

Abstract
Members of the genus Bifidobacterium are common inhabitants of the human gastrointestinal tract. Previously it was shown that arabino-oligosaccharides (AOS) might act as prebiotics and stimulate the bifidobacterial growth in the gut. However, despite the rapid accumulation of genomic data, the precise mechanisms by which these sugars are utilized and associated transcription control still remain unclear. In the current study, we used a comparative genomic approach to reconstruct arabinose and AOS utilization pathways in over 40 bacterial species belonging to the Bifidobacteriaceae family. The results indicate that the gene repertoire involved in the catabolism of these sugars is highly diverse, and even phylogenetically close species may differ in their utilization capabilities. Using bioinformatics analysis we identified potential DNA-binding motifs and reconstructed putative regulons for the arabinose and AOS utilization genes in the Bifidobacteriaceae genomes. Six LacI-family transcriptional factors (named AbfR, AauR, AauU1, AauU2, BauR1 and BauR2) and a TetR-family regulator (XsaR) presumably act as local repressors for AOS utilization genes encoding various α- or β-L-arabinofuranosidases and predicted AOS transporters. The ROK-family regulator AraU and the LacI-family regulator AraQ control adjacent operons encoding putative arabinose transporters and catabolic enzymes, respectively. However, the AraQ regulator is universally present in all Bifidobacterium species including those lacking the arabinose catabolic genes araBDA, suggesting its control of other genes. Comparative genomic analyses of prospective AraQ-binding sites allowed the reconstruction of AraQ regulons and a proposed binary repression/activation mechanism. The conserved core of reconstructed AraQ regulons in bifidobacteria includes araBDA, as well as genes from the central glycolytic and fermentation pathways (pyk, eno, gap, tkt, tal, galM, ldh). The current study expands the range of genes involved in bifidobacterial arabinose/AOS utilization and demonstrates considerable variations in associated metabolic pathways and regulons. Detailed comparative and phylogenetic analyses allowed us to hypothesize how the identified reconstructed regulons evolved in bifidobacteria. Our findings may help to improve carbohydrate catabolic phenotype prediction and metabolic modeling, while it may also facilitate rational development of novel prebiotics.
Keywords: bifidobacteria, arabinose, arabino-oligosaccharides, prebiotics, regulatory networks, transcription factors, comparative genomics
Introduction
Bifidobacteria are Gram-positive, non-motile, non-spore-forming, anaerobic saccharolytic microorganisms that represent one of the deepest branches within the Actinobacteria phylum (Ventura et al., 2007; Lugli et al., 2017). They are also characterized by relatively high (∼60%) G+C content of their genomes (Lee and O’Sullivan, 2010). The majority of known Bifidobacterium species inhabit the gastrointestinal tract of various animals (mostly mammals, though also birds and insects), yet some species may be found in different ecological niches ranging from the oral cavity (Okamoto et al., 2008; Ventura et al., 2009) to fermented milk products (Watanabe et al., 2009).
Since their first isolation from infant feces in 1899 by Henri Tissier (van den Broek et al., 2008; Lee and O’Sullivan, 2010), bifidobacteria have continued to be a subject of study, especially in the last two decades due to increased interest in the functionalities of the GM in (human) health and disease. Particular Bifidobacterium species are known to be among the first colonizers of the sterile gastrointestinal tract and predominate in breastfed infants until weaning, partly because of their ability to metabolize human milk oligosaccharides (Favier et al., 2002; Sela et al., 2008; Milani et al., 2017). Despite the fact that their relative contribution to the GM composition decreases up to 2–14% with aging (Odamaki et al., 2016), these microorganisms are considered to be some of the most prevalent and important members of the community. Bifidobacteria are purported to confer several beneficial properties on their host, among others they are believed to elicit anti-carcinogenic, immunostimulatory, and anti-diarrhoeal activities, while they may also prevent intestinal colonization by pathogenic microbiota (Picard et al., 2005; Russell et al., 2011; Arboleya et al., 2016). Due to these claimed beneficial activities, several bifidobacterial species and strains, such as B. animalis subsp. lactis and B. longum subsp. longum, are commonly used as probiotics and are included in functional foods (Stanton et al., 2005; Russell et al., 2011).
Another approach to enhance the abundance and activity of bacteria as a beneficial gut commensal is the use of prebiotics. A prebiotic is a non-digestible compound that, through its metabolization by microorganisms in the gut, increases the number of and/or activity of beneficial components of the GM, thus conferring positive physiological health effects on their host (Bindels et al., 2015). Previously it was shown by in vitro studies that AOS and their polymers, arabinans, derived from sugar beet can selectivity increase the abundance of bifidobacteria (so-called bifidogenic effect) (Al-Tamimi et al., 2006; Holck et al., 2011; Onumpai et al., 2011; Vigsnæs et al., 2011; Sulek et al., 2014; Moon et al., 2015). However, the desire for a rational design of novel and more effective AOS-based prebiotics is impeded due to the lack of full understanding of catabolic pathways involved and associated regulatory networks.
The sequencing of the first bifidobacterial genome revealed the extensive coding potential to produce a very wide variety of GHs, of which 11 were predicted to be involved in AOS utilization (Schell et al., 2002). Since then efforts have been concentrated on the study of bifidobacterial arabinosidases (predominantly α-L-arabinofuranosidases). Currently, substrate specificity and the mode of action have been described for several of these enzymes in B. adolescentis (Van Laere et al., 1997; van den Broek et al., 2005; Lagaert et al., 2010; Suzuki et al., 2013) and B. longum subsp. longum (Margolles and de los Reyes-Gavilán, 2003; Fujita et al., 2011, 2014; Lee et al., 2011). Despite their functional importance, arabinosidases compose only one step in sugar catabolic pathways. The other parts are arabinose/AOS transporters, none of which have been described in bifidobacteria to this date, and downstream enzymes: L-arabinose isomerase (AraA), ribulokinase (AraB) and L-ribulose-5-phosphate 4-epimerase (AraD). Moreover, the process of utilization of a particular sugar also requires complex regulatory systems in order to coordinate gene expression in response to substrate availability. Carbohydrate utilization pathways are often controlled by DNA-binding transcriptional factors (TFs) that act as repressors or activators and that induce gene expression upon the appearance of a sugar effector (Cohn and Horibata, 1959). A regulon of a given TF is a group of genes and operons controlled by this TF (Rodionov, 2007). Thus, to unveil the complexities of the arabinose and AOS utilization pathways, it is especially important to reconstruct transcriptional networks and elucidate functional parts of a pathway.
In our previous work, we conducted a global analysis of transcriptional regulation of carbohydrate utilization in 10 Bifidobacterium genomes by predicting binding sites and reconstructing regulons for 268 TFs (Khoroshkin et al., 2016). The global regulator AraQ was one of the most striking findings since in addition to arabinose utilization genes its regulon contains several genes of central carbohydrate metabolism (Khoroshkin et al., 2016). In the current study, we focused our attention on the arabinose and AOS utilization pathways and associated transcriptional regulation in a broader, non-redundant set of 39 Bifidobacterium strains (both of human and non-human origin) as well as in 6 closely related species from different genera of the Bifidobacteriaceae family. The main aims were to expand existing regulons, describe new ones and also look at possible evolutionary clues. To tackle these tasks, we implemented a previously described subsystem-based comparative genomic approach which had been successfully used to reconstruct sugar utilization in several lineages of bacteria including Shewanella (Rodionov et al., 2010), Staphylococcus (Ravcheev et al., 2011), Bacillus (Leyn et al., 2013), Streptococcus (Ravcheev et al., 2013a), Bacteroides (Ravcheev et al., 2013b) and Thermotoga (Rodionov et al., 2013). Our results include reconstruction of regulons of 9 orthologous groups of TFs belonging to the LacI, TetR and ROK families. We hypothesize that the majority of these regulators act as local repressors, controlling the expression of co-localized arabinosidases and prospective AOS transporters, whereas AraQ functions as a global regulator, whose mode of action (i.e., acting as a transcriptional repressor or activator) depends on the particular target gene. In summary, the collected data allowed us to build several models of arabinose and AOS utilization in bifidobacteria. Such models in turn facilitated assignment of carbohydrate utilization phenotypes to each analyzed strain and tracing of the possible evolutionary routes of the reconstructed TF regulons.
Materials and Methods
Bioinformatics Methods and Databases
The genomic sequences of the analyzed bifidobacteria were obtained from GenBank (Benson et al., 2017). The initial set of available complete and draft genomes of bacterial species from the Bifidobacteriaceae family was filtered to exclude closely related strains and species with a 98.65% threshold on 16S rRNA similarity (Kim et al., 2014). We also included two different subspecies of B. longum and two strains of Gardnerella vaginalis because these closely related strains differed in the presence of the arabinose utilization genes. As a result, we compiled a non-redundant set of 45 genomes representing 38 Bifidobacterium species and 5 species of other genera that belong to the Bifidobacteriaceae family (Supplementary Table S1). Additionally, we analyzed the orthologous AraQ regulon in three other lineages of Actinobacteria, namely Arthrobacter arilaitensis, Geodermatophilus obscurus, and Stackebrandtia nassauensis. The whole-genome phylogeny tree for the selected Bifidobacteriaceae genomes was build using the Phylogenetic Tree service included in the PATRIC genomic platform (Wattam et al., 2017). Phylogenetic trees for the selected protein families were constructed via the PhyML algorithm (Guindon et al., 2010) in the Seaview software (Gouy et al., 2010), using the default parameters and bootstrap values computed with 100 replicates.
The comparative genome analysis and reconstruction of arabinose and AOS utilization pathways were performed using the subsystems approach (Rodionov et al., 2010) included in the SEED genomic platform (Overbeek et al., 2005). For functional gene annotation and building of the arabinose/AOS catabolic subsystem in SEED we combined existing annotations with information from literature accessed via the PaperBLAST tool (Price and Arkin, 2017) and reference databases including SwissProt for characterized protein annotations (The UniProt Consortium, 2017), KEGG for reference metabolic pathways (Kanehisa et al., 2016), TCDB for transporter classification (Saier et al., 2016) and CAZy for classification of Glycosyde Hydrolases (GHs) (Lombard et al., 2014). Orthologs were identified using BLAST-based protein similarity searches and gene neighborhood analysis implemented in SEED. Cellular localization of GHs was predicted with SignalP (Petersen et al., 2011). The obtained functional roles of proteins were projected across the selected Bifidobacteriaceae genomes thus forming the populated arabinose/AOS utilization subsystem (Supplementary Table S3). Based on the identified gene content we assigned each genome with a specific phenotype, representing the predicted ability to metabolize arabinose and different types of AOS.
Regulon Inference
Reconstruction of TF regulons was performed in two steps. First, we reconstructed arabinose/AOS utilization regulons for five TFs with predicted TF-binding sites (TFBSs) as based on our previous study involving 10 Bifidobacterium genomes (Khoroshkin et al., 2016). These TFs include AraQ (example locus tag BL0275); AbfR (BL0543); AauR (BL0185); BauR1 (BL0426) and XsaR (BAD_1207). We then reconstructed regulons for four newly identified TFs with unknown TFBSs that were co-localized on the chromosome with prospective arabinose and AOS utilization genes. These four regulators are AraU (BL0275), AauU1 (BISA_0485), AauU2 (BREU_1998), and BauR2 (BREU_0448). For identification of TFBSs and regulon reconstruction, we used the previously established comparative genomic approach (Rodionov, 2007). Briefly, it includes the following steps: (i) search for a TFBS motif of a given TF; (ii) construction of nucleotide position weight matrices (PWMs) for a discovered (training) set of TFBSs; (iii) genome-wide identification of additional TFBSs in all studied genomes containing a given TF-encoding ortholog utilizing the obtained matrix. For the previously studied five TFs, we implemented PWMs which were built based on information available in the RegPrecise database (Novichkov et al., 2010). For identification of TFBSs for novel regulators, we collected upstream regions of potential TF-regulated genes in each studied genome containing an ortholog of a TF of interest and scanned for potential DNA motifs with palindromic symmetry using the SignalX tool (Mironov et al., 2000). For each identified motif, a PWM was built via the same tool and was subsequently used to search for additional TFBSs and regulon members in GenomeExplorer (Mironov et al., 2000). The search parameters were as follows: (i) a region -350 to +50 bp relative to a predicted start codon; (ii) a TFBS score threshold of 5 for a strong site and 4.75 for weak sites. For the global AraQ regulon, a TFBS score threshold of 4.75 for strong sites and 4.5 for weak sites was employed. To avoid false positive predictions, predicted TFBSs were filtered using the consistency check and phylogenetic footprinting approaches (Rodionov, 2007). Upstream regions of the AraQ-regulated genes were aligned using Pro-coffee (Di Tommaso et al., 2011). Positions of the -10 and -35 promoter elements were determined via PMW-based searches in GenomeExplorer. TFBS motifs were visualized by WebLogo (Crooks et al., 2004). The reconstructed regulons gene content and candidate TFBSs are described in Supplementary Tables S1, S2.
Results and Discussion
Regulation of Arabinose Utilization
L-arabinose utilization is defined as a process that includes the transport of this monosaccharide from the extracellular environment into the cell and its subsequent conversion to D-xylulose 5-phosphate via the successive actions of the AraA, AraB, and AraD enzymes (Chang et al., 2015). In our previous bioinformatics analysis of TF regulons in 10 Bifidobacterium genomes, we identified the LacI-family transcriptional regulator AraQ that is presumed to control transcription of the araBDA operon (Khoroshkin et al., 2016). Using the previously identified PWM for AraQ-binding sites, we checked the conservation of AraQ operators upstream of orthologous araBDA genes in a broader set of genomes from the Bifidobacteriaceae family. The araBDA operon was identified in 36 out of 45 studied genomes, while an araQ ortholog was present in the majority of these genomes with a single exception of Gardnerella vaginalis ATCC 14019. Search for AraQ-binding sites identified high-scored sites upstream of the araBDA operon in each of the above genomes except G. vaginalis (Supplementary Table S1). Notably, in three Bifidobacterium species, the araBDA operon additionally contains the araE gene encoding a putative transporter from the Sugar Porter (SP) family, which shares 47% sequence identity with the arabinose-proton symporter AraE from Bacillus subtilis (Sá-Nogueira and Ramos, 1997). Co-localization and apparent co-regulation of araE with arabinose catabolic genes confirms the proposed by-homology function in arabinose uptake. Interestingly, orthologous araE genes were identified in 8 other Bifidobacterium genomes, but in those cases, they were not a part of the araBDA operon. Candidate AraQ-binding sites were identified upstream of single araE genes in 5 of these genomes (Supplementary Table S1). The initial set of 45 Bifidobacteriaceae genomes did not reveal any other prospective arabinose transporters that might be regulated by AraQ. In summary, based on our bioinformatics analyses we conclude that AraQ is a conserved regulator of the arabinose catabolism and uptake genes in the Bifidobactericeae family.
Additional similarity searches identified araQ orthologs linked to the araBDA genes in several other families of Actinobacteria including Micrococcaceae (representative genome Arthrobacter arilaitensis), Geodermatophilaceae (Geodermatophilus obscurus) and Glycomycetaceae (Stackebrandtia nassauensis). Although average sequence identity between proteins encoded by these genes and AraQ from bifidobacteria is relatively moderate (∼35%), the identity is considerably higher (up to 90%) within the Helix-Turn-Helix motif involved in DNA binding, suggesting conservation of their AraQ-binding site motifs. The reconstructed AraQ regulons in the above three species include the araBDA operon and an additional putative operon, named araFGH, encoding a putative sugar transport system from the ABC superfamily (Supplementary Table S1). The identified araFGH transporter is located adjacent to the araBDA and araQ genes in all three genomes. Based on the genomic context and metabolic reconstruction we propose that the novel AraFGH transporter in Actinobacteria is involved in arabinose uptake.
Orthologs of araFGH from the above three Actinobacteria were also identified in 19 genomes of the Bifidobacteriaceae family. In most of these genomes, araFGH is co-localized and divergently transcribed with a gene named araU, encoding a putative TF from the ROK family (Figure 1). The common upstream regions of the araU and araFGH genes contain a conserved DNA motif, termed AraU-binding motif (Figure 1), which has a similar structure to DNA motifs recognized by other ROK-family regulators (Kazanov et al., 2013). Comparative genomics-based reconstruction of the AraU regulon revealed that the araFGH operon was the most conserved regulon member (Table 1). Although many previously characterized TFs from the ROK family regulate various sugar utilization pathways (Kazanov et al., 2013), the identified AraU regulator in bifidobacteria is the first example a ROK-family TF that is believed to control arabinose utilization genes.
FIGURE 1.
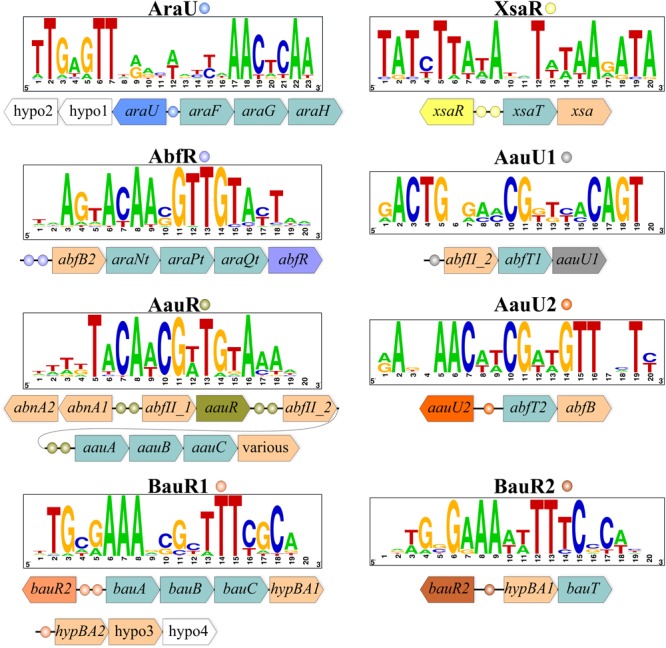
Genomic context and DNA-binding motifs of regulons involved in arabinose/AOS utilization in representative Bifidobacterium genomes. For each TF, a consensus DNA binding motif is shown as a sequence logo. TF genes and their associated binding sites are indicated by matching colors. Other genes are shown as arrows colored by their functional classification: arabinosidases, pale brawn; transporters, light green. Genes encoding hypothetical proteins are shown in white. Detailed information about TF regulon distribution and content is given in Supplementary Table S2.
Table 1.
Overall information about the distribution of TF involved in arabinose/AOS utilization in 45 studied genomes of the Bifidobacteriaceae family and composition of their regulons.
| TF regulon | Example locus taga | TF family | Sugar utilized | No.b | TF regulon contentc |
|---|---|---|---|---|---|
| AraU | BL1693 | ROK | Arabinose | 18 | araU (13), araFGH (16) |
| AbfR | BL0543 | LacI | α-AOS | 12 | abfR (12), abfB2 (12), araNtPtQt (6), afuB-H1 (3), abfII_2 (1) |
| AauR | BL0185 | LacI | α-AOS | 21 | aauR (17), abfII_1 (16), abfII_2 (19), aauABC (19), abnA1 (14), abnA2 (10), afuB-H1 (2), abfBad (2), abfC (1), BDP_0243 (2), BBOMB_ 0938 (1), abfB2 (1), araNtPtQt (1) |
| AauU1 | BISA_0485 | LacI | α-AOS | 3 | aauU1 (3), abfII_2 (3), abfT1 (3) |
| AauU2 | BREU_1998 | LacI | α-AOS | 3 | aauU2 (3), abfB (3), abfT2 (3) |
| XsaR | BAD_1207 | TetR | α-AOS | 5 | xsaR (5), xsA (5), xsaT (5) |
| BauR1 | BL0426 | LacI | β-AOS | 9 | bauR (9), hypBA1 (9), hypBA2 (5), bauABC (9) |
| BauR2 | BREU_0448 | LacI | β-AOS | 5 | bauR2 (4), hypBA1 (4), bauT (3) |
Overall, among 36 studied species of the Bifidobacteriaceae possessing the arabinose catabolic pathway genes, 29 species contain a potential arabinose transporter (araE or araFGH) and thus have a metabolic potential to utilize arabinose (Supplementary Table S3). Interestingly, both predicted arabinose transporters are present in two Bifidobacterium species, B. callitrichos and B. scardovii, where araE appears to be co-regulated with araBDA by AraQ, while araFGH is independently controlled by AraU. The presence of two different regulators for arabinose utilization genes in bifidobacteria may be explained by their response to different molecular effectors, e.g., AraU may potentially respond to arabinose, while AraQ may sense a downstream catabolic intermediate such as L-ribulose-5-P. In summary, bifidobacteria demonstrate not only variable arabinose catabolic and transport capabilities, but also appear to employ distinct routes for transcriptional regulation of arabinose utilization genes. This is in contrast to the previously described arabinose regulons in Escherichia coli and other enterobacteria (Schleif, 2010), the Bacillus/Clostridium group (Zhang et al., 2012), Bacteroides spp. (Chang et al., 2015), and Corynebacterium spp. (Kawaguchi et al., 2009).
Regulation of AOS Utilization
By analogy with arabinose, we defined AOS utilization as a process that includes transporting of an oligosaccharide molecule from the extracellular space into the cell followed by its digestion by various intracellular arabinosidases and the consequent conversion of the released arabinose to D-xylulose 5-phosphate. To identify putative transcriptional regulators of AOS utilization we presumed that local TFs controlling specific sugar catabolic pathway genes are often co-localized with these genes. Consequently, we were able to identify six orthologous groups of TFs that potentially control AOS utilization in bifidobacteria (Table 1), and that have a mosaic distribution in 30 out of 45 studied genomes of the Bifidobactericeae (Figure 2). The identified DNA binding motifs and reconstructed regulons for these local regulators of AOS utilization genes are summarized in Figure 1 and described in details in Supplementary Table S2. Below we provide the description of these novel TF regulons.
FIGURE 2.
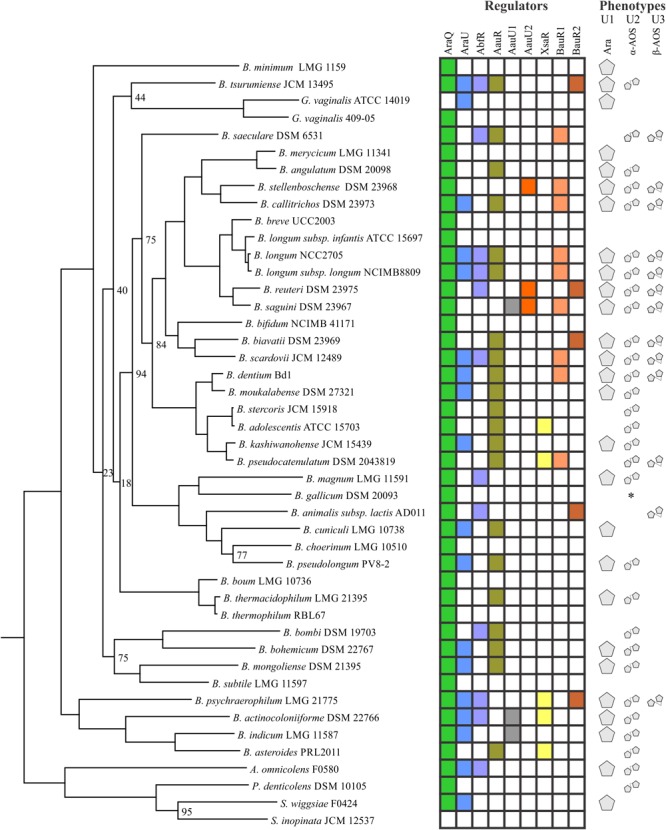
Distribution of arabinose/AOS utilization regulators and predicted growth phenotypes in Bifidobacteriaceae. The presence of a particular TF is shown with a colored rectangle. Predicted arabinose, α- and β-AOS utilization phenotypes are schematically represented as pentagons. The special case of arabinose utilization in B. gallicum is marked with an asterisk and discussed in the text.
The AbfR regulator from the LacI family was found in 11 Bifidobacterium species and Alloscardovia omnicolens (Figure 2), where it was predicted to control one or two genes encoding intracellular α-L-arabinofuranosidases from the GH51 family, named AbfB2 and AfuB-H1. The latter hydrolase and another closely related paralog from the GH51 family had been experimentally characterized in B. longum subsp. longum (locus tags BL1166 and BL1611); both of these enzymes recognize and cleave non-reducing terminal α-1,3- and α-1,5- linked L-arabinofuranose (Af) residues (Margolles and de los Reyes-Gavilán, 2003; Lee et al., 2011). Another putative α-L-arabinofuranosidase from the GH43 family (named AbfII_2) belongs to the reconstructed AbfR-controlled regulon, present only in B. magnum and B. longum strains, while in other bifidobacteria it was predicted to be regulated by another regulator (AauR, see below). Finally, the AbfR-controlled abfB2 loci, being present in six genomes, contain genes encoding components of a putative carbohydrate ABC transport system, named AraNtPtQt (Figure 1). We propose that the AbfR-controlled glycosyl hydrolases and the novel AraNtPtQt transporter are responsible for utilization of α-AOS.
The LacI-family regulator AauR (named for Alpha-Arabino-oligosaccharide Utilization Regulator) is present in 21 Bifidobacterium species, making it the most prevalent among other studied TFs after AraQ (Figure 2). Composition of the reconstructed AauR regulons from various bifidobacterial species is highly variable. The most conserved regulon members are α-L-arabinofuranosidases from the GH43 family (AbfII_1, AbfII_2), endo-α-1,5-L-arabinanases (AbnA1 and AbnA2) and a putative carbohydrate ABC transport system, named AauABC (Figure 1 and Table 1). The phylogenetic analysis of AauR-regulated ABC transporters suggests that they form three distinct orthologous groups that are unevenly distributed across the Bifidobacterium species (Figure 3). In several genomes the aauABC operons contain additional genes encoding α-L-arabinofuranosidases from the GH1 and GH51 families (termed AbfC and AbfBad, respectively), as well as a putative α-galactosidase and a putative endo-1,4-β-xylanase that are absent in other bifidobacteria (Supplementary Table S2). The intracellular AbfC enzyme (BAD_0156 in B. adolescentis) was recently shown to cleave Af from α-1,5-linked AOS in an exo-manner (Suzuki et al., 2013), while AbfBad is a cytoplasmic enzyme that is able to release α-1,3 linked Af residues from double substituted xylose residues in arabinoxylan (Lagaert et al., 2010). The secreted AbnA1 and intracellular AbnA2 arabinases catalyze the internal cleavage of α-AOS and corresponding polysaccharides such as arabinans. The presumably intracellular AbfII enzymes were not experimentally characterized in bifidobacteria; however, their homolog from Streptomyces avermitilis (Araf43A, 51% identity) was shown to cleave only terminal α-1,5- linked Af residues of debranched arabinan (Ichinose et al., 2008). The afuB-H1 and abfB2-araNtPtQt genes were found as a part of AauR regulons in several Bifidobacterium spp., while in other bifidobacteria these genes belonged to the AbfR regulon. In summary, the AauR regulon core is formed by genes potentially involved in the utilization of linear and branched α-AOS and their polymers, such as arabinans.
FIGURE 3.
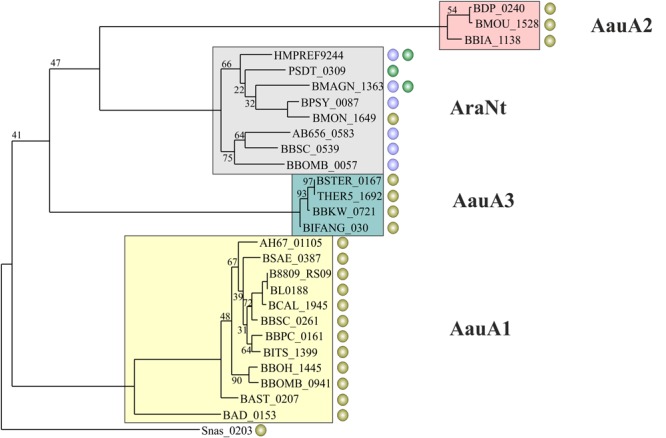
Phylogenetic tree of substrate specific components of ABC transport systems presumably involved in transport of α-AOS. Transporter subunits from the same orthologous group are highlighted with the same background color. The name of each group is given. Circles represent candidate binding sites of various regulators preceding a transporter gene: green – AraQ; purple – AbfR; olive – AauR.
Two other groups of LacI-family regulators, named AauU1 and AauU2, control the putative α-AOS utilization operons in certain Bifidobacterium species (Figure 2). The predicted AauU1 regulon in B. actinocoloniiforme, B. indicum and B. saguini contains the abfII_2-abfT1 operon encoding the GH43-family α-L-arabinofuranosidase and a prospective α-AOS permease from the Oligosaccharide:H+ Symporter (OHS) family (Figure 1). The abfT1 permease is an ortholog (49% identity) of the arabinose-induced transporter CAC1530, which is linked to an arabinosidase gene in Clostridium acetobutylicum (Zhang et al., 2012). The putative AauU2 regulon in B. reuteri, B. saguini and B. stellenboschense contains the abfT2-abfB operon encoding a cytoplasmic α-L-arabinofuranosidase from the GH51 family and a putative permease from the Drug:H+ Antiporter-1 (DHA1) family (Figure 1). We tentatively annotated AbfT1 and AbfT2 as α-AOS transporters that supply substrates for their corresponding α-L-arabinofuranosidases.
Based on our analyses we assigned the BauR1/BauR2 group of TFs to transcriptionally govern genes involved in the utilization of β-AOS that are common in plants and mainly found in various hydroxyproline-rich glycoproteins (Fujita et al., 2014). It was previously proposed that catabolism of β-AOS in B. longum subsp. longum JCM1217 is controlled by a LacI-family regulator (Fujita et al., 2014). Orthologs of this putative TF (named BauR for Beta-Arabino-oligosaccharide Utilization Regulator) are present in 14 studied Bifidobacterium genomes. Detailed analysis of their phylogeny, DNA binding motifs and reconstructed regulons allowed the division of these regulators into two groups, namely BauR1 and BauR2 (Figures 1, 2). The BauR1 regulons include the GH121-family β-L-arabinofuranosidase HypBA1, the GH127-family β-L-arabinobiosidase HypBA2, and a prospective β-AOS ABC transport system (named BauABC). The cytoplasmic HypBA1 hydrolase was shown to release β-1,2- linked Af residues from β-arabinobiose (Fujita et al., 2014), while the surface localized HypBA2 enzyme acts on carrot extensin, potato lectin, and various synthetic substrates liberating β-1,2-arabinobiose (Fujita et al., 2011). The reconstructed BauR2 regulons contain orthologs of the cytoplasmic HypBA1 enzyme and a putative transporter from the Glycoside-Pentoside-Hexuronide (GPH) symporter family (named BauT). We propose that the BauR1/BauR2-control glycosyl hydrolases and the novel BauABC and BauT transporters are responsible for utilization of β-AOS.
Finally, the TetR-family regulator XsaR controls the xsaT-xsa operon in five Bifidobacterium spp. (Figure 1). The putative GH43-family α-L-arabinofuranosidase Xsa is similar (36% identity) to exo-α-L-arabinofuranosidase from B. subtilis, which was shown to release α-1,2- and α-1,3- linked Af residues from branched arabinan and arabinoxylan (Inácio et al., 2008). A putative DHA1-family permease encoded in the same operon with xsa was proposed to be involved in α-AOS uptake. It should be emphasized that XsaR be the first example of a member of the TetR family regulating the process of AOS utilization.
In summary, our bioinformatics analysis of regulons revealed a multitude of regulatory patterns for AOS utilization genes in bifidobacteria (Figure 2 and Supplementary Table S3). First, the identified AOS-related TFs form 6 orthologous groups that exhibit mosaic distribution among the studied genomes; at that a single strain may possess up to three different AOS utilization regulators. Second, several members of the AbfR, AauR, AauU1, and AraQ regulons, namely abfB2, abfII_2 and araNtPtQt, are either regulated by different TFs in various studied genomes (replaced regulons) or co-regulated by two TFs in some genomes (overlapped regulons). Bioinformatics-mediated reconstruction of arabinose /AOS utilization pathways involving the reconstructed TF regulon members is provided in the following section.
Reconstruction of Arabinose/AOS Utilization Pathways
The reconstructed TF regulons included a large set of genes that were potentially involved in the utilization of arabinose and AOS (Supplementary Table S3). We further searched for additional genes involved in these metabolic pathways, but that were not found as a part of the reconstructed TF regulons. First, we added orthologs of two additional α-L-arabinofuranosidases from the GH43 family that had been experimentally described in B. adolescentis, named AbfA and AXH-D3 (Lagaert et al., 2010). AbfA releases α-1,2- or α-1,3- linked Af residues from monosubstituted xylose residues in arabinoxylan, whereas AXH-D3 releases only α-1,3- linked Af residues from double substituted xylose residues. Orthologs of AbfA and AXH-D3 were found in 21 and 9 studied genomes, respectively (Supplementary Table S3). A putative α-L-arabinofuranosidase from the GH51 family (BL1138 in B. longum subsp. longum, named AbfA2) was identified in 9 bifidobacteria. The extracellular α-L-arabinofuranosidase AXH-m23 (BL1543) was identified only in two B. longum subsp. longum strains. A homolog of AXH-m23 in B. subtilis (47% sequence identity) was shown to cleave α-1,2- or α-1,3- linked Af from monosubstituted xylose residues (Bourgois et al., 2007). The expression level of BL1543 significantly increased in the presence of arabinoxylan (Savard and Roy, 2009), therefore emphasizing its role in the degradation of this polysaccharide. Finally, two other prospective GH43-family enzymes, AbfE1 (BL0146) and AbfE2 (BL0158), were identified in B. longum subsp. longum and two other species. These two putative α-L-arabinofuranosidases are characterized by their large size (>950 a.a.) and possible membrane localization. We also predicted that AbfE1 was a part of the expanded AraQ regulon in all four genomes where its ortholog was present (see below).
We further assigned every studied strain with the following predicted phenotypes depending on the presence of genes involved in the transport and catabolism of arabinose and AOS (Figure 2 and Supplementary Table S3). The U1 phenotype was assigned to 27 strains that were predicted to have a capability to utilize arabinose monosaccharide since they contained both the catabolic genes araBDA and the araE/araFGH transporter genes. The U2 and U3 phenotypes were assigned to 29 and 13 strains that were capable to uptake and catabolize α-AOS and β-AOS, respectively. Ten strains are proposed to possess the ability to metabolize both the arabinose monosaccharide as well as each AOS oligosaccharide type (phenotype U123). In contrast, 10 strains are not able to utilize any of these sugars, including 9 ones with phenotype 0, and a single exception of B. gallicum (unique phenotype) that contains the araBDA catabolic genes but lacks any known arabinose or AOS transporter (see below).
The analyzed strains demonstrated a mosaic distribution of the predicted arabinose/AOS utilization phenotypes, and even phylogenetically closely related strains might significantly differ in their utilization capabilities (Supplementary Table S3). The most extreme examples were B. longum subsp. longum and subsp. infantis. The strain ATCC 15697 (B. longum subsp. infantis) lacks the araBDA genes, and is thus not expected to utilize any form of arabinose. In contrast, the NCC2705 and NCIMB8809 strains (B. longum subsp. longum) contain the largest number of genes associated with arabinose/AOS utilization, and are thus assigned the U123 phenotype. In several cases, the related strains were assigned the same phenotype (e.g., U2), yet were shown to possess different α-AOS utilization regulons (e.g., AauU2 in B. stellenboschense and AauR in B. callitrichos) that included different oligosaccharide uptake systems (AbfT2 and AauABC).
To check the robustness of the obtained metabolic reconstructions, we assessed available experimental data on arabinose and AOS utilization in the Bifidobacteriaceae. In the case of arabinose monosaccharide, the predicted phenotypes were in excellent agreement with fermentation data. Thus, we observed concurrence in 20 out of 22 strains, making the accuracy of the prediction of 90% (Supplementary Table S3). All matching cases could be divided into 2 inverse types: (i) when a strain was assigned with the U1 phenotype and had been shown to ferment arabinose, e.g., B. longum subsp. longum NCC2705; (ii) when a strain had been shown not to utilize arabinose and was assigned with phenotype 0. The only strains with inconsistency between the predicted phenotype and experimental data were B. pseudocatenulatum and Parascardovia denticolens since they lacked any known arabinose transporter genes (Supplementary Table S3). One possible explanation for this discrepancy may be that the arabinose uptake in these organisms is carried out by a different yet unknown transport system. Unlike arabinose, there are few reports regarding α-AOS utilization by specific strains of bifidobacteria. In one of them it was shown that linear α-AOS, as well as linear arabinan, supports growth of B. longum subsp. longum ATCC 15707, but not B. bifidum ATCC 29521 or B. longum subsp. infantis ATCC 15697 (Moon et al., 2015). Only the latter strain is included in our reconstruction, and the absence of growth is indeed in agreement with the predicted phenotype (Supplementary Table S3).
Correlation Between Growth Phenotypes and Strain Isolation Sites
Using published data about isolation sites of the analyzed bifidobacteria we compared them with the predicted abilities of microorganisms to utilize arabinose/AOS (Supplementary Table S3). For instance, strains with the highest utilization capabilities (phenotype U123) were isolated from omnivore animals, such as pigs, marmoset and tamarin monkeys, and humans. These results are in agreement with the previous observation that bifidobacteria of monkey/human origin exhibit a higher number of GH genes in their genomes, which is likely caused by their adaptation to a diversified diet of their hosts (Lugli et al., 2017). Additionally, since the primary source of arabinose/AOS are various plant polysaccharides it is expected that strains isolated from herbivorous organisms would also have rich arabinose/AOS utilization phenotypes. Among the studied bifidobacteria, B. saeculare was the only one strain of herbivore origin that was capable of utilizing both arabinose and α- and β-AOS. Two other herbivore-associated strains, B. magnum and B. merycicum, utilize arabinose/α-AOS and arabinose, respectively. In contrast, B. boum could utilize neither arabinose nor AOS.
Among bifidobacteria of human origin, three strains with phenotype 0 (i.e., ones that do not have araBDA) were isolated from infants (Supplementary Table S3). This observation is in line with the fact that the majority of such strains predominate in the infant’s gut due to their specialization on utilizing non-digestible human milk oligosaccharides (Sela et al., 2008; Pokusaeva et al., 2011). The ability of 5 other infant-derived strains to somehow utilize arabinose or/and AOS, might reflect their niche-specific adaptation, i.e., specialization to consuming of sugars other than HMO to avoid competition. One may speculate that with growing of the host and change of the diet, only Bifidobacterium strains that can utilize plant-derived sugars such as AOS can survive. The transition to arabinose/AOS utilization may also be dictated by other members of GM. This notion is based on the discrepancy between the number of intracellular and secreted arabinosidases, and the dearth of latter ones suggests that bifidobacteria rely on other bacteria that can digest polysaccharides to oligosaccharides (Supplementary Table S3). In summary, it would be interesting to check whether utilization phenotypes are to a higher extent associated with the diet rather than with the taxonomy of the host; however, additional data such as diet composition of hosts from which these bacteria were isolated are needed to check this hypothesis.
Models of Arabinose and AOS Utilization in Bifidobacteria
Based on the inferred reconstructions we proposed the following models of arabinose and AOS utilization in bifidobacteria (Figure 4). In each of these models, the intracellular arabinose is converted to D-xylulose-5P via the committed catabolic pathway involving the AraA isomerase, AraB kinase, and AraD epimerase enzymes. The latter product is further metabolized through the bifid shunt (Pokusaeva et al., 2011). The obtained metabolic reconstruction suggests three major models and one additional scenario for utilization of arabinose from various exogenous sources in bifidobacteria.
FIGURE 4.

Models of arabinose and AOS utilization in bifidobacteria. (A) Arabinose utilization via arabinose transporters. (B) Arabinose utilization in B. gallicum. (C) α-AOS utilization. (D) β-AOS utilization. Arabinosidases are shown by light brown ovals. Predicted sugar uptake ABC transporters and permeases are shown as light blue complex and rectangular shapes, respectively. The arabinose monosaccharide catabolic enzymes are shown in green boxes. ‘Hypo GH’ denotes hypothetical glycosyl hydrolases that are presumed to be involved in AOS utilization. Pentagon represents a hydroxyproline residue of a glycosylated protein.
In the first model (Figure 4A), arabinose is transported into the cell via AraE (permease) or AraFGH (ABC-type) transporters. The release of terminal arabinose residues (mainly Af) of various oligosaccharides such as AOS and AXOS is performed by various extracellular GH (mainly α-L-arabinofuranosidases). Our genomic analysis suggests that only two strains of B. longum subsp. longum contain the extracellular arabinofuranosidase AXH-m23, which is presumably implicated in the AXOS utilization. In confirmation, B. longum subsp. longum NCC2705 was shown to consume only arabinose substituents of AXOS and not the xylose backbone (Rivière et al., 2014, 2015). AbfE1 and AbfE2 are two other candidates for arabinose-cleaving activity outside the cell due to their predicted membrane localization. Remarkably, these two enzymes were also rare among the studied species and were found in B. longum subsp. longum and 3 other species of bifidobacteria (Supplementary Table S3). These observations indicate that only a few Bifidobacterium species are capable of cleaving and utilizing extracellular arabinose, while the majority of them utilize free arabinose that is released from oligo- and polysaccharides by other microbial community members. The suitable candidates for such reliance in GM are the Bacteroides species due to a significant diversity of their arsenal of secreted GH (Ravcheev et al., 2013b).
In the second model (Figure 4C), branched and debranched arabinan and long α-AOS molecules are degraded by extracellular endo-α-1,5-L-arabinanases, followed by cellular uptake of the generated α-AOS by means of ABC- or permease-type uptake systems, after which the internalized oligosaccharides are hydrolyzed to arabinose using a variety of intracellular arabinofuranosidases targeting terminal α-1,5-, α-1,2-, and α-1,3- bound Af residues. Using regulon analysis, we identified seven groups of putative α-AOS transporters, which included AraNtPtQt, three groups of AauABC ABC transport systems and three permease-type systems, AbfT1, AbfT2 and XsaT (Supplementary Table S3). Genome context analysis in combination with available data on the specificity of the arabinofuranosidases allowed us to assign the most likely substrates of these transporters. Thus, we deducted that AauABC(1-3) and AbfT1 presumably transported linear α-AOS based on their co-occurrence and co-regulation with α-L-arabinofuranosidases releasing α-1,5-linked Af (AbfII_1, AbfII_2, AbfC). However, the AauABC(3) system may also transport branched α-AOS, since in some species genes of this transporter are coupled with AbfBad, which releases only α-1,3- linked Af residues. For AraNtPtQt and AbfT2, we predict uptake of both branched and linear α-AOS based on their genomic coupling with AbfB2 and AbfB, respectively. Finally, XsaT presumably transports only branched α-AOS. Diversification of α-AOS transporters based on substrate length, a degree of branching and modifications such as feruloylation (Holck et al., 2011) and their mosaic distribution among species of bifidobacteria imply possible co-degradation of AOS by various species that alleviates competition through substrate specialization.
The third model describes utilization of β-AOS released from glycosylated proteins (Figure 4D). The first step is the release of the terminal Af residue from a β-AOS chain bound to a hydroxyproline residue, followed by the release of β-arabinobiose. In B. longum subsp. longum, it was shown experimentally that the second step of this process is carried by the membrane-bound β-L-arabinobiosidase HypBA2, while the first step was predicted to be performed by a hypothetical GH encoded by BLLJ_0213, which is located in the same operon with hypBA2 (Fujita et al., 2014). The liberated disaccharides are transported into the cell via one of the two predicted transport systems, BauT (a permease) and BauABC (an ABC transporter). Intracellular β-arabinobiose is further hydrolyzed by HypBA1, thus producing two arabinose residues (Fujita et al., 2014). Since hypBA2 is missing in several Bifidobacterium strains possessing other β-AOS utilization genes (Supplementary Table S3), we speculate that other unidentified GHases may be involved in these extracytoplasmic reactions. Alternatively, these strains may depend on enzymatic capabilities of other GM species for release of and cross-feed on β-AOS disaccharides.
An additional scenario for utilization of arabinose released from degradation of AXOS was proposed for B. gallicum (Figure 4B). This strain is unable to utilize arabinose from arabinose/xylose mixture (Rivière et al., 2014), which is in agreement with the absence of any predicted arabinose transporter genes in its genome (Supplementary Table S3). B. gallicum is the only studied species whose genome contains the araBDA operon yet does not specify any known pathway for AOS utilization (Figure 2). We hypothesize that this strain not only internalizes xylooligosaccharides (XOS) but also AXOS via the prospective transporter XosABC (BIFGAL_03672-3669-3667). The AXOS are then further broken down by AbfA/XynB (BIFGAL_03676) and a GH43-family enzyme (BIFGAL_03666). This alternative strategy of arabinose utilization may also be operational in other species possessing araBDA, xosABC, and abfA/xynB orthologs.
Global AraQ Regulon
Our results suggest that AraQ is a master regulator of the arabinose catabolic pathway genes araBDA and the arabinose transporter gene araE in the Bifidobacteraceae family. However, AraQ was also found in seven Bifidobacterium strains (e.g., B. breve UCC2003 and B. longum subsp. infantis ATCC 15697) and a single strain of G. vaginalis that lacked the arabinose utilization genes, suggesting it is involved in transcriptional control of other metabolic pathways. Scardovia inopinata and another strain of G. vaginalis (ATCC 14019) were the only two strains from the Bifidobacteraceae family that lacked an araQ ortholog (Figure 2). The comparative genomics reconstruction of orthologous AraQ regulons allowed identification of the conserved core regulon genes and genome-specific regulated genes in the studied 43 genomes of Bifidobacteraceae (Table 2 and Supplementary Table S1). The core regulon members were defined as genes preceeded by potential AraQ-binding sites that were conserved in 20 or more genomes. The core regulon includes the araBDA and araQ genes and seven genes involved in the central carbohydrate metabolism (gap, eno, pyk, tkt-tal, galM) and lactate fermentation (ldh). At that, the pyruvate kinase pyk belongs to the AraQ regulon in all studied Bifidobacterium genomes and in G. vaginalis, while the enolase eno appears to be under AraQ control in 35 genomes.
Table 2.
Composition of the AraQ regulons in 45 studied genomes of the Bifidobacteriaceace family.
| Gene/operon | Functional role(s) | No. a | Mode b |
|---|---|---|---|
| araQ | Global transcriptional regulator AraQ | 20 | R |
| araBDA | Ribulokinase; L-ribulose-5-phosphate 4-epimerase; L-arabinose isomerase | 35 | R |
| araE | Arabinose-proton symporter | 8 | R |
| abfE1 | α-L-arabinofuranosidase AbfE1, GH43 | 4 | ND |
| abfB2 | α-L-arabinofuranosidase AbfB2, GH51 | 3 | ND |
| araNtPtQt | α-AOS ABC transport system | 3 | ND |
| gap | Glyceraldehyde 3-phosphate dehydrogenase | 24 | A |
| eno | Enolase | 35 | A |
| pyk | Pyruvate kinase | 40 | A |
| tkt – tal | Transketolase; transaldolase | 24 | A |
| ldh | Lactate dehydrogenase | 20 | A |
| galM | Galactose mutarotase | 22 | R |
| pta – ackA | Phosphotransacetylase; acetate kinase | 8 | R |
| pflB – pflA | Pyruvate formate lyase | 10 | A |
| BL1113 | Aldo/keto reductase | 6 | R |
| malE | Maltose-binding periplasmic protein | 9 | A |
| malQ1 | 4-α-glucanotransferase | 4 | A? |
| maa | Maltose O-acetyltransferase | 7 | R |
| fucO | Lactaldehyde reductase | 7 | ND |
| glgB – Blon_ 1762–1763 | 1,4-α-glucan branching enzyme GlgB; two-component regulatory system | 6 | R |
| carD – ispF | CarD transcriptional regulator; 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase | 6 | R |
| birA | Biotin-(acetyl-CoA-carboxylase) ligase | 6 | R |
| sixA | Phosphohistidine phosphatase | 5 | R |
| glk | Glucokinase, ROK family | 7 | A |
| bfrBCD – X – bfrR – bfrA | Fructosides ABC transport system; predicted transcriptional regulator; β-fructosidase | 4 | A |
aNumber of genomes in which a gene is regulated by AraQ. Detailed distribution of AraQ-regulated genes and AraQ-binding sites is given in Supplementary Table S1. bPredicted mode of AraQ-dependent regulation: A, activation; R, repression; ND, not determined.
The less conserved members of the AraQ regulon include the acetate and formate fermentation genes (pta-ackA and pflB-pflA), a lactaldehyde reductase (fucO), the arabinose transporter (araE), various arabinofuranosidases and arabinoside transporters, a glucokinase, an aldo/keto reductase and the fructosides catabolism operon bfr. Other putative members of the AraQ regulon such as malE, glgB, carD-ispF are conserved in a narrow taxonomic group of bifidobacteria including B. longum and B. breve, while maa is preceded by AraQ sites predominantly in the B. dentium group. It may be speculated that the latter cases illustrate relatively recent genome- or group-specific regulon expansions.
The observed diversity of functional roles and pathways constituting the reconstructed AraQ regulons suggests that AraQ potentially functions as a dual regulator for various genes. To predict the activation or repression mechanism of AraQ action, we investigated multiple alignments of upstream gene regions containing AraQ-binding sites and predicted the conserved -35 and -10 promoter elements (Supplementary Figure S1). The negative mode of regulation was proposed for genes preceded by an AraQ site, which was either located downstream of the -10 element or if it overlapped with the -10/-35 elements. The positive mode of regulation was assumed for AraQ sites that were situated upstream of the -35 element. The overall results of this analysis are presented in Table 2 and Figure 5. The repression mechanism of AraQ action was proposed for the arabinose utilization genes araBDA, the regulatory gene araQ, the acetate fermentation operon pta-ackA, and other genome-specific regulon members including the galM, BL1113, maa, sixA, birA and glgB genes. In contrast, the activation mechanism of AraQ action was proposed for the majority of core regulon genes involved in the central carbohydrate metabolism (gap, eno, pyk, tkt-tal, ldh), as well as for the maltose and glucose utilization genes (malE, malQ1, glk) and the formate fermentation operon pflBA. The predicted promoters of two genes, namely eno in B. bifidum and ldh in B. longum, were validated using data on experimentally mapped transcription start sites (Minowa et al., 1989; Sun et al., 2014). The obtained results suggest that AraQ may function both as a repressor and an activator of different sets of genes involved in the central carbohydrate metabolism and fermentation.
FIGURE 5.
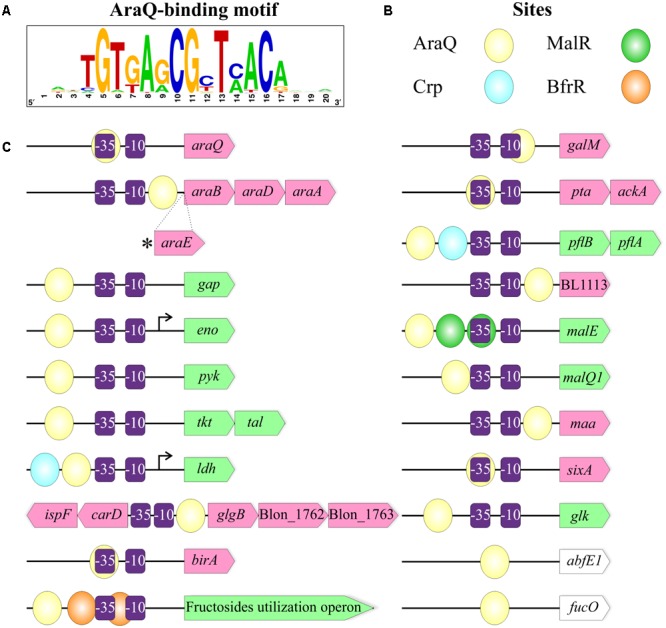
The overall content of the AraQ regulons in bifidobacteria. (A) Consensus DNA motif for AraQ-binding sites represented as a sequence logo build based on all identified sites in 45 studied genomes. (B) A scheme representing the coloring of different TF-binding sites. Each sphere corresponds to a specific TFBS. (C) The overall content of the AraQ regulon. The predicted mechanism of the AraQ action on the expression of a gene of interest is shown by specific coloring: repressed genes are in pink, activated – in green, the ones for which the mode of action was not determined – in white. Prospective –10 and –35 sequences of a bacterial promoter are shown as purple rectangles. Experimentally described transcription start sites are indicated by arrows. The picture represents maximum content of the AraQ regulon, detailed information about its composition in every strain can be found in Supplementary Table S1.
We also observed that some of the AraQ-regulated genes were potentially controlled by additional TFs (Figure 5). The upstream regions of ldh and pflB-pflA also contain putative binding sites of Crp, another prospective global regulator in bifidobacteria (Khoroshkin et al., 2016). The upstream positions of both of these operator sites relative to the -35 promoter element suggest that both AraQ and Crp might act as transcriptional activators for these fermentation genes. The malE gene is additionally controlled by the potential maltose repressor MalR, while the bfr operon is co-regulated by the putative beta-fructoside repressor BfrR (Khoroshkin et al., 2016) (Supplementary Figure S1).
Evolutionary Scenario of the Reconstructed Regulons
Analysis of TF distribution and tentative reconstruction of the regulatory networks allowed us to propose a hypothesis about the possible evolution of the arabinose and AOS regulons in the Bifidobacteriaceae genomes. The global AraQ regulon containing central carbohydrate metabolism genes in addition to the arabinose utilization genes was identified in the majority of the Bifidobacterium species (Supplementary Table S1). G. vaginalis is closely related to the Bifidobacterium species according to the whole-genome phylogeny tree (Figure 2) and the previous phylogenomic analysis of the Bifidobacteriaceae family (Lugli et al., 2017). Among two analyzed strains of G. vaginalis, the AraQ regulon in the 409-05 strain contains the central carbohydrate metabolism genes, whereas the AraQ regulator is missing from another strain, ATCC 14019, likely due to a recent gene loss. In contrast, the local AraQ regulons containing only genes involved in arabinose utilization were identified in three other species from the Bifidobacteriaceae family (A. omnicolens, P. denticolens, Scardovia wiggsiae), as well in Arthrobacter arilaitensis, Geodermatophilus obscurus, and Stackebrandtia nassauensis representing different taxa of Actinobacteria. These observations lead to the hypothesis that the last universal common ancestor of the Bifidobacteriaceae family used AraQ for the control of arabinose utilization genes araBDA, whereas the AraU regulator controlled the arabinose ABC transporter genes araFGH. In the common ancestor of Bifidobacterium/Gardenella, the AraQ regulon expanded to include the central carbohydrate metabolism genes, at that some species lost the arabinose utilization genes but retained AraQ. In some bifidobacteria, e.g., in the B. longum subsp. longum and subsp. infantis, the regulon expansion went to the extreme extent, increasing its size up to approximately 20 genes. This is the first report of regulon expansion in bifidobacteria, although lineage-specific expansions of other central carbohydrate metabolism regulons were documented for other microorganisms, for example, HexR in Shewanella (Leyn et al., 2011) and FruR in Enterobacteria (Ravcheev et al., 2014).
The majority of the identified AOS utilization regulators have a narrow taxonomic distribution being present only in the Bifidobacterium genus (Figure 2 and Supplementary Table S2). However, the AbfR regulon was also found in A. omnicolens, suggesting its appearance in the common ancestor of Bifidobacterium and Alloscardovia genera. Interestingly, the AauU2 regulon was only found in species isolated from tamarins and marmoset monkeys, suggesting its late taxon-specific appearance. The AbfR and AraQ regulons show several interconnections including the cases of mosaic regulation and potential co-regulations (Supplementary Table S3). First, the α-AOS utilization genes in P. denticolens are controlled by AraQ, whereas in members of the Bifidobacterium genus their regulation is intercepted by AbfR. Second, the α-AOS utilization operons in A. omnicolens and B. magnum are preceded by both AraQ- and AbfR-binding sites suggesting both regulators are involved in the simultaneous control of these genes. Finally, the variable composition of AauR regulons in the Bifidobacterium genus was likely shaped by numerous horizontal gene transfer and regulon expansion events. Overall, we believe that the emergence of AOS utilization regulons in bifidobacteria reflects adaptation of these species to the gastrointestinal tract and the lifestyle of feeding on non-digestible plant sugars that are derived from the diet of their host.
Conclusion and Future Perspectives
This study significantly broadens our understanding of arabinose and AOS metabolism and its transcriptional regulation in bifidobacteria. It was shown in previous works that (i) many members of the Bifidobacterium genus are able to ferment arabinose; and (ii) AOS and their polymers of plant origin are causing the bifidogenic effect in vitro and therefore can potentially be used as prebiotics. However, a knowledge gap exists with respect to arabinose/AOS utilization and associated transcriptional regulation in bifidobacteria. In the current work, we used comparative genomics approaches to reconstruct metabolic pathways and transcriptional regulons for arabinose and AOS utilization in 39 Bifidobacterium strains as well as in 6 strains of closely related genera. Overall, our analysis revealed a complex regulatory network operated by 9 TFs from LacI, TetR and ROK families (Figure 1 and Table 1). The reconstructed TF regulons allowed us to predict several prospective transporters for arabinose and α- and β-AOS. As a result, we describe here a comprehensive collection of genes encoding arabinose/AOS catabolic enzymes and transporters (Supplementary Table S3), predicted sugar utilization phenotypes for each studied strain (Figure 2), and propose models for arabinose/AOS utilization in bifidobacteria (Figure 4). The observed diversity of arabinose/AOS utilization pathways can be explained by an adaptation of bifidobacteria to survive in the competitive gut environment via their specialization to grow on different sugars (e.g., different AOS). Our results also suggest that the majority of bifidobacteria rely on the ability of other GM bacteria to break down the complex arabinose-containing polysaccharides such as arabinans. The inferred arabinose/AOS utilization phenotypes bring us closer to the ultimate goal of the rational design of AOS-based prebiotics that might positively affect human health and alleviate diseases. This notion is supported by several publications indeed reporting that these sugars are not digested by human secretions (Suzuki et al., 2004; Moon et al., 2015). However, it should be pointed out that the process of prebiotic development should consider metabolic connections of bifidobacteria with other members of the GM, for example if they are dependent on GH activities produced by other bacteria in order to gain access to certain carbohydrates.
Unlike other investigated TFs AraQ is conserved in all studied members of the Bifidobacterium genus including species with missing araBDA operon. The inferred AraQ regulon suggests that AraQ acts as a global regulator of the central carbohydrate metabolism genes in bifidobacteria. Comparative analysis of AraQ-regulated promoter regions among Bifidobacterium strains suggests that AraQ can potentially act as a bi-functional regulator, which represses the araBDA and several other genes, while it activates many other genes involved in the central glycolytic pathways (Figure 5). The obtained bioinformatics predictions are sufficiently interesting to warrant further experimental verification of AraQ-dependent regulation using both in vitro and in vivo techniques. Other novel regulators of arabinose and AOS utilization predicted for the first time in this work also await experimental validations, as well as testing of the ability of different Bifidobacterium strains to utilize various AOS to confirm predicted utilization phenotypes.
Author Contributions
AA and DR conceived and designed the research project. AA performed the comparative genomic analysis, reconstruction of TF regulons and building of models of arabinose and AOS utilization. DR provided the quality control of the obtained data. AA, DR, and DS wrote the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Pavel Shelyakin and Matvei Khoroshkin for their help with selection and downloading of genomes and to Andrei Osterman for the useful discussions.
Abbreviations
- Af
arabinofuranose
- AOS
arabino-oligosaccharides
- AXOS
arabino-xylooligosaccharides
- GH
glycoside hydrolase
- GM
gut microbiota
- PWM
position weight matrix
- TF
transcription factor
- TFBS
transcription factor-binding site
Footnotes
Funding. This research was supported by the Russian Science Foundation grant no. 14-14-00289. DS is a member of the APC Microbiome Institute funded by Science Foundation Ireland through the Irish Government’s National Development Plan (Grant No. SFI/12/RC/2273).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00776/full#supplementary-material
References
- Al-Tamimi M. A. H. M., Palframan R. J., Cooper J. M., Gibson G. R., Rastall R. A. (2006). In vitro fermentation of sugar beet arabinan and arabino-oligosaccharides by the human gut microflora. J. Appl. Microbiol. 100 407–414. 10.1111/j.1365-2672.2005.02780.x [DOI] [PubMed] [Google Scholar]
- Arboleya S., Watkins C., Stanton C., Ross R. P. (2016). Gut bifidobacteria populations in human health and aging. Front. Microbiol. 7:1204 10.3389/fmicb.2016.01204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D. J., Ostell J., et al. (2017). GenBank. Nucleic Acids Res. 45 D37–D42. 10.1093/nar/gkw1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels L. B., Delzenne N. M., Cani P. D., Walter J. (2015). Towards a more comprehensive concept for prebiotics. Nat. Rev. Gastroenterol. Hepatol. 12 303–310. 10.1038/nrgastro.2015.47 [DOI] [PubMed] [Google Scholar]
- Bourgois T. M., Van Craeyveld V., Van Campenhout S., Courtin C. M., Delcour J. A., Robben J., et al. (2007). Recombinant expression and characterization of XynD from Bacillus subtilis subsp. subtilis ATCC 6051: a GH 43 arabinoxylan arabinofuranohydrolase. Appl. Microbiol. Biotechnol. 75 1309–1317. 10.1007/s00253-007-0956-2 [DOI] [PubMed] [Google Scholar]
- Chang C., Tesar C., Li X., Kim Y., Rodionov D. A., Joachimiak A. (2015). A novel transcriptional regulator of L-arabinose utilization in human gut bacteria. Nucleic Acids Res. 43 10546–10559. 10.1093/nar/gkv1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn M., Horibata K. (1959). Inhibition by glucose of the induced synthesis of the β-galactoside-enzyme system of Escherichia coli. Analysis of maintenance. J. Bacteriol. 78 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J.-M., et al. (2011). T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 39 W13–W17. 10.1093/nar/gkr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier C. F., Vaughan E. E., De Vos W. M., Akkermans A. D. L. (2002). Molecular monitoring of succession of bacterial communities in human neonates. Appl. Environ. Microbiol. 68 219–226. 10.1128/AEM.68.1.219-226.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K., Sakamoto S., Ono Y., Wakao M., Suda Y., Kitahara K., et al. (2011). Molecular cloning and characterization of a β-L-arabinobiosidase in Bifidobacterium longum that belongs to a novel glycoside hydrolase family. J. Biol. Chem. 286 5143–5150. 10.1074/jbc.M110.190512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K., Takashi Y., Obuchi E., Kitahara K., Suganuma T. (2014). Characterization of a novel β-L-arabinofuranosidase in Bifidobacterium longum. J. Biol. Chem. 289 5240–5249. 10.1074/jbc.M113.528711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M., Guindon S., Gascuel O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27 221–224. 10.1093/molbev/msp259 [DOI] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Holck J., Lorentzen A., Vigsnæs L. K., Licht T. R., Mikkelsen J. D., Meyer A. S. (2011). Feruloylated and nonferuloylated arabino-oligosaccharides from sugar beet pectin selectively stimulate the growth of Bifidobacterium spp. in human fecal in vitro fermentations. J. Agric. Food Chem. 59 6511–6519. 10.1021/jf200996h [DOI] [PubMed] [Google Scholar]
- Ichinose H., Yoshida M., Fujimoto Z., Kaneko S. (2008). Characterization of a modular enzyme of exo-1,5-alpha-L-arabinofuranosidase and arabinan binding module from Streptomyces avermitilis NBRC14893. Appl. Microbiol. Biotechnol. 80 399–408. 10.1007/s00253-008-1551-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inácio J. M., Correia I. L., de Sá-Nogueira I. (2008). Two distinct arabinofuranosidases contribute to arabino-oligosaccharide degradation in Bacillus subtilis. Microbiology 154 2719–2729. 10.1099/mic.0.2008/018978-0 [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 D457–D462. 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi H., Sasaki M., Vertès A. A., Inui M., Yukawa H. (2009). Identification and functional analysis of the gene cluster for L-arabinose utilization in Corynebacterium glutamicum. Appl. Environ. Microbiol. 75 3419–3429. 10.1128/AEM.02912-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanov M. D., Li X., Gelfand M. S., Osterman A. L., Rodionov D. A. (2013). Functional diversification of ROK-family transcriptional regulators of sugar catabolism in the Thermotogae phylum. Nucleic Acids Res. 41 790–803. 10.1093/nar/gks1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoroshkin M. S., Leyn S. A., Van Sinderen D., Rodionov D. A. (2016). Transcriptional regulation of carbohydrate utilization pathways in the Bifidobacterium genus. Front. Microbiol. 7:120 10.3389/fmicb.2016.00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Oh H.-S., Park S.-C., Chun J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64 346–351. 10.1099/ijs.0.059774-0 [DOI] [PubMed] [Google Scholar]
- Lagaert S., Pollet A., Delcour J. A., Lavigne R., Courtin C. M., Volckaert G. (2010). Substrate specificity of three recombinant α-l-arabinofuranosidases from Bifidobacterium adolescentis and their divergent action on arabinoxylan and arabinoxylan oligosaccharides. Biochem. Biophys. Res. Commun. 402 644–650. 10.1016/j.bbrc.2010.10.075 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Hyun Y.-J., Kim D.-H. (2011). Cloning and characterization of α-L-arabinofuranosidase and bifunctional α-L-arabinopyranosidase/β-D-galactopyranosidase from Bifidobacterium longum H-1. J. Appl. Microbiol. 111 1097–1107. 10.1111/j.1365-2672.2011.05128.x [DOI] [PubMed] [Google Scholar]
- Lee J.-H., O’Sullivan D. J. (2010). Genomic insights into bifidobacteria. Microbiol. Mol. Biol. Rev. 74 378–416. 10.1128/MMBR.00004-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyn S. A., Kazanov M. D., Sernova N. V., Ermakova E. O., Novichkov P. S., Rodionov D. A. (2013). Genomic reconstruction of the transcriptional regulatory network in Bacillus subtilis. J. Bacteriol. 195 2463–2473. 10.1128/JB.00140-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyn S. A., Li X., Zheng Q., Novichkov P. S., Reed S., Romine M. F., et al. (2011). Control of proteobacterial central carbon metabolism by the HexR transcriptional regulator. J. Biol. Chem. 286 35782–35794. 10.1074/jbc.M111.267963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42 D490–D495. 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli G. A., Milani C., Turroni F., Duranti S., Mancabelli L., Mangifesta M., et al. (2017). Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC Genomics 18:568. 10.1186/s12864-017-3955-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolles A., de los Reyes-Gavilán C. G. (2003). Purification and functional characterization of a novel α-L-arabinofuranosidase from Bifidobacterium longum B667. Appl. Environ. Microbiol. 69 5096–5103. 10.1128/AEM.69.9.5096-5103.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C., Duranti S., Bottacini F., Casey E., Turroni F., Mahony J., et al. (2017). The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 81:e00036-17. 10.1128/MMBR.00036-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa T., Iwata S., Sakai H., Masaki H., Ohta T. (1989). Sequence and characteristics of the Bifidobacterium longum gene encoding L-lactate dehydrogenase and the primary structure of the enzyme: a new feature of the allosteric site. Gene 85 161–168. 10.1016/0378-1119(89)90476-9 [DOI] [PubMed] [Google Scholar]
- Mironov A. A., Vinokurova N. P., Gelfand M. S. (2000). Software for analysis of bacterial genomes. Mol. Biol. 34 222–231. 10.1007/BF02759643 [DOI] [PubMed] [Google Scholar]
- Moon J. S., Shin S. Y., Choi H. S., Joo W., Cho S. K., Li L., et al. (2015). In vitro digestion and fermentation properties of linear sugar-beet arabinan and its oligosaccharides. Carbohydr. Polym. 131 50–56. 10.1016/j.carbpol.2015.05.022 [DOI] [PubMed] [Google Scholar]
- Novichkov P. S., Laikova O. N., Novichkova E. S., Gelfand M. S., Arkin A. P., Dubchak I., et al. (2010). RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res. 38 D111–D118. 10.1093/nar/gkp894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odamaki T., Kato K., Sugahara H., Hashikura N., Takahashi S., Xiao J.-Z., et al. (2016). Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 16:90. 10.1186/s12866-016-0708-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M., Benno Y., Leung K.-P., Maeda N. (2008). Bifidobacterium tsurumiense sp. nov., from hamster dental plaque. Int. J. Syst. Evol. Microbiol. 58 144–148. 10.1099/ijs.0.65296-0 [DOI] [PubMed] [Google Scholar]
- Onumpai C., Kolida S., Bonnin E., Rastall R. A. (2011). Microbial utilization and selectivity of pectin fractions with various structures. Appl. Environ. Microbiol. 77 5747–5754. 10.1128/AEM.00179-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R., Begley T., Butler R. M., Choudhuri J. V., Chuang H.-Y., Cohoon M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33 5691–5702. 10.1093/nar/gki866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen T. N., Brunak S., von Heijne G., Nielsen H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8 785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- Picard C., Fioramonti J., Francois A., Robinson T., Neant F., Matuchansky C. (2005). Review article: bifidobacteria as probiotic agents – physiological effects and clinical benefits. Aliment. Pharmacol. Ther. 22 495–512. 10.1111/j.1365-2036.2005.02615.x [DOI] [PubMed] [Google Scholar]
- Pokusaeva K., Fitzgerald G. F., van Sinderen D. (2011). Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 6 285–306. 10.1007/s12263-010-0206-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Arkin A. P. (2017). PaperBLAST: Text mining papers for information about homologs. mSystems 2 e39–e17. 10.1128/mSystems.00039-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravcheev D. A., Best A. A., Sernova N. V., Kazanov M. D., Novichkov P. S., Rodionov D. A. (2013a). Genomic reconstruction of transcriptional regulatory networks in lactic acid bacteria. BMC Genomics 14:94. 10.1186/1471-2164-14-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravcheev D. A., Best A. A., Tintle N., DeJongh M., Osterman A. L., Novichkov P. S., et al. (2011). Inference of the transcriptional regulatory network in Staphylococcus aureus by integration of experimental and genomics-based evidence. J. Bacteriol. 193 3228–3240. 10.1128/JB.00350-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravcheev D. A., Godzik A., Osterman A. L., Rodionov D. A. (2013b). Polysaccharides utilization in human gut bacterium Bacteroides thetaiotaomicron: comparative genomics reconstruction of metabolic and regulatory networks. BMC Genomics 14:873. 10.1186/1471-2164-14-873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravcheev D. A., Khoroshkin M. S., Laikova O. N., Tsoy O. V., Sernova N. V., Petrova S. A., et al. (2014). Comparative genomics and evolution of regulons of the LacI-family transcription factors. Front. Microbiol. 5:294 10.3389/fmicb.2014.00294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière A., Gagnon M., Weckx S., Roy D., De Vuyst L. (2015). Mutual cross-feeding interactions between Bifidobacterium longum subsp. longum NCC2705 and Eubacterium rectale ATCC 33656 explain the bifidogenic and butyrogenic effects of arabinoxylan oligosaccharides. Appl. Environ. Microbiol. 81 7767–7781. 10.1128/AEM.02089-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière A., Moens F., Selak M., Maes D., Weckx S., De Vuyst L. (2014). The ability of bifidobacteria to degrade arabinoxylan oligosaccharide constituents and derived oligosaccharides is strain dependent. Appl. Environ. Microbiol. 80 204–217. 10.1128/AEM.02853-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D. A. (2007). Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107 3467–3497. 10.1186/1471-2164-14-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D. A., Rodionova I. A., Li X., Ravcheev D. A., Tarasova Y., Portnoy V. A., et al. (2013). Transcriptional regulation of the carbohydrate utilization network in Thermotoga maritima. Front. Microbiol. 4:244. 10.3389/fmicb.2013.00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D. A., Yang C., Li X., Rodionova I. A., Wang Y., Obraztsova A. Y., et al. (2010). Genomic encyclopedia of sugar utilization pathways in the Shewanella genus. BMC Genomics 11:494. 10.1186/1471-2164-11-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell D. A., Ross R. P., Fitzgerald G. F., Stanton C. (2011). Metabolic activities and probiotic potential of bifidobacteria. Int. J. Food Microbiol. 149 88–105. 10.1016/j.ijfoodmicro.2011.06.003 [DOI] [PubMed] [Google Scholar]
- Saier M. H., Reddy V. S., Tsu B. V., Ahmed M. S., Li C., Moreno-Hagelsieb G. (2016). The transporter classification database (TCDB): recent advances. Nucleic Acids Res. 44 D372–D379. 10.1093/nar/gkv1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sá-Nogueira I., Ramos S. S. (1997). Cloning, functional analysis, and transcriptional regulation of the Bacillus subtilis araE gene involved in L-arabinose utilization. J. Bacteriol. 179 7705–7711. 10.1128/jb.179.24.7705-7711.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savard P., Roy D. (2009). Determination of differentially expressed genes involved in arabinoxylan degradation by Bifidobacterium longum NCC2705 Using Real-Time RT-PCR. Probiotics Antimicrob. Proteins 1:121. 10.1007/s12602-009-9015-x [DOI] [PubMed] [Google Scholar]
- Schell M. A., Karmirantzou M., Snel B., Vilanova D., Berger B., Pessi G., et al. (2002). The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. U.S.A. 99 14422–14427. 10.1073/pnas.212527599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleif R. (2010). AraC protein, regulation of the L-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol. Rev. 34 779–796. 10.1111/j.1574-6976.2010.00226.x [DOI] [PubMed] [Google Scholar]
- Sela D. A., Chapman J., Adeuya A., Kim J. H., Chen F., Whitehead T. R., et al. (2008). The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. U.S.A. 105 18964–18969. 10.1073/pnas.0809584105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton C., Ross R. P., Fitzgerald G. F., Van Sinderen D. (2005). Fermented functional foods based on probiotics and their biogenic metabolites. Curr. Opin. Biotechnol. 16 198–203. 10.1016/j.copbio.2005.02.008 [DOI] [PubMed] [Google Scholar]
- Sulek K., Vigsnaes L. K., Schmidt L. R., Holck J., Frandsen H. L., Smedsgaard J., et al. (2014). A combined metabolomic and phylogenetic study reveals putatively prebiotic effects of high molecular weight arabino-oligosaccharides when assessed by in vitro fermentation in bacterial communities derived from humans. Anaerobe 28 68–77. 10.1016/j.anaerobe.2014.05.007 [DOI] [PubMed] [Google Scholar]
- Sun Z., Westermann C., Yuan J., Riedel C. U. (2014). Experimental determination and characterization of the gap promoter of Bifidobacterium bifidum S17. Bioengineered 5 371–377. 10.4161/bioe.34423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Murakami A., Yoshida K. (2013). Motif-guided identification of a glycoside hydrolase gamily 1 α-L-arabinofuranosidase in Bifidobacterium adolescentis. Biosci. Biotechnol. Biochem. 77 1709–1714. 10.1271/bbb.130279 [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Tanaka K., Amano T., Asakura T., Muramatsu N. (2004). Utilization by intestinal bacteria and digestibility of arabino-oligosaccharides in vitro. J. Jpn. Soc. Hortic. Sci. 73 574–579. 10.2503/jjshs.73.574 [DOI] [Google Scholar]
- The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45 D158–D169. 10.1093/nar/gkw1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Broek L. A. M., Hinz S. W. A., Beldman G., Vincken J.-P., Voragen A. G. J. (2008). Bifidobacterium carbohydrases-their role in breakdown and synthesis of (potential) prebiotics. Mol. Nutr. Food Res. 52 146–163. 10.1002/mnfr.200700121 [DOI] [PubMed] [Google Scholar]
- van den Broek L. A. M., Lloyd R. M., Beldman G., Verdoes J. C., McCleary B. V., Voragen A. G. J. (2005). Cloning and characterization of arabinoxylan arabinofuranohydrolase-D3 (AXHd3) from Bifidobacterium adolescentis DSM20083. Appl. Microbiol. Biotechnol. 67 641–647. 10.1007/s00253-004-1850-9 [DOI] [PubMed] [Google Scholar]
- Van Laere K. M., Beldman G., Voragen A. G. (1997). A new arabinofuranohydrolase from Bifidobacterium adolescentis able to remove arabinosyl residues from double-substituted xylose units in arabinoxylan. Appl. Microbiol. Biotechnol. 47 231–235. 10.1007/s002530050918 [DOI] [PubMed] [Google Scholar]
- Ventura M., Canchaya C., Tauch A., Chandra G., Fitzgerald G. F., Chater K. F., et al. (2007). Genomics of actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 71 495–548. 10.1128/MMBR.00005-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M., Turroni F., Zomer A., Foroni E., Giubellini V., Bottacini F., et al. (2009). The Bifidobacterium dentium Bd1 genome sequence reflects its genetic adaptation to the human oral cavity. PLoS Genet. 5:e1000785. 10.1371/journal.pgen.1000785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigsnæs L. K., Holck J., Meyer A. S., Licht T. R. (2011). In vitro fermentation of sugar beet arabino-oligosaccharides by fecal microbiota obtained from patients with ulcerative colitis to selectively stimulate the growth of Bifidobacterium spp. and Lactobacillus spp. Appl. Environ. Microbiol. 77 8336–8344. 10.1128/AEM.05895-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Makino H., Sasamoto M., Kudo Y., Fujimoto J., Demberel S. (2009). Bifidobacterium mongoliense sp. nov., from airag, a traditional fermented mare’s milk product from Mongolia. Int. J. Syst. Evol. Microbiol. 59 1535–1540. 10.1099/ijs.0.006247-0 [DOI] [PubMed] [Google Scholar]
- Wattam A. R., Davis J. J., Assaf R., Boisvert S., Brettin T., Bun C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45 D535–D542. 10.1093/nar/gkw1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Leyn S. A., Gu Y., Jiang W., Rodionov D. A., Yang C. (2012). Ribulokinase and transcriptional regulation of arabinose metabolism in Clostridium acetobutylicum. J. Bacteriol. 194 1055–1064. 10.1128/JB.06241-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.