

Abstract
Adjustment of cell cycle progression is crucial for bacterial survival and adaptation under adverse conditions. However, the understanding of modulation of cell cycle control in response to environmental changes is rather incomplete. In α-proteobacteria, the broadly conserved cell cycle master regulator CtrA underlies multiple levels of control, including coupling of cell cycle and cell differentiation. CtrA levels are known to be tightly controlled through diverse transcriptional and post-translational mechanisms. Here, small RNA (sRNA)-mediated post-transcriptional regulation is uncovered as an additional level of CtrA fine-tuning. Computational predictions as well as transcriptome and proteome studies consistently suggested targeting of ctrA and the putative cold shock chaperone cspA5 mRNAs by the trans-encoded sRNA (trans-sRNA) GspR (formerly SmelC775) in several Sinorhizobium species. GspR strongly accumulated in the stationary growth phase, especially in minimal medium (MM) cultures. Lack of the gspR locus confers a fitness disadvantage in competition with the wild type, while its overproduction hampers cell growth, suggesting that this riboregulator interferes with cell cycle progression. An eGFP-based reporter in vivo assay, involving wild-type and mutant sRNA and mRNA pairs, experimentally confirmed GspR-dependent post-transcriptional down-regulation of ctrA and cspA5 expression, which most likely occurs through base-pairing to the respective mRNA. The energetically favored secondary structure of GspR is predicted to comprise three stem-loop domains, with stem-loop 1 and stem-loop 3 targeting ctrA and cspA5 mRNA, respectively. Moreover, this work reports evidence for post-transcriptional control of ctrA by CspA5. Thus, this regulation and GspR-mediated post-transcriptional repression of ctrA and cspA5 expression constitute a coherent feed-forward loop, which may enhance the negative effect of GspR on CtrA levels. This novel regulatory circuit involving the riboregulator GspR, CtrA, and a cold shock chaperone may contribute to fine-tuning of ctrA expression.
Keywords: non-coding RNAs, cell cycle, post-transcriptional control, RNA binding proteins, CtrA, Sinorhizobium meliloti
Introduction
Bacteria, just like any other type of propagating cells, require robust mechanisms to faithfully replicate their genetic material and partition it to their progeny, even in fluctuating environments. To meet this challenge, bacteria have evolved diverse resilient regulatory mechanisms that guarantee a tight control of cell cycle progression while allowing for integration of environmental cues. In α-proteobacteria, cell cycle architecture shows conserved features, but also a high diversification (Brilli et al., 2010). The essential cell cycle regulator CtrA is broadly conserved in this group of bacteria and has a key role in governing cell cycle progression (Quon et al., 1996; Brilli et al., 2010). The role of CtrA has been best studied in Caulobacter crescentus. CtrA negatively controls initiation of DNA replication, but also transcriptionally regulates genes primarily involved in late cell cycle events, motility, and polar morphogenesis in this asymmetric bacterium, which divides in a stalked and a swarmer cell.
CtrA underlies multiple levels of transcriptional and post-translational regulation, the latter including phosphorylation and proteolysis (Domian et al., 1997; Reisenauer and Shapiro, 2002; Biondi et al., 2006; Iniesta et al., 2006; Heinrich et al., 2016). Transcriptional control of ctrA is based on a regulatory circuit mainly involving DnaA and GcrA (Holtzendorff et al., 2004; McAdams and Shapiro, 2009). DnaA initiates replication and activates a multitude of genes related to nucleotide biogenesis, polar morphogenesis and cell division, including gcrA. GcrA regulates genes related to DNA metabolism and segregation, and activates transcription of ctrA, thus providing a feedback mechanism. After translation, CtrA needs to be activated by phosphorylation, which is mainly controlled by the response regulator DivK. Altogether, these diverse mechanisms build an extensive circuitry that coordinates the cyclic occurrence and activity state of CtrA throughout the cell cycle.
While the core functionalities of this circuitry are understood in much molecular detail, considerably less is known about regulatory mechanisms tuning this cell cycle control to adapt to environmental conditions. Recent studies have unveiled the role of other regulators, such as the second messenger c-di-GMP and the alarmone (p)ppGpp in controlling CtrA levels in C. crescentus (Smith et al., 2014; Sanselicio et al., 2015). In Sinorhizobium meliloti, a plant symbiotic α-proteobacterium, the small non-coding RNA EcpR1 post-transcriptionally modulates expression of dnaA and gcrA under stress conditions (Robledo et al., 2015). This finding added an additional layer to the mechanisms that contribute to interlinking stress responses with the cell cycle machinery in bacteria. Untranslated small RNAs (sRNAs) are widespread post-transcriptional regulators that modulate fundamental processes of bacterial physiology in response to environmental conditions. Bacterial trans-encoded sRNAs (trans-sRNAs) usually modulate mRNA translation and stability by pairing to the 5′-untranslated region (UTR), usually occluding the ribosome binding site (Waters and Storz, 2009). This interaction is often assisted by RNA binding proteins (Gottesman and Storz, 2011) and contributes to fine-tune the intracellular levels of proteins, thereby facilitating bacterial adaptation to changing niches.
Although our knowledge of sRNA functions in S. meliloti is still in its infancy, this non-coding transcriptome is one of the best known among α-proteobacteria in terms of structure, conservation, and functional characterization (Jiménez-Zurdo et al., 2013; Becker et al., 2014; Jiménez-Zurdo and Robledo, 2015). In this study, we took advantage of the comprehensive catalog of trans-sRNAs in this organism (Schlüter et al., 2010, 2013) and screened for sRNAs targeting the ctrA mRNA. Here, we report the trans-sRNA GspR to inhibit cell proliferation and demonstrate that it is able to post-transcriptionally modulate expression of the cell cycle master regulatory gene ctrA, and cspA5, which codes for a cold shock chaperone. We also show that CspA5 positively influences ctrA expression, thereby enabling a feed-forward loop composed of CtrA, CspA5, and the stress-induced trans-sRNA GspR, which may contribute to the regulation of CtrA levels.
Materials and Methods
Prediction of sRNA-mRNA Interactions
Computational predictions of sRNA-mRNA interactions, either using mRNA or sRNA as query sequences, was performed at a genome-wide scale applying IntaRNA and CopraRNA with standard parameters (Wright et al., 2014). The full-length GspR homologous sequences from S. meliloti 1021 (NC_003047, our reference genome), S. meliloti BL225C (NC_015590), Sinorhizobium medicae WSM419 (NC_009636), and Sinorhizobium fredii NGR234 (NC_012587) were used as query for CopraRNA. sRNA secondary structures were predicted with RNAfold (Gruber et al., 2008) and represented with VARNA (Darty et al., 2009).
Bacterial Strains and Cultivation
Supplementary Table S1 lists all bacterial strains and plasmids used in this study. E. coli strains were grown at 37°C in LB medium and rhizobia either in complex tryptone yeast (TY) medium (Beringer, 1974), defined minimal medium (MM; Robertsen et al., 1981), or low phosphate (0.1 mM) MOPS-buffered MM (Zhan et al., 1991) at 30°C with agitation (200 rpm). Solid media were supplemented with antibiotics when required to the following final concentrations (mg/ml): streptomycin (Sm) 600; tetracycline (Tc) 10; gentamycin (Gm) 40; and kanamycin (Km) for E. coli and Agrobacterium 50 and for Sinorhizobium strains 180. The antibiotic concentration was reduced to 50% in liquid cultures. Unless other conditions are indicated, 1 mM IPTG was added to exponential phase cultures (OD600 of 0.4–0.45). Stress conditions were applied to exponentially growing cultures as previously described (Schlüter et al., 2013) and cells were harvested 1 h later. For growth assays, bacteria carrying the corresponding sRNA overproduction construct were grown to the indicated OD600 and 100 μl of IPTG-treated and untreated cultures were transferred to a 96 well microtiter plate to measure OD600 in a VICTOR Multilabel Plate Reader (PerkinElmer). Error bars indicate the standard deviation of at least 10 replicates growing in the same plate. Growth curves were repeated at least two times with similar results. Symbiotic assays with Medicago sativa plants were basically performed as described before (Robledo et al., 2015).
Strain Construction
Primers used for cloning were designed based on the S. meliloti strain Rm1021 genome data (Galibert et al., 2001) and listed in Supplementary Table S2. Transcriptional fusions of promoterless egfp to the putative promoters of gspR and SmelC776 (up to 188 and 229 nt upstream of the predicted TSS, respectively) were constructed by inserting the promoter regions as a SpeI-XbaI fragment in the replicative middle copy plasmid pBBegfp (Robledo et al., 2017). Translational fusion of egfp to predicted target genes were constructed by inserting PCR amplified fragments comprising the 5′ region from the native TSS (Schlüter et al., 2013) to the start codon and up to 33 further codons as XbaI (BglII)-NheI fragments into plasmid pR_EGFP.
Marker-free deletion of the gspR-smelC776 locus by SOEing and construction of the IPTG inducible sinR-sinI based overexpression system of GspR and SmelC776 were basically performed as described before (Robledo et al., 2015). The deletion mutant lacks a 273 nucleotide stretch from 99 nt upstream the gspR TSS to 2 nt upstream the start of SmelC776. For induced overproduction of either SmelC776 or GspR, the encoding DNA regions were cloned into the middle-copy number plasmid pSRKKm under control of the SinR-controlled PsinI promoter in the sinR sinI Rm2011 mutant strain Sm2B2019, which was used to avoid interference with endogenous sinR and sinI alleles involved in quorum sensing. Induction of the PsinI promoter was controlled by IPTG-inducible expression of sinR included in the overexpression plasmid. Since sRNA genes were directly fused to the TSS of PsinI, sRNA transcription started with activation of PsinI (Becker et al., 2014; Robledo et al., 2015). In all sRNA overexpression assays, IPTG-driven transcription of the unrelated SmelC812 sRNA gene from plasmid pSKControl+ was used as negative control (Robledo et al., 2015). SmelC812 is encoded antisense to the 5′ UTR of a transposase gene and, to our knowledge, overproduction of this sRNA has no negative side effects (Schlüter et al., 2010). For cspA5 complementation, the full-length gene was placed under control of the IPTG inducible Plac promoter in plasmid pSRKGm.
RNA Isolation and Northern Blot Hybridization
Total RNA including the sRNA fraction (50-250 nt) was isolated from bacterial cultures using the miRNeasy mini Kit (Qiagen). Initially, cell pellets were resuspended in 700 μl QIAzol Lysis Reagent (Qiagen) and then transferred to grinding tubes (soilGen). Cell destruction occurred via mechanical grinding using the X-Ribolyzer system (MP Biomedicals). After centrifugation, the supernatant was used for further treatment according to the manufacturer’s instructions (miRNeasy mini Kit, Qiagen). DNaseI digestion was applied according to the user manual instructions (Fermentas). RNA quality and concentration was measured using the NanoDrop2000 (Peqlab).
For northern blot, RNAs samples were separated on 6% polyacrylamide/7 M urea gels and transferred to nylon membranes by semi-dry electroblotting. 5′-end radiolabeled oligonucleotide probes (Supplementary Table S2) were used for hybridization as described (Robledo et al., 2018). To estimate RNA size, an RNA molecular weight marker (NEB) was included.
Microscopy
Bacteria were examined using a Nikon Eclipse Ti-E by differential interference contrast and epifluorescence basically as described before (Frage et al., 2016).
cDNA Synthesis and Microarray Hybridization
cDNA synthesis, microarray processing, sample hybridization, and image acquisition were performed as described previously applying the Sm14kOLI microarray that carries 50 –70 mer oligonucleotide probes directed against coding regions and both strands of the intergenic regions (IGR) (Sinorhizobium meliloti Rm1021 Sm14kOLI; Bahlawane et al., 2008). Probes in the IGR were separated by approximately 50–100 nt. The analysis of microarray images was performed with ImaGene 6.0 software (BioDiscoveries). Lowess normalization and significance test (p-value adjustment based on fdr) were performed with the EMMA software (Dondrup et al., 2009). The M-value represents the logarithmic ratio between both channels. The A-value represents the binary logarithm of the product of the intensities of both channels. Transcriptome data are available at ArrayExpress (Accession No. E-MTAB-3775).
Proteome Sample Preparation
Bacteria were grown in MOPS medium with 21 mM of NH4Cl as nitrogen source instead of sodium glutamate. Notably, for all cultivations of GspR+ cells the 15N isotope of NH4Cl (Cambridge Isotope Laboratories) was used. Equal amounts of Control+ and GspR+ cells were pooled, harvested, and finally pelletized. Pellets were resuspended in 5–10 ml lysis buffer (10 mM MgCl2, 1 mM CaCl2, 50 μg/ml DNaseI, 50 μg/ml RNAseA, 20 mM Tris–HCl, pH = 8). Mechanical cell disruption was applied via three passages through a French press followed by centrifugation at 2,000 g for 2 min at 4°C. Ultracentrifugation of the supernatant was performed at 160,000 g for 1 h at 4°C to separate membrane (pellet) and cytoplasmic (supernatant) fractions. Pellets were resuspended in ∼5 ml lysis buffer. The resolved membrane and the cytoplasmic fraction were lyophilized. Dried protein samples (1 mg each) were resolved in 40–80 μl Tris loading buffer with 50 mM DTT, incubated at 70 °C for 10 min and treated with 120 mM Iodoacetamide 20 min in the dark at room temperature. Treated samples were separated in a SDS gel and each lane (200 μg protein) was sliced in ∼24 pieces (Sobrero et al., 2012). Gel-slices were dried in a SpeedVac (∼15 min/45°C). Trypsin treatment (0.1 μg trypsin per gel-slice) was applied overnight at 37 °C. After treatment, 200 μl MeCN was added to the sample for 30 min in an ultra-sonic bath. The supernatant was concentrated to dryness (Speedvac, 45°C) and finally dissolved in 25 μl 10% MeCN/0.1% TFA.
Mass Spectrometry
Mass spectrometric analysis of the samples was performed using an Orbitrap Velos Pro mass spectrometer (ThermoScientific). An Ultimate nanoRSLC-HPLC system, equipped with a C18 nano RP column (particle size 1.8 μm) was connected online to the mass spectrometer through a Proxeon nanospray source. Depending on the concentration of the samples, 1–15 μl of the tryptic digest was injected onto a 2 cm × 300 μm PepMap C18 pre-concentration column. Automated trapping and desalting of the sample was performed at a flow rate of 6 μl/min using water/0.05% formic acid as solvent for 5 min. Separation of the tryptic peptides was achieved with the following gradient of water/0.045% formic acid (solvent A) and 80 % acetonitrile/0.05 % formic acid (solvent B) at a flow rate of 300 nl/min: holding 4 % B for 5 min, followed by a linear gradient to 45% B within 30 min and linear increase to 95% solvent B in an additional 5 min. The column was connected to a stainless steel nanoemitter and the eluent sprayed directly towards the heated capillary of the mass spectrometer using a potential of 2,300 V. A survey scan with a resolution of 60,000 within the Orbitrap mass analyzer was combined with at least 10 data-dependent MS/MS scans with dynamic exclusion for 30 s either using CID with the linear ion-trap or using HCD and orbitrap detection at a resolution of 7,500.
Protein Identification and Quantification
Data sets were imported into the internet application QuPE (Albaum et al., 2009, 2011). A MascotTM (Perkins et al., 1999) search was conducted using a database that contained the complete proteome information of S. meliloti Rm1021 as well as an equally sized set of randomized amino acid sequences allowing for the later calculation of false discovery rates as suggested before (Reidegeld et al., 2008). Peptide tolerance was set to 10.0 ppm, MS/MS tolerance to 1,000.0 mmu, and two missed cleavage sites were allowed. Oxidation of methionine was allowed as a variable modification, and furthermore, a modification of arginine and lysine was introduced to account for a possible selected non-monoisotopic peak of a 15N-labeled precursor with a weight of approximately 1 Da (Zhang et al., 2009). Only hits having a score above Mascot’s own significance threshold (P < 0.05) were kept. In addition, false discovery rates were calculated in QuPE and required to be below P < 0.05. In total, 206,840 peptides were identified corresponding to 1,674 proteins. Proteome data were quantified using QuPE’s built-in algorithm using an 15N incorporation level of 98%. Rather strict parameters were employed (r > 0.4, isotopic distribution similarity >0.9) and results were filtered for a signal-to-noise value of at least 3.0. In total, 46,900 peptides were quantified accounting for 1,508 proteins. After quantification, all proteins were kept in the final result set which were represented either by at least two peptides with different amino acid sequences/charges or by peptides with the same amino acid composition but from two different samples. In summary, 512 proteins passed this filter criterion.
Fluorescence Assays
Bacterial cells carrying the promoter test plasmids with transcriptional gspR promoter-egfp or SmelC776 promoter-egfp fusions were grown and measured as described before (Robledo et al., 2015, 2018). Reporter plasmids carrying translational fusions of the 5′ UTR and the first codons of target genes to egfp were transferred by conjugation to Sm2B2019DD harboring plasmids pSKControl+ or pSKGspR+. Three double transconjugants for each combination of inducible sRNA overproduction construct and target-egfp fusion were grown to exponential phase (OD600 of 0.5–0.6) and half volume of each culture were treated with 0.5 mM IPTG for 6 h. For estimation of the relative fitness, Rm2011 and 2011ecpR1 were labeled with mCherry or eGFP by single integration of either plasmid pKOSm or pKOSe (Robledo et al., 2015). Starter cultures were individually grown in MM overnight, diluted to OD600 of 0.005 and mixed at a 1:1 ratio in 5 ml of MM. Every 3 days, the eGFP and mCherry fluorescence of the cultures was measured and the mixed population was diluted 1,000-fold in fresh media. Treated and control cultures (100 μl) were transferred to a 96-well microtiter plate to measure OD600, mCherry and/or eGFP-mediated fluorescence in the Infinite M200 Pro microplate reader (Tecan).
Results
sRNA GspR Is Predicted to Interact With the 5′ UTR of ctrA mRNA
In this study, we have taken a computational approach to identify sRNA candidates for post-transcriptional regulation of ctrA in S. meliloti. A stretch of the ctrA mRNA, encompassing the 5′ region from the most distal transcription start site (TSS) to the 50th codon (nucleotide positions -291 to +150 relative to the start codon), was used as query sequence to predict sRNAs as putative interaction partners. Hypothesizing that sRNAs controlling cell cycle-related target genes are phylogenetically conserved, we screened chromosomally encoded trans-sRNAs which have been previously found to be conserved in the order Rhizobiales (Reinkensmeier et al., 2011). This computational screen predicted the S. meliloti trans-sRNAs SmelC775 and SmelC291 (EcpR1) as potential regulators of ctrA (Figures 1A,B; Robledo et al., 2015). The putative interaction of SmelC291 (EcpR1) with the ctrA mRNA was previously investigated, although not experimentally confirmed (Robledo et al., 2015). Here, we focused on SmelC775.
FIGURE 1.
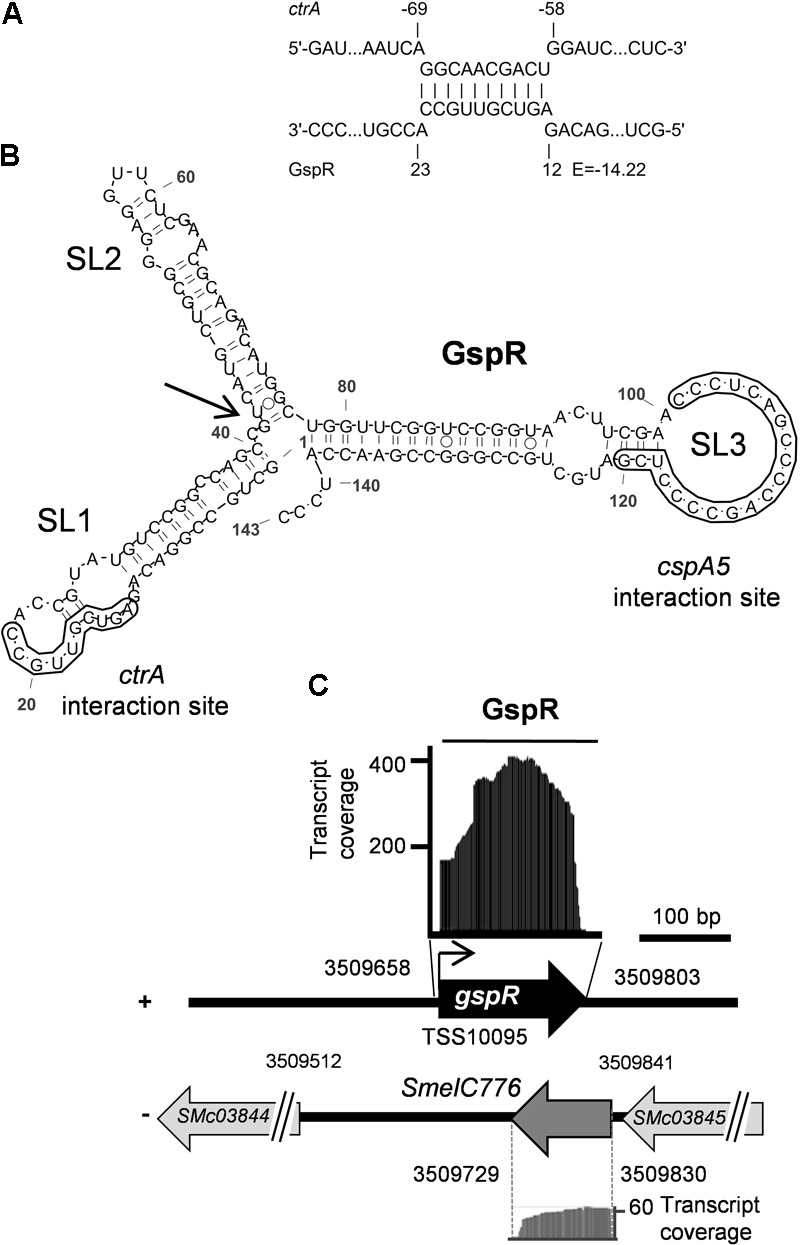
ctrA 5′ UTR is predicted to bind to GspR (SmelC775). (A) Predicted thermodynamically favored antisense interactions between GspR and ctrA mRNA. Numbers denote nucleotide positions relative to the TSS of gspR and the start codon of the putative target mRNAs. The predicted energy score (E) is indicated in kcal/mol. (B) Predicted secondary structure of the full-length GspR transcript with a minimum free energy of -66.30 kcal/mol exhibiting three independent stem loop (SL1 to SL3) structures. Nucleotide positions relative to the 5′-end are indicated. The SL1 and SL3 regions predicted to interact with the ctrA and the cspA5 mRNAs, respectively, are boxed. (C) gspR genomic locus. Genes predicted on the complementary strand (-), including the SmelC776 locus are shown. Genome coordinates are indicated. RNAseq coverage profiles of both GspR and SmelC776 in S. meliloti Rm1021 are depicted. Black and gray areas represent coverage of GspR (SmelC775) and SmelC776, respectively, from samples enriched in processed transcripts (Schlüter et al., 2010). The angled arrow marks the GspR 5′-end (TSS10095) and the horizontal bar indicates the full-length 144 nt GspR sequence used for structure prediction.
SmelC775 was first identified by RNAseq in S. meliloti type strain Rm1021 as a 143-nt trans- sRNA (Figures 1B,C) and validated by northern blot (Schlüter et al., 2010). In this study, we renamed this sRNA GspR (Growth Stop Phenotype RNA) because of the phenotype caused by SmelC775 overproduction (see below). GspR is encoded approximately 145 kb distant from the chromosomal origin of replication in the intergenic region flanked by SMc03844 and SMc03845 (Figure 1C), both encoding conserved hypothetical proteins. RNAseq data identified the 100-nt transcript SmelC776 antisense to gspR. Both RNAs overlap by 74 nucleotides (Schlüter et al., 2010; Figure 1C). The predicted energetically most stable secondary structure of GspR consists of three stem-loop domains (SL1 to SL3, Figure 1B). The sRNA sequence predicted to base-pair with the ctrA mRNA comprises a continuous stretch of 10 nucleotides (E = -14.2 kcal/mol) mapping to GspR SL1 (nucleotide positions 13 to 22 of GspR) and to nucleotide positions -59 to -68 relative to the start codon of the ctrA mRNA (Figure 1A).
The GspR coding region was found to be conserved in various Sinorhizobium/Ensifer species including S. meliloti, S. medicae, S. fredii, E. sojae, E. adherens, and S. americanum. While SL2 shows slight sequence differences, SL1 and SL3 motifs are identical in all GspR homologs (Reinkensmeier et al., 2011), suggesting that these sequences may be involved in conserved interactions with target mRNAs. Therefore, the full-length sequences of GspR homologs from different Sinorhizobium species were also scanned for putative interactions with mRNAs as described in section “Materials and Methods.” This screen returned the ctrA mRNA as conserved GspR target. Identical GspR-ctrA mRNA interaction sites (Figure 1A) were predicted in the different Sinorhizobium species. This suggested functional conservation, which motivated us to further investigate this sRNA.
GspR Accumulated in the Stationary Phase of Bacterial Growth
Previous northern blot hybridizations, detecting GspR with a double-stranded DNA probe, showed gspR expression in all the conditions tested (i.e., exponential growth phase in TY and GMX media, and cold, heat, and salt stresses) with just small variations in expression levels in the S. meliloti 2011 wild type (Schlüter et al., 2010). Since sRNA SmelC776, transcribed antisense to gspR (Figure 1C), may interfere with GspR hybridization, we used strand-specific antisense oligonucleotides to detect each sRNA independently in S. meliloti Rm2011. This wild-type strain is a close relative of the type strain S. meliloti Rm1021. SmelC776 hybridization signals were not clearly detected in any of the conditions tested (Supplementary Figure S1). However, northern hybridizations identified a dominant ∼143-nt GspR transcript and a shorter variant of ∼100 nucleotides, together with less abundant shorter GspR-derived RNA species under several growth conditions (Figure 2A). In light of previous RNAseq data (Figure 1C; Schlüter et al., 2010), the ∼100-nt processed form may constitute a GspR variant lacking SL1.
FIGURE 2.
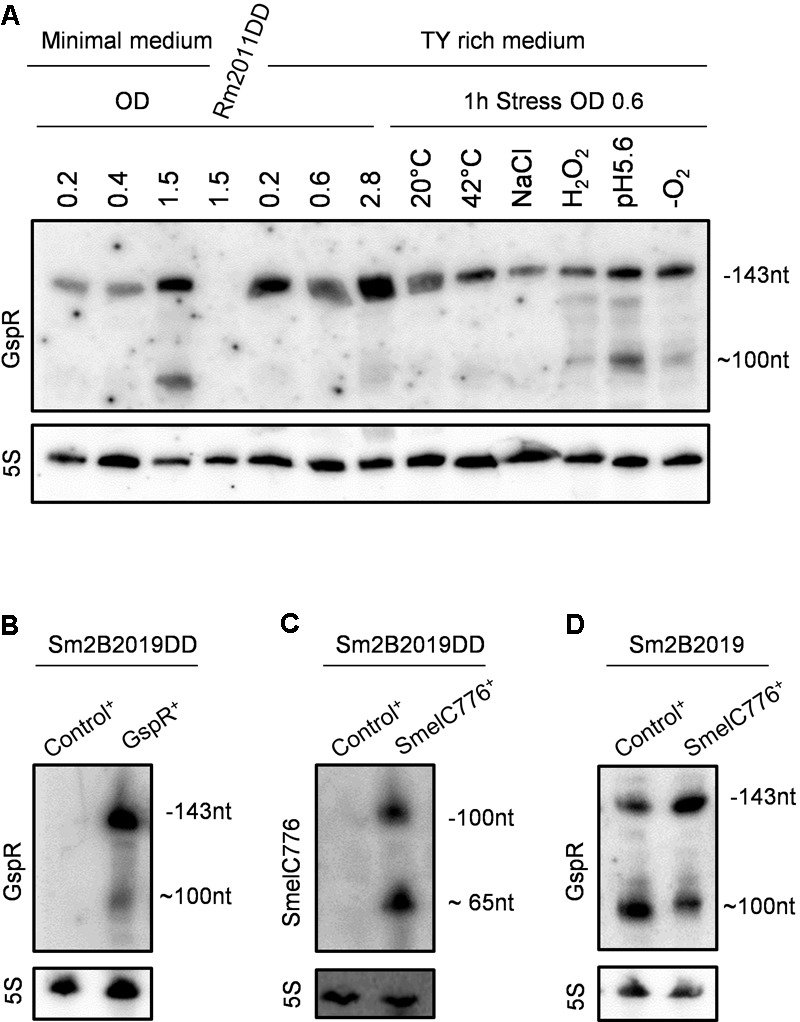
Accumulation of GspR is independent of SmelC776 expression. (A) Expression profiling of GspR. Northern blot detection of GspR abundance in the S. meliloti Rm2011 wild type under different growth and stress conditions in minimal and TY-rich media in exponential and stationary growth phase and in TY-rich medium at 20°C, cold stress; 42°C, heat stress; NaCl, 0.4 mM sodium chloride (osmotic stress); H2O2, 10 mM hydrogen peroxide (oxidative stress); pH 5.6, 20 mM MES (acidic stress); -O2, microoxic conditions. Exposure times were optimized for each panel. 5S rRNA probing was used as RNA loading control. (B–D) Northern blot detection of GspR and SmelC776 RNA variants in Sm2B2019DD or Sm2B2019 strains carrying either pSKControl+ (Control+), pSKSmelC776+ (SmelC776+), or pSKGspR+ (GspR+), 4 h after induction with IPTG.
GspR-derived RNA species were detected in exponentially growing S. meliloti Rm2011 and accumulated during the stationary phase of growth, especially in MM cultures. However, the amount of GspR did not increase significantly in different stress conditions, i.e., cold, heat, salt, oxidative, acidic, and microoxic stresses, compared with the exponential growth condition in TY-rich medium (Figure 2A).
Previous analysis of dRNAseq data identified a primary 5′-end of GspR (TSS_10095, Figure 1C) but not of SmelC776 (Schlüter et al., 2013). The putative promoter regions of gspR and SmelC776, comprising nucleotide positions -159 to +29 and -221 to +8, respectively, relative to the corresponding TSS were fused to egfp. When cultured in TY or minimal media, Rm2011 carrying the SmelC776 upstream region-reporter fusion did not exceed background fluorescence derived from the control plasmid lacking a promoter upstream of egfp. These results suggest that the fused region has no promoter activity under the conditions tested and that SmelC776 is probably not a primary transcript. In agreement with the failure to detect a primary 5′ end by dRNAseq (Schlüter et al., 2010), this sRNA may rather be generated by post-transcriptional processing from the 3′ UTR of the upstream SMc03845 mRNA.
When grown in TY rich or minimal media, wild-type strain Rm2011 carrying the PgspR-egfp promoter reporter fusion showed activities in exponentially growing bacteria (OD600 of 0.4 or 0.6). These activities slightly increased in the early stationary growth phase, more visible in minimal than in rich medium, matching the trend towards higher levels of GspR in the stationary growth phase, as revealed by northern hybridizations (Supplementary Figure S2A). The GspR 5′-end was not preceded by known S. meliloti promoter signatures. However, the gspR upstream region is 100% conserved in S. meliloti and S. medicae and 84% in S. fredii NGR234 strain (Supplementary Figure S2B).
GspR Expression and Processing Are Likely Independent of SmelC776
The gspR and SmelC776 coding regions are inversely oriented and overlap in their 3′ regions by 74 nucleotides (Figure 1C), suggesting mutual interference between the GspR and SmelC776 sRNAs. This hypothesis was tested by studying the effect of increased levels of one of these sRNAs on the other. To this end, the abundance of either of these sRNAs was increased by plasmid-based IPTG-induced overproduction, either in Sm2B2019 with the wild type gspR/SmelC776 chromosomal locus, or in the gspR/SmelC776 markerless deletion mutant Sm2B2019DD. This mutant, lacking the putative gspR promoter and the coding regions of both full-length sRNAs, was used as genetic background to avoid interference with the chromosomal locus. The SmB2019 background (ΔsinRI) was required to exclude interference with the plasmid-based inducible overexpression system (see section “Materials and Methods”). The control sRNA gene SmelC812 similarly cloned in plasmid pSKControl+ was used as control in overexpression assays (Robledo et al., 2015).
Accordingly, gspR was not detected by northern hybridization of RNA from stationary phase cultures of Rm2011DD and Sm2B2019DD in MM (Figures 2A,B). Northern hybridizations detected two signals corresponding to 100-nt and ∼65-nt SmelC776 species upon IPTG-induced ectopic overexpression of SmelC776 in Sm2B2019DD (Figure 2C). This processing pattern is unlikely to be driven by a possible antisense interaction with GspR, since it occurred independently of the presence of the chromosomal gspR/SmelC776 locus (Figure 2C). The 100-nt processed form of GspR was also detected regardless the presence of the chromosomal locus (Figure 2B), further suggesting that mechanisms of GspR and SmelC776 biogenesis are unrelated. Hybridization with two pairs of riboprobes, each targeting a region within the overlapping stretch of GspR and SmelC776, and another specific to one of these transcripts, rendered the same pattern (data not shown). Finally, the overproduction of SmelC776 in Sm2B2019 cells did not alter processing or stability of GspR (Figure 2D). Taken all together, the biogenesis of these two sRNAs is probably mutually independent.
The gspR Locus Confers a Fitness Advantage in S. meliloti
To study the biological function of GspR, motility, growth, morphology, and symbiotic phenotypes were monitored in S. meliloti strains lacking gspR. Both the gspR/SmelC776 deletion mutant Rm2011DD and the gspR/SmelC776/ecpR1 triple mutant (i.e., containing an additional deletion in the locus coding for the cell-cycle related EcpR1 sRNA; Robledo et al., 2015) were not impaired in motility and they exhibited wild type-like growth in TY rich, MOPS (both defined or nutrient-limited) or MM. Cell viability (CFU/ml) after growth in MM until late stationary phase was also unaffected with respect to the wild type. Furthermore, the markerless gspR/SmelC776 deletion mutant was proficient in symbiosis with its host plant Medicago sativa, indistinguishable from the symbiotic phenotypes of the parental strain. Transposon insertions in SMc03844 or SMc03845 flanking the gspR/SmelC776 locus also showed wild type-like growth (data not shown).
In contrast, deletion of gspR/SmelC776 attenuates competitiveness as determined by a fitness growth assay against the Rm2011 wild type. For this experiment, strains were chromosomally tagged to constitutively express egpf or mcherry as previously done (Robledo et al., 2015). 2011 egfp cells were mixed in a ratio of 1:1 with either 2011 mCherry or 2011DD mCherry cultures in MM and grown until the late stationary phase, where the highest levels of GspR expression were detected (Figure 2A). Every 3 days of incubation, the eGFP:mCherry fluorescence ratio of the stationary cultures was measured (Supplementary Figure S3) and the mixed population was diluted 1,000-fold in fresh MM. After 3 days of cultivation, the ratios of fluorescent signals representing the proportions of the mixed strains were similar in all cultures. The wild-type control culture further maintained a similar ratio even after three consecutive sub-cultivations. However, the proportion of the 2011 egfp strain progressively increased over the 2011DD mCherry mutant, showing a 52% increment of the fluorescence ratio after the third sub-cultivation. These results indicate a lower fitness of the mutant in competition with the wild type.
GspR Overproduction Hampers Cell Growth in Sinorhizobium
To obtain further clues to the cellular role of the GspR sRNA, the GspR-overproducing S. meliloti strain was phenotypically characterized. IPTG-induced overexpression of plasmid-borne gspR in strain Sm2B2019DD, confirmed by northern hybridization, negatively affected growth (Figures 3A,B). While Sm2B2019DD carrying the gspR overexpression plasmid pSKGspR+ showed normal growth in absence of the inducer, growth was increasingly hampered in the presence of rising concentrations of the inducer and completely inhibited at 0.5 mM IPTG (Figure 3B). The delay in growth started approximately 4 h after induction, which corresponds to two S. meliloti cell cycles. Growth differences between induced and uninduced cultures were more pronounced when IPTG was added at lower cell densities and were less dramatic when induction was started at higher initial cell densities (Supplementary Figures S4A,B). Overexpression of other S. meliloti sRNAs (e.g., SmelC776, SmelCR01029, SmelCR01763, SmelB008 and SmelC045) using the same strategy did not affect growth (data not shown).
FIGURE 3.
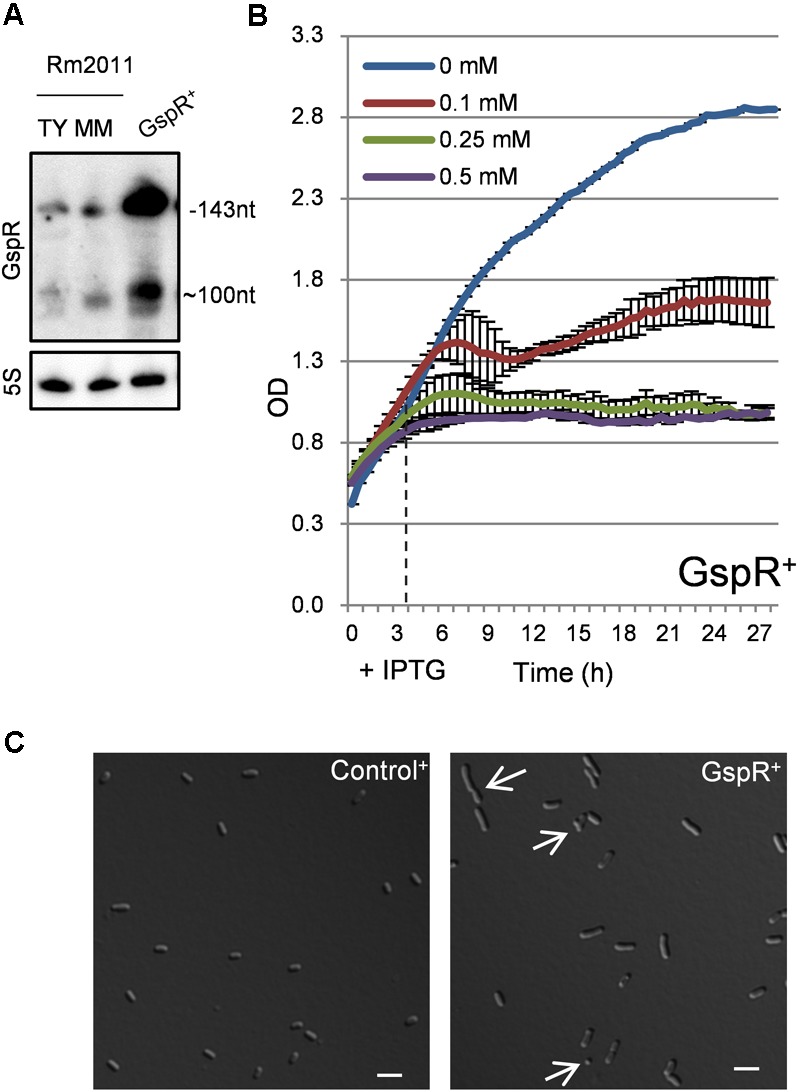
Growth stop phenotype associated to gspR overexpression. (A) Northern blot detection of GspR RNA variants in the S. meliloti Rm2011 wild type strain at the stationary growth phase in rich TY or minimal media (left) or in the Sm2B2019DD strain carrying pSKGspR+ (GspR+) 4 h upon induction with 0.5 mM IPTG. 5S hybridization signals are shown as loading controls. (B) Growth kinetics after addition of different IPTG concentrations to OD600 0.4 TY-rich medium cultures of S. meliloti Sm2B2019DD cells carrying pSKGspR+ (0 h). Horizontal dashed line shows the start point of significant growth differences following addition of 0.5 mM IPTG (4 h). (C) Morphology of S. meliloti Sm2B2019DD strains overproducing GspR or the SmelC812 control antisense RNA 30 h post-induction with 0.5 mM IPTG. Arrows mark cells with abnormal morphology in GspR+ cultures (see text for details). Bars correspond to 2 μm.
24 h post-induction with 0.5 mM IPTG, control cells showed the typical morphology of stationary phase cells, 1 to 2 μm in length, while GspR overproducing cells were heterogeneous in morphology (Figure 3C); 12.7% of the gspR overexpressing cells were abnormally long (>2 μm), 6.0% showed a round morphology (length <1 μm) and an additional 1.0% showed a branched morphology (Figure 3C, white arrows; sampling of 1,000 cells). In contrast, less than 0.5% of the control population showed abnormal cell morphologies. Time-lapse microscopy revealed that 50% of the cells inhibited in growth by gspR overexpression (induced by 0.5 mM IPTG) proceeded to cell division and resumed growth when transferred to fresh medium lacking the inductor. In comparison, 96% of equally treated Control+ cells resumed growth. Overproduction of S. meliloti GspR also led to cell growth arrest in S. fredii and S. medicae, both encoding GspR homologs, but not in Agrobacterium tumefaciens, which lacks a GspR homolog (Supplementary Figure S4C).
GspR sRNA has been reported to be Hfq-independent (Torres-Quesada et al., 2014). Growth arrest upon induced GspR overexpression was independent of the RNA chaperone Hfq and the C-terminal domain of the ribonuclease RNase E, i.e., gspR blocked growth in both the hfq deletion mutant Rm2011hfq and Rm2011rne675 encoding an RNase E variant lacking the C-terminal domain (Baumgardt et al., 2014; Supplementary Figures S4D,E).
GspR Regulates cspA5 and ctrA mRNAs Through Distinct Stem-loops
Bacterial sRNAs typically target multiple mRNAs. To identify other putative GspR target genes, transcriptome and proteome profiling were performed 30, 60, and 90 min post-induction of GspR overproduction (Supplementary Figure S5, and Supplementary Data Sheet S1). The microarray experiments distinguished 56 (30 upregulated/26 downregulated) differentially expressed protein coding genes in the GspR+ strain in comparison to the control strain, together with eleven 5′-UTRs, two mRNA fragments, and several sRNAs. GspR-dependent changes in protein synthesis were observed for 19 (8/11) candidates. Together, transcriptome and proteome analysis identified 68 (34 up, 34 down) protein coding genes showing altered gene expression profiles in the pSKGspR+ strain (M value: ≥1, ≤-1). Global functional predictions showed that the majority of identified genes (∼34%, 23) are related to cellular metabolism of S. meliloti with a clear dominance in transport/metabolism of amino acids (7) and inorganic ions (8).
The following putative target genes were consistently identified by both computational predictions (CopraRNA prediction in Sinorhizobium with P < 0.05) and expression profiling experiments (significant decrease (M ≤ -1) in transcript or protein abundance, at least at one of the tested time points following gspR overexpression (Supplementary Data Sheet S1): ctrA [CopraRNA prediction ctrA 5′-UTR: P < 0.042; transcriptome profiling ctrA: M = -0.41 (60 min) and -0.50 (90 min); proteome profiling: M = -0.56 (30 min) and -1.14 (60 min)], cspA5 [CopraRNA prediction cspA5 5′-UTR: P < 0.00024; transcriptome profiling cspA5 5′-UTR: M = -0.65 (30 min), -1.36 (60 min), and -0.67 (90 min)], and SMc02819 [CopraRNA prediction SMc02819 : P < 0.00014; proteome profiling SMc02819: M = -1.50 (30 min)].
GspR-mediated translational control of these candidate target mRNAs was tested by a two-plasmid assay (Torres-Quesada et al., 2013; Robledo et al., 2015). To this end, the 5′-UTR of the putative target gene (Schlüter et al., 2013), containing the predicted GspR interaction sequence, and a short stretch of the corresponding coding region, was translationally fused to egfp under the control of the constitutive synthetic PSyn promoter (Giacomini et al., 1994) to generate reporter constructs in plasmid pR_EGFP. A gcrA reporter construct (Robledo et al., 2015), not regulated by GspR, was used as negative control (Supplementary Figure S6). These reporter plasmids were mobilized to Sm2B2019DD GspR+ and S. meliloti Sm2B2019DD Control+, carrying plasmid-borne inducible gspR and SmelC812 (control sRNA gene; Robledo et al., 2015) expression constructs, respectively. Post-transcriptional effects were assessed by determining fluorescence of the reporter fusion protein upon induced sRNA expression. All assays were performed under conditions ensuring comparable growth of the reporter strains (see section “Materials and Methods”).
SMc02819
Its protein product is homologous to endoribonuclease T2. A 15-nt stretch overlapping the coding sequence (positions +36 to +51) was predicted to interact with GspR SL3 (E = -18.09 kcal/mol). We were unable to test SMc02819 for GspR-mediated regulation because the reporter construct pSMc02819-136+99-egfp displayed hardly detectable fluorescence.
cspA5
Homologs of cspA5 encode cold shock proteins, which are generally, but not exclusively, induced upon a temperature downshift affecting mRNA structure stability and RNase recognition to counteract the physiological effects of temperature changes (Barria et al., 2013). Discontinuous base-pairing of GspR SL3 with a 27-nt stretch overlapping the Shine-Dalgarno and the start codon sequences of the cspA5 mRNA (nt position -16 to +13 relative to the AUG) was predicted (E = -17.75 kcal/mol; Figures 4A,B). Four different TSSs have been assigned to the cspA5 mRNA (Schlüter et al., 2013). To design a reporter construct, we selected the second TSS, which is located 53 nt upstream of the AUG and preceded by a RpoD promoter signature (Schlüter et al., 2013). The reporter construct comprised the cspA5 5′-UTR starting with TSS2 and the first 48 nt of the coding region fused to egfp (plasmid pcspA5-53+45-egfp; Figure 4B). This construct was used to assess the regulatory effect of GspR and a series of mutant variants on cspA5 by the double-plasmid assay. Induced overexpression of gspR reduced pcspA5-53+45-egfp mediated fluorescence by 36% compared to the control. Overproduction of GspR mutant variants carrying changes of 2 or 4 nt in SL1 similarly repressed activity of this cspA5 reporter construct, while GspR-3.4, carrying mutations in SL3, did not influence reporter activity (Figure 4C). This further supports that the GspR-cspA5 mRNA interaction region resides in SL3 of the sRNA. Accordingly, changes of 2 nt in the predicted target region within the cspA5 5′-UTR of the reporter construct (pcspA5-BS-egfp) abolished the negative effect caused by gspR overexpression (Figure 4D). Therefore, GspR SL3 seems to canonically regulate cspA5 by antisense pairing with a nucleotide stretch located closely upstream of the start codon in the mRNA.
FIGURE 4.
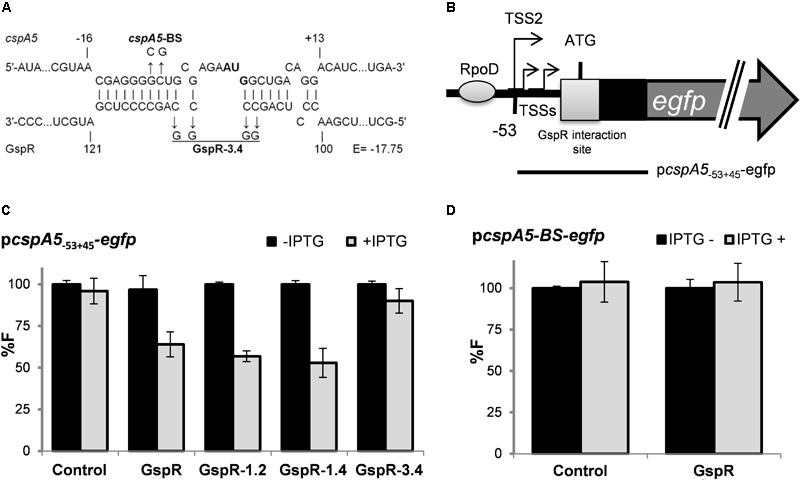
GspR SL3 represses cspA5 post-transcriptionally. (A) Predicted duplexes between GspR and cspA5 mRNA. Positions are denoted relative to the AUG; A is +1. The predicted energy score (E) is indicated in kcal/mol. The nucleotide exchanges in the predicted interaction regions of cspA5 (cspA5-BS) and GspR (GspR 3.2 and GspR 3.4) mRNAs are indicated in bold. (B) Schematic representations of the genomic regions and the fragment (indicated by bars) translationally fused to egfp. The potential cspA RpoD-dependent promoter is indicated by a gray circle and GspR-interaction site by a box. (C,D) Means of relative fluorescence intensity values of Sm2B2019DD co-transformed with overexpression plasmids carrying control SmelC812, gspR, or its mutant variants; and PcspA5-53+45-egfp C) or PcspA5-BS-egfp. (D) Reporter plasmids carrying either the native 5′UTR encoding sequences derived from cspA5 or a variant with 2 mutations in the predicted GspR binding sites (BS). The standard deviation represents at least three independent determinations of three double transconjugants grown in three independent cultures. Specific activities were normalized to the levels of the strain carrying the vector with the control RNA gene without IPTG added to yield percent relative fluorescence (% F).
ctrA
For the ctrA mRNA, continuous base pairing involving a 10-nt stretch of the target and GspR SL1 was predicted (Figures 5A,B). Multiple TSSs have been reported for ctrA. Two of these, TSS2 (Figure 5B) and TSS6 (position -291) are preceded by consensus promoter motifs and associated with strongly accumulating transcripts (Schlüter et al., 2013). The putative interaction sequence of ctrA mRNA with GspR SL1 starts 1 nt after TSS2 (Figures 5A,B). The post-transcriptional effect of GspR on ctrA expression was analyzed by different reporter constructs (Figure 5B). Compared to the control, gspR overexpression resulted in 44% and 47% decreased activity of the reporter constructs ctrA-69+93 (TSS2) and ctrA-112+3 (TSS4), respectively, both including the putative interaction site with GspR (Figures 5C,D). However, activity of reporter construct ctrA-56+93, which does not harbor the predicted interaction sequence, did not significantly change upon induced gspR overexpression, further supporting the prediction (Figure 5E). Moreover, 1, 2, and 4 nt changes in GspR SL1 (gspR-1.1, gspR-1.2, and gspR-1.4; Figure 5A) progressively mitigated the GspR-mediated repression of reporter expression in the same strain background and culture conditions previously used in the assays with wild-type GspR (Figure 5D). In contrast, mutations in GspR SL3 did not affect GspR-mediated repression of ctrA (Figure 5D). Concomitantly, introduction of 2 or 4 nt changes into the predicted binding site within the ctrA mRNA in the reporter constructs, leading to ctrA-BS.2 and ctrA-BS.4 (Figure 5A), also abolished the negative regulatory effect of GspR on reporter activity (Figures 5F,G). Combined compensatory mutations of ctrA-BS.2 or ctrA-BS.4 and GspR1.2 or GspR1.4, respectively, partially restored the regulation by GspR (Figures 5F,G). Altogether, these data validate ctrA mRNA as target of GspR and strongly suggest that this regulation is mediated by base pairing with complementary nucleotides within the SL1 single stranded region of GspR.
FIGURE 5.
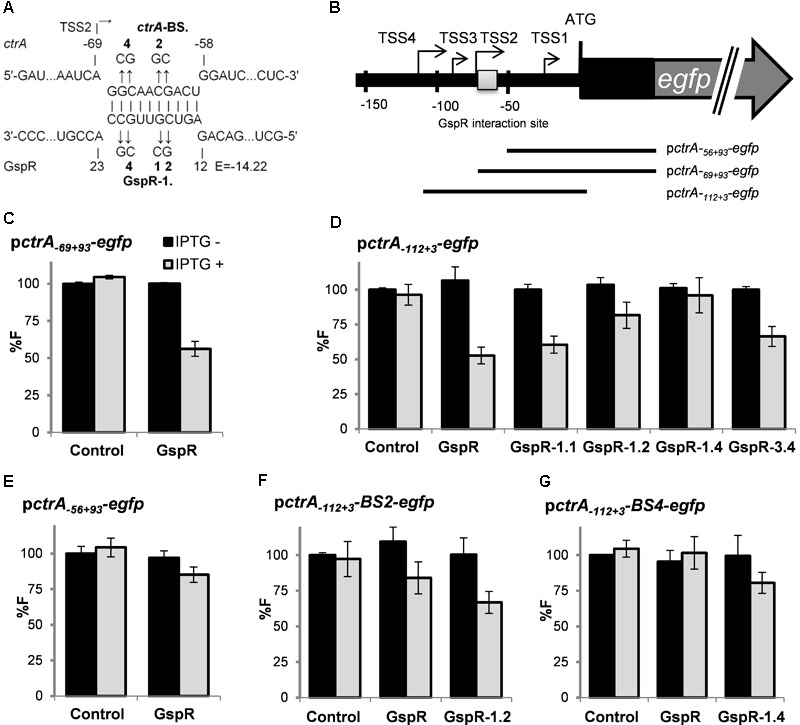
GspR SL1 represses ctrA post-transcriptionally. (A) Predicted duplexes between GspR and ctrA mRNA. The nucleotide exchanges in the predicted interaction regions of ctrA (ctrA-BS.2 and BS.4) and GspR (GspR 1.1, 1.2, 1.4, and 3.4) mRNAs are indicated in bold. (B) Schematic representations of the genomic regions and the fragments translationally fused to egfp. (C–G) Means of relative fluorescence intensity values of Sm2B2019DD co-transformed with overexpression plasmids carrying control SmelC812, gspR, or its mutant variants and the indicated ctrA translational fusion. Fluorescence measurements have been performed as described in Figure 4.
We also tested the GspR mutant variants GspR-1.4 and GspR-3.4 for their potential to repress growth of S. meliloti cultures. While overproduction of wild-type GspR or GspR-1.4 negatively affected growth, overproduction of GspR-3.4 or the control RNA SmelC812 did not significantly slow down growth (Supplementary Figure S4F). This indicates that the growth phenotype caused by elevated levels of GspR is associated with SL3, and thus involves regulation of other target mRNAs.
CspA5 Enhances ctrA mRNA Translation Efficiency
CspA5 homologs act as nucleic acid chaperones, preventing the formation of mRNA secondary structures, thereby facilitating mRNA transcription and/or translation (Barria et al., 2013; Keto-Timonen et al., 2016). The ctrA mRNA transcribed from TSS4 is predicted to fold into a secondary structure formed by two stem loops (SLA and SLB, Figure 6A). We therefore wondered whether CspA5 post-transcriptionally influences ctrA expression.
FIGURE 6.
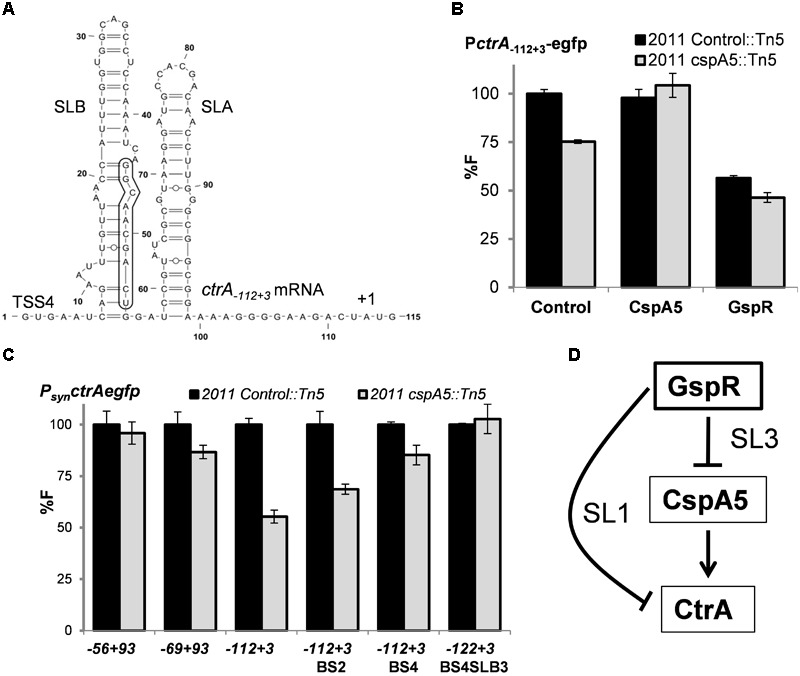
CspA5 facilitates ctrA translation. (A) RNAfold predicted secondary structure of ctrA-112+3 mRNA transcripts exhibiting two stem loop (SL) structures. Nucleotide positions relative to the 5′-end of TSS4 are indicated. The 10-nt GspR interaction region on ctrA mRNA SLB is boxed. Means of relative fluorescence intensity values in the Rm2011 Control::Tn5 or Rm2011 cspA5::Tn5 background cells: (B) co-transformed with ctrA-112+3 translational fusion and control plasmid pSRKGm, PlaccspA5 or pSKGspR+ in the presence of IPTG; (C) transformed with the indicated ctrA native translational fusions or its mutant variants. Fluorescence measurements have been performed as described in Figure 4. (D) Schematic representation of the coherent feed forward loop circuit formed by the sRNA GspR and its ctrA and cspA5 mRNA targets, which are negatively regulated through GspR SL1 and SL3, respectively.
Fluorescence values yielded by different ctrA-egfp translational fusions were compared in the background of a S. meliloti cspA5::mini-Tn5 mutant and the control strain Rm2011 Control::Tn5, carrying a mini-Tn5 insertion in the IGR between sodB and SMc00107 (Schlüter et al., 2013). This strain has been reported not to be impaired in growth under several culture and stress conditions (Pobigaylo et al., 2006). The reporter construct ctrA-112+3 (TSS4) showed lower activity in cells lacking functional CspA5 than in control cells (Figures 6B,C). This phenotype was complemented by ectopic expression of full-length cspA5 from the Plac promoter (Figure 6B). Besides that, GspR overproduction resulted in a decrease in ctrA-egfp reporter activity in the cspA5 mutant compared to the control strain, suggesting that both post-transcriptional mechanisms of control are not mutually exclusive (Figure 6C). This decrease in activity was not evident for reporter constructs encoding shorter portions of the ctrA mRNA (-69/+3 and -56/+3 relative to the ctrA start codon; Figure 6C). The predicted secondary RNA structures of these constructs lack the longer SLB of the ctrA mRNA (Supplementary Figures S7A,B), which may represent the inhibitory structure melted by CspA5. The ctrA-112+3 reporter construct carrying 2 or 4 nt changes in the SLB stem (ctrA-BS.2 and ctrA-BS.4) still showed less ctrA-egfp derived fluorescence in the cspA5 mutant (Figure 6C). These constructs are predicted to fold similar to the ones with the native 5′-UTR sequence (Supplementary Figures S7C,D). However, the combination of the ctrA-BS.4 changes with three additional mutations in SLB, resulting in an altered predicted secondary structure (Supplementary Figure S7E), showed similar levels of reporter activity and was not affected by the cspA5 mutation (Figure 6C). In summary, these results indicate that CspA5 positively regulates CtrA, probably by uncoiling SLB formed in the long ctrA 5′ UTR.
Discussion
Cellular levels of master transcriptional regulators governing key physiological processes must be strictly controlled to ensure adaptation to environmental changes and bacterial survival. In α-proteobacteria, the master cell cycle regulator CtrA controls expression of more than a hundred genes and underlies multiple levels of regulation, which guarantee coordinated progression of fundamental cell cycle processes and adaptation to stress conditions. Regulatory mechanisms acting at the transcriptional level or influencing activity and levels of CtrA by phosphorylation and proteolytic degradation have been extensively studied (Domian et al., 1997; Reisenauer and Shapiro, 2002; Holtzendorff et al., 2004; Iniesta et al., 2006; Smith et al., 2014; Heinrich et al., 2016). In this work, we showed that the trans-sRNA GspR and the RNA chaperone CspA5 expand the diverse regulation of CtrA by mechanisms addressing the ctrA mRNA in S. meliloti. This additional level of regulation probably contributes to further control CtrA and thereby reinforces resilience and robustness of the mechanisms governing α-proteobacterial cell cycle progression in different growth phases and changing environments.
GspR Targets and Functional Role
Compared to the wild type, loss of CtrA in Rhodobacter capsulatus led to the recent identification of 18 differentially expressed and so far functionally uncharacterized sRNAs (Grüll et al., 2017). Conversely, trans-sRNA mediated post-transcriptional modulation of genes related to cell cycle progression has previously been reported in S. meliloti. Here, the EcpR1 sRNA post-transcriptionally modulates expression of dnaA and gcrA (Robledo et al., 2015). The discovery of GspR to influence stability or translation of the ctrA mRNA adds this level of regulation to another master regulator of the complex circuitry governing cell cycle progression in S. meliloti. While EcpR1 is functionally conserved in several members of the Rhizobiales (Reinkensmeier et al., 2011; Robledo et al., 2015), GspR was only identified in species belonging to the Sinorhizobium genus. This suggests GspR-mediated regulation to have evolved as a specific adaptation within this genus. Moreover, cell growth arrest upon gspR overexpression was specifically observed in various Sinorhizobium strains, suggesting functional conservation. Although we were unable to detect significant cell growth or morphology phenotypes in a gspR deletion mutant, this mutant was moderately outcompeted by the wild type in fitness assays. Because of functional redundancies in regulatory mechanisms, i.e., the mechanisms controlling CtrA levels, bacterial sRNA knock-out mutants frequently lack strong phenotypes under experimental conditions (Storz et al., 2011).
Sinorhizobium meliloti is able to establish a root nodule symbiosis with leguminous host plants. In the course of this interaction, the bacteria invade the root nodules and terminally differentiate to polyploid nitrogen-fixing bacteroids (Oke and Long, 1999; Oldroyd et al., 2011). Several studies propose that CtrA depletion is an important feature in S. meliloti bacteroid differentiation, but the underlying mechanisms remain veiled (Penterman et al., 2014; Roux et al., 2014; Pini et al., 2015). While GspR was dispensable for a functional symbiosis, it cannot be ruled out that GspR contributes to fine-tune the establishment of this interaction. Supporting this possibility, a transcriptome study of individual root nodule zones detected high levels of GspR in the regions of plant cell infection and bacteroid differentiation, but not in the nitrogen-fixation zone containing mature bacteroids (Roux et al., 2014).
The functions of the two experimentally confirmed GspR targets, ctrA encoding a transcriptional regulator and cspA5 coding for a non-specific RNA chaperone, suggest secondary downstream effects of GspR-mediated regulation. In agreement with this expectation, we identified members of the CtrA regulon (Pini et al., 2015) among the genes showing altered expression upon GspR overproduction (Supplementary Data Sheet S1). These include btaA, coding for a protein involved in cell envelope synthesis, and the motility genes flaA, flaC, and flgG; btaA being upregulated and the motility genes downregulated both under GspR overproduction and CtrA depletion conditions (Pini et al., 2015). Furthermore, the 5′-UTR of the minC mRNA, but not the coding region, was significantly less abundant in gspR overexpressing cells. In E. coli, MinC, MinD, and MinE act in concert to control formation and positioning of the FtsZ-ring, which drives septation (Cheng et al., 2007). In S. meliloti, minCDE is dispensable, but either minC-minD overexpression or disruption of minE cause severe growth phenotypes (Cheng and Walker, 1998). Interestingly, minC is repressed by CtrA, while the majority of genes was found to be activated by this regulator (Pini et al., 2015).
Two Single-Stranded Loop Regions of GspR Bind Different mRNA Targets
The confirmed interaction regions of GspR with the cspA5 and ctrA mRNAs reflect different mechanisms of inhibition. GspR SL3 seems to mediate canonical translational repression by targeting the translation initiation region in cspA5 mRNA, whereas GspR SL1 most probably binds a region -69 to -58 nucleotides upstream of the start codon in the ctrA mRNA. Repression of translation at target sequences located upstream of the ribosome binding site has already been reported for other sRNA–mRNA pairs, e.g., GcvB-gltl and EcpR1-gcrA (Sharma et al., 2007; Robledo et al., 2015), but the underlying mechanisms are largely unknown. Two to four nucleotide changes in GspR SL1 were necessary to abolish its regulatory activity on ctrA, suggesting a high strength of sRNA-mRNA binding, similar to the EcpR1-gcrA pairing (Robledo et al., 2015).
While some sRNAs bind multiple targets through the same single-stranded domain (Balbontín et al., 2010; Robledo et al., 2015), others use distinct functional domains for multiple targeting (Overlöper et al., 2014; Feng et al., 2015). GspR matches the second group, with two confirmed targets pairing to SL1 and SL3 domains. Overproduction of GspR mutant variants carrying changes in SL1 or SL3 did not show off-target regulation, supporting both GspR stem loops as independent functional domains with respect to regulation of ctrA and cspA5. Intriguingly, northern and RNAseq data indicated two coexisting GspR RNAs: the full-length transcript and a shorter, less abundant variant lacking SL1 and therefore being unable to regulate ctrA. Different stabilities of the two GspR variants may influence the regulatory activity and specificity of GspR.
Although, gspR overexpression appeared to significantly reduce translation of the ctrA mRNA and both CtrA-depleted (Pini et al., 2015) and GspR-overproducing cells showed growth arrest phenotypes, these phenotypes cannot be associated to GspR-mediated regulation of ctrA through SL1. Our mutational studies link growth repression caused by GspR to SL3, which targets cspA5 mRNA. However, a cspA5 insertion mutant was not hampered in growth, indicating that GspR-mediated regulation of yet uncharacterized targets, individually or in combination, causes this phenotype.
GspR Controls a Feed-Forward Loop Involving CspA5 and CtrA
Cold shock proteins have been reported to facilitate mRNA translation under different stress conditions, including cold, thereby contributing to bacterial adaptation to changing environments (Keto-Timonen et al., 2016). In this study, we revealed regulation of ctrA expression by CspA5 in S. meliloti. The role of CspAs has not been addressed yet in rhizobacteria and its homology to cold shock proteins does not necessarily imply that they have a role at low temperatures. Eight CspA homologs were found in S. meliloti: chromosomally encoded CspA1 to 5, and pSymA-encoded CspA6 to 8. Similarly, E. coli carries nine csp genes and at least four have to be deleted to generate a growth phenotype (Xia et al., 2001). Therefore, the lack of a growth phenotype of the S. meliloti cspA5 mutant is not surprising.
Bacteria use different network motifs involving a variable number of regulators (Beisel and Storz, 2010). Recent studies in enterobacteria have stressed the role of sRNAs together with transcriptional factors in regulatory networks (Beisel and Storz, 2011; Papenfort et al., 2015). Feed-forward loops are composed of one regulator controlling a second regulator and a target gene controlled by both these regulators (Beisel and Storz, 2010). When both arms of the loop act jointly, the loop is classified as coherent. Here, we show that GspR sRNA post-transcriptionally represses the mRNAs of the cold shock chaperone homolog CspA5 and the cell cycle master regulator CtrA, which is post-transcriptionally controlled by CspA5 (Figure 6D), thus forming a coherent feed-forward loop. This regulatory circuit may enhance the negative effect of GspR on ctrA expression. On the one hand, it directly represses ctrA and, on the other hand, it negatively influences its activator CspA5. Intriguingly, the cspA5 promoter region harbors a putative CtrA binding motif (Schlüter et al., 2013), but direct regulation has not been proved under the conditions tested. This may hint to a further link in this network. To the best of our knowledge, GspR is the first sRNA reported to post-transcriptionally control via separate seed-pairing domains both targets in a feed-forward loop.
Author Contributions
AB, MR, J-PS, and JJ-Z conceived and designed the experiments. MR and J-PS performed the main experiments. J-PS, LL, UL, and SA carried out the transcriptome and proteome profiling. MR and AB analyzed the data. MR, AB, and JJ-Z wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Barbara Herte, Tina Krieg, and Vicenta Millán for the technical assistance and the core facilities of EEZ-CSIC for routine sequencing of plasmid inserts.
Footnotes
Funding. This research was supported by funding from the LOEWE Program of the state of Hesse (SYNMIKRO) to AB and MR, the German Network for Bioinformatics Infrastructure (de.NBI, BMBF-funded project “Bielefeld-Gießen Center for Microbial Bioinformatics – BiGi,” Grant No. 031A533) to SPA, the German Research Foundation (CRC/TR 174 and INST 160/536-1 FUGG) to AB, and the Spanish Ministerio de Economía y Competitividad ERDF-cofinanced grant BFU2013-48282-C2-2-P to JJ-Z and Program of “Formación Post-doctoral” contract to MR.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00763/full#supplementary-material
References
- Albaum S. P., Hahne H., Otto A., Haußmann U., Becher D., Poetsch A. (2011). A guide through the computational analysis of isotope-labeled mass spectrometry-based quantitative proteomics data: an application study. Proteome Sci. 9:30. 10.1186/1477-5956-9-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaum S. P., Neuweger H., Fränzel B., Lange S., Mertens D., Trötschel C. (2009). Qupe–a rich internet application to take a step forward in the analysis of mass spectrometry-based quantitative proteomics experiments. Bioinformatics 25 3128–3134. 10.1093/bioinformatics/btp568 [DOI] [PubMed] [Google Scholar]
- Bahlawane C., McIntosh M., Krol E., Becker A. (2008). Sinorhizobium meliloti regulator MucR couples exopolysaccharide synthesis and motility. Mol. Plant Microbe Interact. 21 1498–1509. 10.1094/MPMI-21-11-1498 [DOI] [PubMed] [Google Scholar]
- Balbontín R., Fiorini F., Figueroa-Bossi N., Casadesús J., Bossi L. (2010). Recognition of heptameric seed sequence underlies multi-target regulation by RybB small RNA in Salmonella enterica. Mol. Microbiol. 78 380–394. 10.1111/j.1365-2958.2010.07342.x [DOI] [PubMed] [Google Scholar]
- Barria C., Malecki M., Arraiano C. M. (2013). Bacterial adaptation to cold. Microbiology 159(Pt 12) 2437–2443. 10.1099/mic.0.052209-0 [DOI] [PubMed] [Google Scholar]
- Baumgardt K., Charoenpanich P., McIntosh M., Schikora A., Stein E., Thalmann S. (2014). RNase E affects the expression of the acyl-homoserine lactone synthase gene sinI in Sinorhizobium meliloti. J. Bacteriol. 196 1435–1447. 10.1128/JB.01471-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A., Overlöper A., Schlüter J. P., Reinkensmeier J., Robledo M., Giegerich R. (2014). Riboregulation in plant-associated α-proteobacteria. RNA Biol. 11 550–562. 10.4161/rna.29625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel C. L., Storz G. (2010). Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol. Rev. 34 866–882. 10.1111/j.1574-6976.2010.00241.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel C. L., Storz G. (2011). The base-pairing RNA spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol. Cell 41 286–297. 10.1016/j.molcel.2010.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringer J. E. (1974). R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84 188–198. 10.1099/00221287-84-1-188 [DOI] [PubMed] [Google Scholar]
- Biondi E. G., Reisinger S. J., Skerker J. M., Arif M., Perchuk B. S., Ryan K. R. (2006). Regulation of the bacterial cell cycle by an integrated genetic circuit. Nature 444 899–904. 10.1038/nature05321 [DOI] [PubMed] [Google Scholar]
- Brilli M., Fondi M., Fani R., Mengoni A., Ferri L., Bazzicalupo M. (2010). The diversity and evolution of cell cycle regulation in alpha-proteobacteria: a comparative genomic analysis. BMC Syst. Biol. 4:52. 10.1186/1752-0509-4-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H. P., Walker G. C. (1998). Succinoglycan is required for initiation and elongation of infection threads during nodulation of alfalfa by Rhizobium meliloti. J. Bacteriol. 180 5183–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Sibley C. D., Zaheer R., Finan T. M. (2007). A Sinorhizobium meliloti minE mutant has an altered morphology and exhibits defects in legume symbiosis. Microbiology 153(Pt 2) 375–387. 10.1099/mic.0.2006/001362-0 [DOI] [PubMed] [Google Scholar]
- Darty K., Denise A., Ponty Y. (2009). VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25 1974–1975. 10.1093/bioinformatics/btp250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domian I. J., Quon K. C., Shapiro L. (1997). Cell type-specific phosphorylation and proteolysis of a transcriptional regulator controls the G1-to-S transition in a bacterial cell cycle. Cell 90 415–424. 10.1016/S0092-8674(00)80502-4 [DOI] [PubMed] [Google Scholar]
- Dondrup M., Albaum S. P., Griebel T., Henckel K., Jünemann S., Kahlke T. (2009). EMMA 2–a MAGE-compliant system for the collaborative analysis and integration of microarray data. BMC Bioinformatics 10:50. 10.1186/1471-2105-10-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Rutherford S. T., Papenfort K., Bagert J. D., van Kessel J. C., Tirrell D. A. (2015). A qrr noncoding RNA deploys four different regulatory mechanisms to optimize quorum-sensing dynamics. Cell 160 228–240. 10.1016/j.cell.2014.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frage B., Döhlemann J., Robledo M., Lucena D., Sobetzko P., Graumann P. L. (2016). Spatiotemporal choreography of chromosome and megaplasmids in the Sinorhizobium meliloti cell cycle. Mol. Microbiol. 100 808–823. 10.1111/mmi.13351 [DOI] [PubMed] [Google Scholar]
- Galibert F., Finan T. M., Long S. R., Puhler A., Abola P., Ampe F. (2001). The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293 668–672. 10.1126/science.1060966 [DOI] [PubMed] [Google Scholar]
- Giacomini A., Ollero F. J., Squartini A., Nuti M. P. (1994). Construction of multipurpose gene cartridges based on a novel synthetic promoter for high-level gene expression in gram-negative bacteria. Gene 144 17–24. 10.1016/0378-1119(94)90197-X [DOI] [PubMed] [Google Scholar]
- Gottesman S., Storz G. (2011). Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 3:a003798. 10.1101/cshperspect.a003798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber A. R., Lorenz R., Bernhart S. H., Neuböck R., Hofacker I. L. (2008). The Vienna RNA website. Nucleic Acids Res. 36 W70–W74. 10.1093/nar/gkn188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüll M. P., Peña-Castillo L., Mulligan M. E., Lang A. S. (2017). Genome-wide identification and characterization of small RNAs in Rhodobacter capsulatus and identification of small RNAs affected by loss of the response regulator CtrA. RNA Biol. 14 914–925. 10.1080/15476286.2017.1306175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich K., Sobetzko P., Jonas K. (2016). A Kinase-Phosphatase switch transduces environmental information into a bacterial cell cycle circuit. PLoS Genet. 12:e1006522. 10.1371/journal.pgen.1006522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzendorff J., Hung D., Brende P., Reisenauer A., Viollier P. H., McAdams H. H. (2004). Oscillating global regulators control the genetic circuit driving a bacterial cell cycle. Science 304 983–987. 10.1126/science.1095191 [DOI] [PubMed] [Google Scholar]
- Iniesta A. A., McGrath P. T., Reisenauer A., McAdams H. H., Shapiro L. (2006). A phospho-signaling pathway controls the localization and activity of a protease complex critical for bacterial cell cycle progression. Proc. Natl. Acad. Sci. U.S.A. 103 10935–10940. 10.1073/pnas.0604554103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Zurdo J. I., Robledo M. (2015). Unraveling the universe of small RNA regulators in the legume symbiont Sinorhizobium meliloti. Symbiosis 67 43–54. 10.1007/s13199-015-0345-z [DOI] [Google Scholar]
- Jiménez-Zurdo J. I., Valverde C., Becker A. (2013). Insights into the noncoding RNome of nitrogen-fixing endosymbiotic α-proteobacteria. Mol. Plant Microbe Interact. 26 160–167. 10.1094/MPMI-07-12-0186-CR [DOI] [PubMed] [Google Scholar]
- Keto-Timonen R., Hietala N., Palonen E., Hakakorpi A., Lindström M., Korkeala H. (2016). Cold shock proteins: a minireview with special emphasis on Csp-family of enteropathogenic Yersinia. Front. Microbiol. 7:1151. 10.3389/fmicb.2016.01151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdams H. H., Shapiro L. (2009). System-level design of bacterial cell cycle control. FEBS Lett. 583 3984–3991. 10.1016/j.febslet.2009.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oke V., Long S. R. (1999). Bacteroid formation in the Rhizobium-legume symbiosis. Curr. Opin. Microbiol. 2 641–646. 10.1016/S1369-5274(99)00035-1 [DOI] [PubMed] [Google Scholar]
- Oldroyd G. E., Murray J. D., Poole P. S., Downie J. A. (2011). The rules of engagement in the legume-rhizobial symbiosis. Annu. Rev. Genet. 45 119–144. 10.1146/annurev-genet-110410-132549 [DOI] [PubMed] [Google Scholar]
- Overlöper A., Kraus A., Gurski R., Wright P. R., Georg J., Hess W. R. (2014). Two separate modules of the conserved regulatory RNA AbcR1 address multiple target mRNAs in and outside of the translation initiation region. RNA Biol. 11 624–640. 10.4161/rna.29145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K., Espinosa E., Casadesús J., Vogel J. (2015). Small RNA-based feedforward loop with AND-gate logic regulates extrachromosomal DNA transfer in Salmonella. Proc. Natl. Acad. Sci. U.S.A. 112 E4772–E4781. 10.1073/pnas.1507825112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penterman J., Abo R. P., De Nisco N. J., Arnold M. F., Longhi R., Zanda M. (2014). Host plant peptides elicit a transcriptional response to control the Sinorhizobium meliloti cell cycle during symbiosis. Proc. Natl. Acad. Sci. U.S.A. 111 3561–3566. 10.1073/pnas.1400450111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20 3551–3567. [DOI] [PubMed] [Google Scholar]
- Pini F., De Nisco N. J., Ferri L., Penterman J., Fioravanti A., Brilli M. (2015). Cell cycle control by the master regulator CtrA in Sinorhizobium meliloti. PLoS Genet. 11:e1005232. 10.1371/journal.pgen.1005232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobigaylo N., Wetter D., Szymczak S., Schiller U., Kurtz S., Meyer F. (2006). Construction of a large signature-tagged mini-Tn5 transposon library and its application to mutagenesis of Sinorhizobium meliloti. Appl. Environ. Microbiol. 72 4329–4337. 10.1128/AEM.03072-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quon K. C., Marczynski G. T., Shapiro L. (1996). Cell cycle control by an essential bacterial two-component signal transduction protein. Cell 84 83–93. 10.1016/S0092-8674(00)80995-2 [DOI] [PubMed] [Google Scholar]
- Reidegeld K. A., Eisenacher M., Kohl M., Chamrad D., Körting G., Blüggel M. (2008). An easy-to-use decoy database builder software tool, implementing different decoy strategies for false discovery rate calculation in automated MS/MS protein identifications. Proteomics 8 1129–1137. 10.1002/pmic.200701073 [DOI] [PubMed] [Google Scholar]
- Reinkensmeier R., Schlüter J. P., Giegerich R., Becker A. (2011). Conservation and occurrence of trans-encoded sRNAs in the Rhizobiales. Genes 2 925–956. 10.3390/genes2040925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisenauer A., Shapiro L. (2002). DNA methylation affects the cell cycle transcription of the CtrA global regulator in Caulobacter. EMBO J. 21 4969–4977. 10.1093/emboj/cdf490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertsen B. K., Aman P., Darvill A. G., McNeil M., Albersheim P. (1981). Host-Symbiont interactions: V. The structure of acidic extracellular polysaccharides secreted by Rhizobium leguminosarum and Rhizobium trifolii. Plant Physiol. 67 389–400. 10.1104/pp.67.3.389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robledo M., Frage B., Wright P. R., Becker A. (2015). A stress-induced small RNA modulates alpha-rhizobial cell cycle progression. PLoS Genet. 11:e1005153. 10.1371/journal.pgen.1005153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robledo M., García-Tomsig N. I., Jiménez-Zurdo J. I. (2018). Primary characterization of small RNAs in symbiotic nitrogen-fixing bacteria. Methods Mol. Biol. 1734 277–295. 10.1007/978-1-4939-7604-1_22 [DOI] [PubMed] [Google Scholar]
- Robledo M., Peregrina A., Millán V., García-Tomsig N. I., Torres-Quesada O., Mateos P. F. (2017). A conserved α-proteobacterial small RNA contributes to osmoadaptation and symbiotic efficiency of rhizobia on legume roots. Environ. Microbiol. 19 2661–2680. 10.1111/1462-2920.13757 [DOI] [PubMed] [Google Scholar]
- Roux B., Rodde N., Jardinaud M. F., Timmers T., Sauviac L., Cottret L. (2014). An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 77 817–837. 10.1111/tpj.12442 [DOI] [PubMed] [Google Scholar]
- Sanselicio S., Bergé M., Théraulaz L., Radhakrishnan S. K., Viollier P. H. (2015). Topological control of the Caulobacter cell cycle circuitry by a polarized single-domain PAS protein. Nat. Commun. 6:7005. 10.1038/ncomms8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter J. P., Reinkensmeier J., Barnett M. J., Lang C., Krol E., Giegerich R. (2013). Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 14:156. 10.1186/1471-2164-14-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter J. P., Reinkensmeier J., Daschkey S., Evguenieva-Hackenberg E., Janssen S., Jänicke S. (2010). A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 11:245. 10.1186/1471-2164-11-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma C. M., Darfeuille F., Plantinga T. H., Vogel J. (2007). A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 21 2804–2817. 10.1101/gad.447207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. C., Joshi K. K., Zik J. J., Trinh K., Kamajaya A., Chien P. (2014). Cell cycle-dependent adaptor complex for ClpXP-mediated proteolysis directly integrates phosphorylation and second messenger signals. Proc. Natl. Acad. Sci. U.S.A. 111 14229–14234. 10.1073/pnas.1407862111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrero P., Schlüter J. P., Lanner U., Schlosser A., Becker A., Valverde C. (2012). Quantitative proteomic analysis of the Hfq-regulon in Sinorhizobium meliloti 2011. PLoS One 7:e48494. 10.1371/journal.pone.0048494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G., Vogel J., Wassarman K. M. (2011). Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell 43 880–891. 10.1016/j.molcel.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Quesada O., Millán V., Nisa-Martínez R., Bardou F., Crespi M., Toro N. (2013). Independent activity of the homologous small regulatory RNAs AbcR1 and AbcR2 in the legume symbiont Sinorhizobium meliloti. PLoS One 8:e68147. 10.1371/journal.pone.0068147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Quesada O., Reinkensmeier J., Schlüter J. P., Robledo M., Peregrina A., Giegerich R. (2014). Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol. 11 563–579. 10.4161/rna.28239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters L. S., Storz G. (2009). Regulatory RNAs in bacteria. Cell 136 615–628. 10.1016/j.cell.2009.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. R., Georg J., Mann M., Sorescu D. A., Richter A. S., Lott S. (2014). CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 42 W119–W123. 10.1093/nar/gku359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B., Ke H., Inouye M. (2001). Acquirement of cold sensitivity by quadruple deletion of the cspA family and its suppression by PNPase S1 domain in Escherichia coli. Mol. Microbiol. 40 179–188. 10.1046/j.1365-2958.2001.02372.x [DOI] [PubMed] [Google Scholar]
- Zhan H. J., Lee C. C., Leigh J. A. (1991). Induction of the second exopolysaccharide (EPSb) in Rhizobium meliloti SU47 by low phosphate concentrations. J. Bacteriol. 173 7391–7394. 10.1128/jb.173.22.7391-7394.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. Z., Sun Z. C., Fu X. R., Nan F. F., Fan Q. X., Wu X. A. (2009). Analysis of serum proteome profiles of non-Hodgkin lymphoma for biomarker identification. J. Proteomics 72 952–959. 10.1016/j.jprot.2009.03.009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.