model to predict survival in mantle-cell lymphoma using frozen or formalin- fixed, paraffin-embedded tissue. J Clin Oncol. 2008;26(30):4966–4972. [DOI] [PubMed] [Google Scholar]
- 19.Ek S, Bjorck E, Porwit-MacDonald A, Nordenskjold M, Borrebaeck CA. Increased expression of Ki-67 in mantle cell lymphoma is associated with de-regulation of several cell cycle regulatory components, as identified by global gene expression analysis. Haematologica. 2004;89(6):686–695. [PubMed] [Google Scholar]
- 20.Rosenwald A, Wright G, Wiestner A, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3(2):185–197. [DOI] [PubMed] [Google Scholar]
- 21.Mozos A, Royo C, Hartmann E, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica. 2009;94(11):1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navarro A, Clot G, Royo C, et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012;72(20):5307–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majlis A, Pugh WC, Rodriguez MA, Benedict WF, Cabanillas F. Mantle cell lymphoma: correlation of clinical outcome and biologic features with three histologic variants. J Clin Oncol. 1997;15(4):1664–1671. [DOI] [PubMed] [Google Scholar]
- 24.Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood. 2007;109(11):4599–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new targeted therapeutics. Nat Rev Cancer. 2007;7(10):750–762. [DOI] [PubMed] [Google Scholar]
- 26.Determann O, Hoster E, Ott G, et al. Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti- CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group. Blood. 2008; 111(4):2385–2387. [DOI] [PubMed] [Google Scholar]
- 27.Perez-Galan P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013; 12(3):229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akhter A, Street L, Ghosh S, et al. Concomitant high expression of Toll-like receptor (TLR) and B-cell receptor (BCR) signalling molecules has clinical implications in mantle cell lymphoma. Hematol Oncol. 2015;35(1):79–86. [DOI] [PubMed] [Google Scholar]
- 30.Saba NS, Liu D, Herman SE, et al. Pathogenic role of B-cell receptor signaling and canonical NF-kappaB activation in mantle cell lymphoma. Blood. 2016; 128(1):82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cortellazzo S, Martelli M, Ladetto M, Ferrero S, Ciccone G, Evangelista A, et al. High dose sequential chemotherapy with rituximab and ASCT as first line therapy in adult MCL patients: clinical and molecular response of the MCL-0208 trial, a FIL study. Haematologica. 2015;100(s1):3.25552677 [Google Scholar]
- 33.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization (WHO) classification of lymphoid neoplasms. Blood. 2016; 127(20):2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bomben R, Gobessi S, Dal BM, et al. The miR-17-92 family regulates the response to Toll-like receptor 9 triggering of CLL cells with unmutated IGHV genes. Leukemia. 2012;26(7):1584–1593. [DOI] [PubMed] [Google Scholar]
- 35.Dal Bo M, D’Agaro T, Gobessi S, et al. The SIRT1/TP53 axis is activated upon B-cell receptor triggering via miR-132 up-regulation in chronic lymphocytic leukemia cells. Oncotarget. 2015;6(22):19102–19117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Draghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA. Global functional profiling of gene expression. Genomics. 2003;81(2):98–104. [DOI] [PubMed] [Google Scholar]
- 37.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott DW, Abrisqueta P, Wright GW, et al. New Molecular Assay for the Proliferation Signature in Mantle Cell Lymphoma Applicable to Formalin-Fixed Paraffin- Embedded Biopsies. J Clin Oncol. 2017; 35(15):1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nera KP, Kohonen P, Narvi E, et al. Loss of Pax5 promotes plasma cell differentiation. Immunity. 2006;24(3):283–293. [DOI] [PubMed] [Google Scholar]
- 40.Hadzidimitriou A, Agathangelidis A, Darzentas N, et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood. 2011;118(11):3088–3095. [DOI] [PubMed] [Google Scholar]
- 41.Rickert RC. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol. 2013; 13(8):578–591. [DOI] [PubMed] [Google Scholar]
- 42.Wang ML, Blum KA, Martin P, et al. Long- term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood. 2015; 126(6):739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92. [DOI] [PubMed] [Google Scholar]
- 44.Lenz G, Balasubramanian S, Goldberg J, Rizo A, Schaffer M, Phelps C, et al. Sequence variants in patients with primary and acquired resistance to ibrutinib in the phase 3 MCL3001 (RAY) trial. Haematologica. 2016;101(s1):155. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.