

Abstract
Mitochondrial calcium overload is a crucial event in determining the fate of neuronal cell survival and death, implicated in pathogenesis of neurodegenerative diseases. One of the driving forces of calcium influx into mitochondria is mitochondria membrane potential (ΔΨm). Therefore, pharmacological manipulation of ΔΨm can be a promising strategy to prevent neuronal cell death against brain insults. Based on these issues, we investigated here whether nobiletin, a Citrus polymethoxylated flavone, prevents neurotoxic neuronal calcium overload and cell death via regulating basal ΔΨm against neuronal insult in primary cortical neurons and pure brain mitochondria isolated from rat cortices. Results demonstrated that nobiletin treatment significantly increased cell viability against glutamate toxicity (100 µM, 20 min) in primary cortical neurons. Real-time imaging-based fluorometry data reveal that nobiletin evokes partial mitochondrial depolarization in these neurons. Nobiletin markedly attenuated mitochondrial calcium overload and reactive oxygen species (ROS) generation in glutamate (100 µM)-stimulated cortical neurons and isolated pure mitochondria exposed to high concentration of Ca2+ (5 µM). Nobiletin-induced partial mitochondrial depolarization in intact neurons was confirmed in isolated brain mitochondria using a fluorescence microplate reader. Nobiletin effects on basal ΔΨm were completely abolished in K+-free medium on pure isolated mitochondria. Taken together, results demonstrate that K+ influx into mitochondria is critically involved in partial mitochondrial depolarization-related neuroprotective effect of nobiletin. Nobiletin-induced mitochondrial K+ influx is probably mediated, at least in part, by activation of mitochondrial K+ channels. However, further detailed studies should be conducted to determine exact molecular targets of nobiletin in mitochondria.
Keywords: Calcium, Mitochondrial calcium, Mitochondrial K+ channels, Mitochondrial membrane potential, Nobiletin
INTRODUCTION
The main function of mitochondria is adenosine triphosphate (ATP) production. Electron transport chains of mitochondria generate H+ electrochemical gradient as electrons pass through complex I, III and IV, activating ATP synthase. The total process is called as oxidative phosphorylation. However, since the 20th century, many studies have focused on the regulatory function of mitochondria in determining the fate of cellular survival and death. Especially, mitochondrial calcium plays a key role in the regulatory mechanism of cell survival and death. Overload of mitochondria calcium during brain insults results in oxidative stress, impaired mitochondrial function, neuronal cell death and neuroinflammation, implicated in pathogenesis of many neurodegenerative diseases [1,2].
Gunter and Pfeiffer [3] described driving forces of Ca2+ into mitochondria by an equation: ΔµCa=RT ln ([Ca2+]out/[Ca2+]in)+2F(Ψout-#x03A8;in). According to this equation, there are two different driving forces, i) Ca2+ concentration gradient between cytosol calcium ([Ca2+]c) and mitochondrial calcium([Ca2+]m), ii) mitochondria membrane potential (ΔΨm) gradient [3]. Based on correlation between [Ca2+]m and ΔΨm, it has been proposed that pharmacological manipulation of ΔΨm can be a key strategy to prevent neurotoxic mitochondrial overload and neuronal cell death by reducing driving forces of Ca2+ into mitochondria [4]. Regarding these ideas, it has been reported that mild mitochondrial depolarization reduced mitochondrial calcium overload in several studies conducted in different cell types [5,6].
In our previous study, we demonstrated that ethanolic peel extract of Citrus sunki Hort. ex Tanaka (CPE) induces mild mitochondrial depolarization intrinsically [7]. Based on this property, CPE significantly attenuated mitochondrial calcium overload and cell death against H2O2 toxicity in HT-22 cells. Among various flavonoid compounds in CPE (i.e., rutin, hesperidin, sinensetin, tangeretin and nobiletin) belongs to polymethoxylated flavone (PMF) group [7]. Nobiletin exerts several beneficial effects in improving cognitive function or motor deficits in several animal models such as cerebral ischemia [8], Parkinson's and Alzheimer's diseases [9,10]. In addition, nobiletin induced neurites outgrowth in PC12 cells [11] and significantly suppressed microglial activation and neuroinflammation [12]. These evidences suggest that nobiletin is a promising candidate as a neuroprotective agent and deserves to be thoroughly explored.
Based on these issues, we investigated here whether nobiletin prevents glutamate toxicity-induced neurotoxic mitochondrial calcium overload and neuronal cell death through K+ influx and ΔΨm across mitochondrial inner membrane in primary cerebrocortical neurons or isolated brain mitochondria.
METHODS
Reagents
Tetramethylrhodamine ethyl ester (TMRE), rhod-2 acetoxymethyl ester (Rhod-2 AM), MitoSOX Red, Fura-2 acetoxymethyl ester (Fura-2 AM) were purchased from Invitrogen. Minimal essential medium (MEM), Neurobasal medium, fetal bovine serum (FBS), B-27 serum-free supplement, glutamine and penicillin/streptomycin were purchased from Gibco BRL. Nobiletin was isolated and purified from the peel of Citrus sunki Hort. ex Tanaka in Jeju National University, Department of Biology as described previously [13]. All other reagents were obtained from Sigma-Aldrich, unless otherwise indicated.
Primary culture of cortical neurons
Primary cortical neurons were prepared from cerebral cortices of postnatal 1-day-old Sprague-Dawley rats. The neonatal brain were dissected and the cortices were transferred to plating medium (containing MEM supplemented with 10% FBS, 25 mM glucose, 1 mM sodium pyruvate, 25 mM glutamine and 1% penicillin/streptomycin) and dissociated by trituration using fire-polished glass Pasteur pipettes. Then, cells were plated onto poly-L-lysine-coated round glass coverslips placed in 12-well plates at a density of 1.3×105/well. After 6 h, plating medium was replaced to Neurobasal media supplemented with 2% B-27, 50 mM glutamine and 1% penicillin/streptomycin, and a half of culture medium was replaced every four days. Cultured neurons were incubated at 37℃ in a humidified 5% CO2/ 95% air atmosphere. The cells were used after 7 days in vitro (DIV). The study was approved by Animal Care and Use Committee of Jeju National University and we performed all experiments in accordance with the guidelines.
Dual real-time imaging-based fluorometry of both cytosolic and mitochondrial calcium levels in the same cortical neurons
Fura-2 and Rhod-2 were used to measure cytosolic and mitochondrial calcium levels. Cell-permeable acetoxymethyl ester (AM) forms were used for these two probes. Fura-2 is a ratiometric fluorescent indicator to measure [Ca2+]c using the ratio of emitted fluorescence intensity at excitation wavelengths of 340 and 380 nm [14]. Another calcium indicator Rhod-2, used for a selective [Ca2+]m probe, exhibits charge-driven uptake into the mitochondria and evokes increase of fluorescence upon Ca2+ binding [7]. The cortical neurons cultured on a coverslip were loaded with 10 µM Fura-2 AM and 0.1% Pluronic F127 for 45 min at 37℃. And then, cells were loaded with 2 µM Rhod-2 AM for 30 min at 4℃. After washing three times, a coverslip was transferred to the recording chamber. Cells were continuously superfused with normal Tyrode solution (in mM): NaCl 145, KCl 5, CaCl2 2, MgCl2 1.3, HEPES 10, glucose 10, pH 7.4 with NaOH. The fluorescence was measured with alternative excitation wavelengths of 340 and 380 nm and an emission wavelength of 510 nm for Fura-2, and an excitation wavelength of 540 nm and an emission wave-length of 605 nm for Rhod-2. High-speed filter switching device (Sutter Instrument, Lambda, DG-4) was used for dual recording of Fura-2 and Rhod-2. The fluorescence images were acquired at 6 s intervals using an inverted microscope Olympus IX71 (Olympus) and a cooled-charged device (CCD) camera (Cascade, Roper Scientific). The data were analyzed using Metafluor software (Molecular Devices).
Real-time imaging-based fluorometry of ΔΨm and mitochondrial ROS in intact cortical neurons
The cationic fluorescent probe TMRE is sequestered by mitochondria in proportion to ΔΨm [15]. The cationic MitoSOX Red is also selectively targeted to the mitochondria. Once MitoSOX Red is oxidized by superoxide anions which are the predominant ROS in mitochondria, it exhibits red fluorescence. The cortical neurons were loaded for 15 min at 37℃ with 25 nM TMRE and 5 µM MitoSOX Red for ΔΨm and mitochondrial superoxide, respectively. After then, the neurons on the cover glass were washed three times with normal Tyrode solution. The cover glass was transferred to the recording chamber which was continuously superfused with normal Tyrode solution. Digitized fluorescence images were acquired at 30 s intervals using an inverted microscope Olympus IX71 (Olympus) with a cooled-charged device (CCD) camera (Cascade, Roper Scientific), and analyzed in a personal computer using Metafluor software (Molecular Devices).
Preparation of pure mitochondria isolated from rat brain cortices
The isolated mitochondria were obtained from rat brain cortices, as previously described [16]. In short, the cortices were removed from the brains of 9–16 day-old rats. The fragmented cortices were placed in EGTA-containing isolation buffer (IB) and homogenized in the Dounce-type tissue grinders (Kimble chase). Pestle A and B are used sequentially. The clearance of pestle A and B are 0.0035–0.0065 mm and 0.0010-0.0030 mm respectively. The IB contained (in mM): Mannitol 225, Sucrose 75, HEPES 5, ECTA 3, BSA 0.1%, titrated with KOH to pH 7.4. The homogenates were centrifuged at 600×g for 10 min. The supernatant was transferred to a new tube and then centrifuged again at 600×g for 10 min. Then, supernatants were centrifuged at 12,000×g for 10 min. The pellets were resuspended in IB buffer without EGTA and homogenized using Dounce-type tissue grinders. The clearance of pestle A and B used in this stage were 0.0028–0.0047 mm and 0.0008–0.0022 mm respectively. The homogenates were centrifuged at 12,000×g for 10 min. All the above procedures were carried out at 4℃. The isolated mitochondrial proteins were quantified using Bio-Rad protein assay dye. Electron microscopy (EM) and oxygen consumption rate (OCR) measurement were conducted to evaluate morphology and metabolic activity of isolated mitochondria.
Measurement of ΔΨm and ROS in pure isolated mitochondria using a microplate reader
After preparation of pure mitochondria isolated from rat brain cortices, an isolated brain mitochondrial model was set up. The mitochondria suspension (500 µg of protein/ml) was incubated in recording buffer for 10 min at 37℃ with 25 nM TMRE and 50 µM DCF-DA for ΔΨm and mitochondrial ROS, respectively [7,15,17]. The recording buffer contained (in mM): KCl 100, HEPES 20, Tris 20, NaCl 10, succinate 5, KH2PO4 1, EGTA 0.02, rotenone 0.002, oligomycin 0.001 and CaCl2 0.0001. The loaded mitochondria in suspension were treated with nobiletin or other reagents in 96 well plates. And then, fluorescence intensities were measured using a fluorescence microplate reader (SPECTRA FluoR, Tecan).
Measurement of oxygen consumption rate (OCR)
The OCR of isolated mitochondria was measured using a Seahorse XF-24 extracellular flux analyzer (Seahorse Bioscience) following the manufacturer's protocol. Briefly, 5 µg of isolated mitochondria were suspended in 50 µL of assay medium and transferred to each well for OCR measurement. Mitochondrial assay medium contained (in mM): mannitol 220, sucrose 70, KH2PO4 10, MgCl2 5, HEPES 2, EGTA 1 and fatty acid-free BSA 0.2% (w/v), pH 7.2. One day before the experiment, sensor cartridge was placed into calibration buffer and incubated overnight in a non-CO2 condition at 37℃. The reagents listed below were added sequentially according to the manufacturer's protocol: ADP (2 mM), oligomycin (2 µg/ml) as an inhibitor of mitochondrial ATP synthase, FCCP (2 µM) as an electron transport chain accelerator, rotenone/antimycin A (0.5 µM) as a complex I and III inhibitor. The OCR was recorded by sensor cartridge and analyzed using Seahorse XF-24 software.
Electron microscopy
Electron microscopy for morphological analysis of mitochondria isolated from rat brain cortices was performed as described previously [18]. In short, mitochondria samples obtained through serial centrifugation were fixed overnight with 2.5% glutaraldehyde in 0.15 M cacodylated buffer (pH 7.3) at 4℃, after than samples were centrifuged for 60 s at 3,000 rpm. After washing with the same buffer, samples were post-fixed with 2% osmium tetroxide for 30 min and en bloc stained with 0.1% uranyl acetate solution for 1 h, embedded in 3% agarose gel. Embedded samples were dehydrated through an ascending ethanol series and embedded with an Epon mixture. Thin sections of 70 nm were collected on 200-mesh cooper grid and stained with ueanyl acetate followed by lead citrate. Electron microscopic observation was performed with a Hitachi H-7500 (Hitachi, Japan) transmission electron microscope with 80 kV acceleration voltages.
DPPH free radical scavenging assay
DPPH (1,1-diphenyl-2-picrylhydrazyl) was used to investigate free radical scavenging activity of nobiletin in the solution state, as previously described [19]. The mixture of DPPH and nobiletin was incubated in dark at room temperature for 1 h, and the absorbance was read at 517 nm using a microplate reader (Tecan, Sunrise, AT, USA).
Cell viability assay
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] was used to investigate cytoprotective effect of nobiletin on cell viability, as previously described [19]. Absorbance was subsequently read at 540 nm using a microplate reader (Model 550, Bio-Rad, USA).
Statistical analysis
Data were expressed as mean values±standard error of the mean (SEM). The significance of data was analyzed using Student's t-test using the sigma plot 8.0 software. The differences between groups were considered to be significant when p<0.05.
RESULTS
Nobiletin, the key compound of CPE, exhibits neuroprotective effects based on mild mitochondrial depolarization
We previously reported that CPE evokes partial mitochondrial depolarization intrinsically while carbonyl cyanide m-chlorophenylhydrazone (CCCP, 10 µM), a well-known ΔΨm dissipation-inducer, evokes complete mitochondrial depolarization [7]. Based on this study, we investigated here whether nobiletin, an active compound of CPE, induces partial depolarization of ΔΨm in resting state and reveal neuroprotective activity against glutamate toxicity in primary cortical neurons (Fig. 2).
Fig. 2. The effects of nobiletin on basal ΔΨm and cell viability against glutamate toxicity in primary cortical neurons.

(A, D) Recording traces of ΔΨm using real-time imaging-based fluorometry with TMRE (see ‘METHODS’ for the detailed description). Various concentrations of nobiletin and CPE were superfused over primary cortical neurons on a cover slip in a recording chamber from the arrow point. TMRE fluorescence values from individual cells were normalized to values before drug treatment shown as an arrow. (B, E) Quantification of ΔΨm at the end of experiment for panel A and D. (C, F) Effects of nobiletin and CPE on cell viability against glutamate toxicity (100 µM, 20 min) were investigated using MTT assay. Values are the mean±S.E.M. *p<0.05, **p<0.01, ***p<0.001 as compared with the control group and #p<0.05 as compared with glutamate alone-treated group.
Various concentrations of nobiletin (30, 50, 100 and 200 µM) were superfused over TMRE-loaded cortical neurons on a cover slip in a recording chamber. ΔΨm values were recorded and analyzed using real-time imaging-based fluorometry (see ‘METHODS’ for more detailed description). TMRE fluorescence values from individual cells were normalized to values before drug treatment in Fig. 2A. Recording traces reveal average recordings of TMRE intensities obtained from individual cells. Results demonstrated that nobiletin treatment significantly evoked mild mitochondrial depolarization in dose-dependent manner. Normalized TMRE values at 13-min points were 94.71±5.47%, 73.06±4.79%, 61.47±6.06% and 54.26±10.37% in nobiletin (30, 50, 100 and 200 mM)-treated groups, respectively (Fig. 2B), compared to the control group. Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) is another ΔΨm dissipation-inducer, similar to CCCP. As shown in Fig. 2, FCCP (10 mM) induced a marked decrease of TMRM values, which means total ΔΨm dissipation. Nobiletin-induced partial mitochondrial depolarization in intact neurons was also confirmed in isolated brain mitochondria using fluorescence microplate reader, as shown in Figs. 5B and C.
Fig. 5. The effects of K+ influx on nobiletin-induced partial mitochondrial depolarization in isolated brain mitochondria.

(A) The effect of FCCP on ΔΨm was investigated with TMRE in an isolated brain mitochondrial model (see ‘METHODS’ for the detailed description) using a fluorescence microplate reader, as a positive control. (B, C). The effect of nobiletin on ΔΨm was measured with TMRE in the presence or absence of K+ in the medium using a fluorescence microplate reader in an isolated brain mitochondrial model. To remove K+ in the medium, KCl (100 mM) was replaced to CsCl (100 mM). Dose responses 10 min after nobiletin treatment (B) and time courses (C) were analyzed. Values are the mean±S.E.M. *p<0.05, **p<0.01, ***p<0.001 as compared with the control group.
The neuroprotective effect of nobiletin against glutamate toxicity was examined in primary cortical neurons using MTT assay. It is well established that glutamate toxicity results in massive and global calcium influx into the cytosol and subsequent mitochondrial calcium overload and evokes cell death [2]. Nobiletin (100 µM) was pretreated for 10 min and glutamate (100 µM) was stimulated for 20 min in the presence of nobiletin. As shown in Fig. 2C, nobiletin significantly increased neuronal cell viability to 80.32±4.80% against glutamate toxicity (100 µM, 20 min).
In addition, we found that CPE (500 and 1,000 µg/ml) intrinsically evoked mitochondrial depolarization in a dose-dependent manner (Figs. 2D and E) and significantly enhanced cell viability (Fig. 2F) against glutamate toxicity (100 µM, 20 min), similar to nobiletin. Results suggest that beneficial effects of CPE on ΔΨm and neuronal cell viability are partially, at least, attributed to its key compound nobiletin, as suggested previously [7].
Nobiletin reduces glutamate-induced mitochondrial calcium overload
High concentration of glutamate induces excess Ca2+ influx from extracellular space to cytosol via glutamate receptor/channels and subsequently excess cytosol Ca2+ is moved into mitochondria [2]. Mild mitochondrial depolarization attenuates mitochondrial calcium overload by reducing driving force of Ca2+ uptake into mitochondria [4,5,6]. Therefore, we investigated here whether nobiletin attenuates mitochondrial calcium overload against glutamate toxicity.
Cytosol and mitochondrial Ca2+ levels were simultaneously recorded and analyzed using dual real-time imaging-based fluorometry with Fura-2 AM and Rhod-2 AM (see ‘METHODS’ for more detailed description). Glutamate (100 µM) in the presence or absence of nobiletin (100 µM) was superfused over Fura-2 AM and Rhod-2 AM-loaded cortical neurons on a cover slip in a recording chamber. Cytosol and mitochondrial Ca2+ levels from individual cells, indicated as Fura-2 AM and Rhod-2 AM fluorescence values, were normalized to values before drug treatment. In Fig. 3A and C, cytosolic and mitochondrial Ca2+ level rises spontaneously in control without glutamate treatment in cortical neuros exposed to external environment in recording chamber. Regarding causes of this phenomenon, several explanations are possible (i.e., decrease of temperature or O2 level, physical stress due to flow system through tubing, fluorometry-related chemical and light toxicities).
Fig. 3. The effects of nobiletin on glutamate-induced overload of cytosol and mitochondrial calcium in primary cortical neurons.

(A, C) Dual real-time imaging-based fluorometry of [Ca2+]c and [Ca2+]m were simultaneously conducted in the same neurons (see ‘METHODS’ for the detailed description). Fura-2 and Rohd-2 fluorescence values from individual cells were normalized to values before drug treatment. (B, D) Quantification of Fura-2 and Rohd-2 fluorescence values at the end of experiment for panel A and C. Values are the mean±S.E.M. *p<0.05, **p<0.01 as compared with the control group and #p<0.05 as compared with glutamate alone-treated group. N.S., not statistically significant.
Nobiletin markedly abolished glutamate-induced mitochondrial calcium overload in cortical neurons by 85.56±4.10% (Figs. 3C and D). However, nobiletin did not affect glutamate-induced [Ca2+]c increase (Figs. 3A and B). Taken together from Fig. 3, we demonstrated that nobiletin capable of evoking mild mitochondrial depolarization, potently blocked glutamate-induced mitochondrial calcium overload in primary cortical neurons.
Nobiletin attenuates glutamate-induced mitochondrial ROS generation
Mitochondrial Ca2+ overload can lead to ROS generation and oxidative stress although Ca2+ has no direct effect in electron transport chains or oxidation/reduction in mitochondria [20]. In this regard, we investigated whether nobiletin inhibits glutamate toxicity-induced mitochondrial ROS generation in intact primary cortical neurons (Figs. 4A and B). In addition, this issue was explored in a pure isolated mitochondria model (Fig. 4C).
Fig. 4. The effects of nobiletin on mitochondrial ROS generation in glutamate-stimulated cortical neurons and isolated brain mitochondria exposed to high concentration of Ca2+.

(A) Recording traces of mitochondrial superoxide using real-time imaging-based fluorometry with MitoSOX Red (see ‘METHODS’ for the detailed description). MitoSOX Red fluorescence values from individual cells were normalized to values before drug treatment shown as an arrow. (B) Quantification of MitoSOX Red fluorescence values at the end of experiment for panel A. (C) Effects of nobiletin on mitochondrial ROS generation were measured with DCF-DA indicator using a fluorescence microplate reader in an isolated brain mitochondrial model (See ‘METHODS’ for the detailed description). (D) Free radical scavenging activity of nobiletin was measured using DPPH assay. Values are the mean±S.E.M. **p<0.01, ***p<0.001 as compared with untreated controls and, #p<0.05, ##p<0.01, ###p<0.001 as compared with glutamate or CaCl2 (5 µM)-treated group.
In intact cortical neuros, mitochondrial superoxide levels were recorded and analyzed using real-time imaging-based fluorometry with MitoSOX Red. Glutamate (100 µM) in the presence or absence of nobiletin (100 µM) was superfused over MitoSOX Red-loaded cortical neurons on a cover slip in a recording chamber. Mitochondrial ROS levels from individual cells were normalized to values before drug treatment. Nobiletin almost blocked glutamate-induced mitochondrial ROS in intact cortical neurons (Figs. 4A and B).
In the isolated mitochondrial model (see ‘METHODS’ for more detailed description), experiments were conducted with metabolically active and functioning mitochondria as revealed in OCR data (Supplementary Fig. 1B). The cristae structure is well maintained in isolated mitochondria, as revealed in EM results (Supplementary Fig. 1A). High concentration of CaCl2 (5 µM) was treated to isolated mitochondria to mimic glutamate toxicity model in intact cortical neurons. Glutamate treatment does not work in isolated mitochondria since glutamate receptors are expressed in plasma membrane, not in mitochondria membrane. In isolated mitochondrial model as revealed in Fig. 4C, high concentration of Ca2+ was added in the medium instead of glutamate. Using a fluorescence microplate reader, mitochondrial ROS was measured using DCF-DA. Nobiletin significantly attenuated ROS generation in isolated pure mitochondria exposed to high concentration of Ca2+ in a dose (30, 50 and 100 mM)-dependent manner (Fig. 4C), consistent to ROS data from intact cortical neurons.
Data from DPPH free radical scavenging assay revealed that nobiletin did not exhibit direct free radical-scavenging activity in cell-free and mitochondria-free tube system. N-acetyl cysteine (NAC), a well-known anti-oxidant, was used as a positive control in DPPH assay (Fig. 4D). Taken together, results suggest that nobiletin markedly reduces mitochondrial ROS production against glutamate toxicity and mitochondrial calcium overload in intact primary cortical neurons (Figs. 4A and B) and pure isolated mitochondria (Fig. 4C) although it does not have free radical scavenging activity (Fig. 4D).
Nobiletin induces partial mitochondrial depolarization through K+ influx across mitochondrial inner membrane
Influx of K+ from cytosol into the mitochondrial matrix induces mild uncoupling, resulting in dissipation of ΔΨm [4]. The K+ ions are dominant cations among intracellular ions in resting neurons. Based on this background, we hypothesized that nobiletin-induced mild mitochondrial depolarization is mainly mediated by influx of K+ into mitochondria. To confirm correlation between mitochondrial K+ influx and nobiletin effect on ΔΨm, a pure isolated mitochondrial model was used. The effect of FCCP on ΔΨm was investigated as positive control using a fluorescence microplate reader in pure isolated mitochondrial system. When depolarization was induced by FCCP, TMRE intensity increased in dose-dependent manner (Fig. 5A). KCl (100 mM) was replaced with CsCl (100 mM) to remove K+ effects in the medium and thereby K+ dependence in nobiletin effect on ΔΨm was investigated. Figs. 5B and C demonstrated that nobiletin effects on mild mitochondrial depolarization were not shown in absence of K+ in the medium when KCl (100 mM) was replaced to CsCl (100 mM) in an isolated mitochondria model. Results suggest that nobiletin induces partial membrane depolarization by promoting influx of K+ across mitochondrial inner membrane. It could be suggested that K+ influx into mitochondrial matrix by nobiletin is probably mediated by mitochondrial K+ channels such as mitochondrial large-conductance Ca2+-activated K+ channels (mitoBKCa) and mitochondrial ATP-sensitive K+ channels (mitoKATP). Based on this idea, we further explored molecular targets of nobiletin in mitochondria using pharmacological inhibitors. Some preliminary findings related with mitoBKCa and mitoKATP are shown in ‘Supplementary Fig. 2’. However, further detailed studies should be conducted to determine exact molecular targets of nobiletin in mitochondria.
DISCUSSION
In this study, we demonstrated in primary cultured cortical neurons and pure brain mitochondria isolated from rat cortices that nobiletin prevents neurotoxic mitochondrial calcium overload, excess mitochondrial ROS and neuronal cell death through ΔΨm regulation. Results demonstrated that nobiletin evokes partial mitochondrial depolarization in intact cortical neurons (Figs. 2A and B) and isolated brain mitochondria (Figs. 5B and C). Nobiletin markedly attenuated mitochondrial calcium overload and ROS generation in glutamate (100 µM)-stimulated cortical neurons (Figs. 3 C and D, Fig. 4A) and isolated pure mitochondria exposed to high concentration (5 µM) of Ca2+ (Fig. 4C).
Excess exposure of glutamate evoked marked overload of both mitochondria Ca2+ and ROS. Nobiletin treatment significantly reduced these two reciprocal parameters, as shown in Fig. 3 and 4. The exact mechanism by which mitochondrial Ca2+ stimulates ROS generation inside mitochondria remains elusive. However, several plausible mechanisms have been proposed. Possible mechanisms include mitochondrial Ca2+-induced increase of metabolic rate, mitochondrial Ca2+-stimulated dissociation of cytochrome c and mitochondrial Ca2+-stimulated mitochondrial permeability transition pore opening with cytochrome c release [20]. Mitochondrial ROS can inversely affect Ca2+ dynamics and modulate Ca2+ surge. The reciprocal cross-talk between mitochondrial Ca2+ and ROS may results in a feedforward, self-amplified loop evoking subsequent cellular damage and death [21].
The main findings in this study may be mitochondrial K+ influx by nobiletin. We demonstrated that neuroprotective effect of nobiletin via mild mitochondrial depolarization is largely mediated by influx of K+ into mitochondria (Figs. 5B and C). Nobiletin effects on basal ΔΨm were completely abolished in K+-free medium on pure brain mitochondria isolated from rat cortices. Results suggest that K+ influx into mitochondrial matrix is critically involved in the nobiletin effect on ΔΨm. The mitochondrial K+ influx is probably mediated, at least in part, by activation of mitochondrial K+ channels. However, further detailed studies should be conducted to determine exact molecular targets of nobiletin in mitochondria.
There are several ion channels/transporters and electron transport chains on the inner membrane of mitochondria, which are proposed as possible molecular targets of nobiletin in mitochondria. It does not seem that mitochondrial calcium uniporter (MCU) as the major route for mitochondrial Ca2+ uptake is involved in nobiletin-induced ΔΨm depolarization. If nobiletin might attenuate glutamate-induced mitochondrial Ca2+ overload through inhibition of MCU, it should rather hyperpolarize mitochondrial membrane than depolarize it. Several studies have revealed that K+ channels/transporters are present in mitochondria inner membrane: mitoKATP, mitoBKCa, voltage-gated potassium channel Kv1.3, twin-pore domain TASK-3 potassium channels and K+/H+ exchangers. The mitoBKCa and mitoKATP channels are major mitochondrial K+ channels [22]. We demonstrated using pure brain mitochondria isolated from rat cortices that neuroprotective effect of nobiletin via mild mitochondrial depolarization is largely mediated by influx of K+ into mitochondria (Figs. 5B and C). Based on these results, it could be suggested that nobiletin-induced mitochondrial K+ influx is probably mediated, at least in part, by activation of by mitochondrial K+ channels. Based on this idea, we further explored mitochondrial targets of nobiletin. Some preliminary findings related with mitoBKCa and mitoKATP are shown in ‘Supplementary Fig. 2’. We investigated here whether nobiletin-induced ΔΨm depolarization is blocked by iberiotoxin and 5-hydroxydecanoate (5-HD) as well-known inhibitors of mitoBKCa and mitoKATP channels, respectively. As shown in Supplementary Fig. 2, our preliminary findings indicated that nobiletin-induced ΔΨm depolarization was significantly inhibited in the group treated with iberiotoxin (10 nM). These supplementary results could suggest that nobiletin may induce neuroprotective ΔΨm depolarization by promoting the influx of K+ into mitochondria through activation of mitoBKCa. However, further detailed studies should be conducted to determine exact molecular targets of nobiletin in mitochondria.
Among many flavonoids, naringenin, one of flavanones abundant in genus Citrus such as grapefruit and orange, has been widely studied recently. Activation of mitoBKCa is involved in cardioprotective mechanism of naringenin against myocardial ischemia/reperfusion [23]. Naringenin is proposed as one of BKCa channel openers in vascular smooth muscle cells [24]. An electrophysiological study recently revealed that channel activities of mitoKATP and mitoBKCa were enhanced after treatment of 10 µM naringenin in single channel study using mitoplasts isolated from primary human dermal fibroblast cells [25]. These recent reports focus special attention on intracellular pathways mediated by Citrus flavonoids regarding their beneficial effects on several physiological and pathophysiological conditions.
In conclusion, we reveal here that nobiletin evokes partial mitochondrial depolarization in intact cortical neurons and isolated brain mitochondria, and thereby prevents neurotoxic mitochondrial calcium overload, excess mitochondrial ROS and neuronal cell death. Furthermore, K+ influx into mitochondrial matrix is critically involved in the nobiletin effect on ΔΨm. In addition, it could be suggested that the mitochondrial K+ influx is probably mediated, at least in part, by activation of mitochondrial K+ channels. All these findings reveal a beneficial role of nobiletin-induced partial mitochondrial depolarization in neuroprotection, which is similar to ischemic pre-conditioning (IPC) in endogenous mechanism of cardioprotection. Therefore, pharmacological manipulation of ΔΨm through novel substances (i.e., nobiletin) could be a promising strategy to prevent neuronal cell death against brain insults.
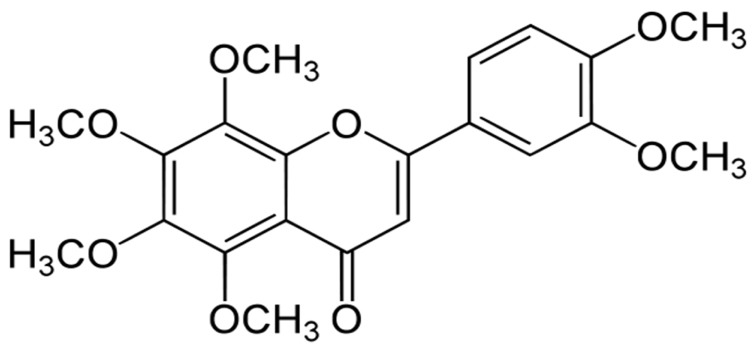
Fig. 1. Chemical skeletal structure of nobiletin (5,6,7,8,3′,4′-hexamethoxyflavone).
ACKNOWLEDGMENTS
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (NRF-2013R1A1A2013585 and NRF-2015R1D1A1A01061010).
Footnotes
Author contributions: J.H.L., K.A., J.J.W., S.C.J. and Y.J.C. performed the experiments and analyzed the data. S.C.J. and D.B.P. contributed for discussion. S.J.K. and S.H.H. made natural materials. H.W.K. and I.J.R. performed EM experiments. S.Y.E. supervised and coordinated the study.
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
SUPPLEMENTARY MATERIALS
Supplementary data including two figures can be found with this article online at http://pdf.medrang.co.kr/paper/pdf/Kjpp/Kjpp022-03-09-s001.pdf.
Morphological and metabolic analysis of mitochondria isolated from rat cortices.
The effects of mitochondrial K+ channel inhibitors on nobiletin-induced ΔΨm changes in intact cortical neurons.
References
- 1.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 2.Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464:111–121. doi: 10.1007/s00424-012-1112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 4.Ishida H, Hirota Y, Genka C, Nakazawa H, Nakaya H, Sato T. Opening of mitochondrial K(ATP) channels attenuates the ouabaininduced calcium overload in mitochondria. Circ Res. 2001;89:856–858. doi: 10.1161/hh2201.100341. [DOI] [PubMed] [Google Scholar]
- 5.Sanz-Blasco S, Valero RA, Rodríguez-Crespo I, Villalobos C, Núñez L. Mitochondrial Ca2+ overload underlies Ab oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One. 2008;3:e2718. doi: 10.1371/journal.pone.0002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valero RA, Senovilla L, Núñez L, Villalobos C. The role of mitochondrial potential in control of calcium signals involved in cell proliferation. Cell Calcium. 2008;44:259–269. doi: 10.1016/j.ceca.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Wu JJ, Cui Y, Yang YS, Jung SC, Hyun JW, Maeng YH, Park DB, Lee SR, Kim SJ, Eun SY. Mild mitochondrial depolarization is involved in a neuroprotective mechanism of citrus sunki peel extract. Phytother Res. 2013;27:564–571. doi: 10.1002/ptr.4745. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto Y, Shioda N, Han F, Moriguchi S, Nakajima A, Yokosuka A, Mimaki Y, Sashida Y, Yamakuni T, Ohizumi Y, Fukunaga K. Nobiletin improves brain ischemia-induced learning and memory deficits through stimulation of CaMKII and CREB phosphorylation. Brain Res. 2009;1295:218–229. doi: 10.1016/j.brainres.2009.07.081. [DOI] [PubMed] [Google Scholar]
- 9.Yabuki Y, Ohizumi Y, Yokosuka A, Mimaki Y, Fukunaga K. Nobiletin treatment improves motor and cognitive deficits seen in MPTPinduced Parkinson model mice. Neuroscience. 2014;259:126–141. doi: 10.1016/j.neuroscience.2013.11.051. [DOI] [PubMed] [Google Scholar]
- 10.Onozuka H, Nakajima A, Matsuzaki K, Shin RW, Ogino K, Saigusa D, Tetsu N, Yokosuka A, Sashida Y, Mimaki Y, Yamakuni T, Ohizumi Y. Nobiletin, a citrus flavonoid, improves memory impairment and Ab pathology in a transgenic mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2008;326:739–744. doi: 10.1124/jpet.108.140293. [DOI] [PubMed] [Google Scholar]
- 11.Nagase H, Omae N, Omori A, Nakagawasai O, Tadano T, Yokosuka A, Sashida Y, Mimaki Y, Yamakuni T, Ohizumi Y. Nobiletin and its related flavonoids with CRE-dependent transcription-stimulating and neuritegenic activities. Biochem Biophys Res Commun. 2005;337:1330–1336. doi: 10.1016/j.bbrc.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Cui Y, Wu J, Jung SC, Park DB, Maeng YH, Hong JY, Kim SJ, Lee SR, Kim SJ, Kim SJ, Eun SY. Anti-neuroinflammatory activity of nobiletin on suppression of microglial activation. Biol Pharm Bull. 2010;33:1814–1821. doi: 10.1248/bpb.33.1814. [DOI] [PubMed] [Google Scholar]
- 13.Choi SY, Hwang JH, Ko HC, Park JG, Kim SJ. Nobiletin from citrus fruit peel inhibits the DNA-binding activity of NF-kappaB and ROS production in LPS-activated RAW 264.7 cells. J Ethnopharmacol. 2007;113:149–155. doi: 10.1016/j.jep.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 14.Eun SY, Jung SJ, Park YK, Kwak J, Kim SJ, Kim J. Effects of capsaicin on Ca2+ release from the intracellular Ca2+ stores in the dorsal root ganglion cells of adult rats. Biochem Biophys Res Commun. 2001;285:1114–1120. doi: 10.1006/bbrc.2001.5272. [DOI] [PubMed] [Google Scholar]
- 15.Scaduto RC, Jr, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J. 1999;76:469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iglesias-González J, Sánchez-Iglesias S, Beiras-Iglesias A, Soto-Otero R, Méndez-Álvarez E. A simple method for isolating rat brain mitochondria with high metabolic activity: effects of EDTA and EGTA. J Neurosci Methods. 2013;213:39–42. doi: 10.1016/j.jneumeth.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Blattner JR, He L, Lemasters JJ. Screening assays for the mitochondrial permeability transition using a fluorescence multiwell plate reader. Anal Biochem. 2001;295:220–226. doi: 10.1006/abio.2001.5219. [DOI] [PubMed] [Google Scholar]
- 18.Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C, Kim H, Kim K, Sesaki H, Rhyu IJ, Kim H, Sun W. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat Commun. 2017;8:15754. doi: 10.1038/ncomms15754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui Y, Park JY, Wu J, Lee JH, Yang YS, Kang MS, Jung SC, Park JM, Yoo ES, Kim SH, Ahn Jo S, Suk K, Eun SY. Dieckol attenuates microglia-mediated neuronal cell death via ERK, Akt and NADPH oxidase-mediated pathways. Korean J Physiol Pharmacol. 2015;19:219–228. doi: 10.4196/kjpp.2015.19.3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- 21.Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci (Landmark Ed) 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szewczyk A, Jarmuszkiewicz W, Kunz WS. Mitochondrial potassium channels. IUBMB Life. 2009;61:134–143. doi: 10.1002/iub.155. [DOI] [PubMed] [Google Scholar]
- 23.Testai L, Martelli A, Marino A, D'Antongiovanni V, Ciregia F, Giusti L, Lucacchini A, Chericoni S, Breschi MC, Calderone V. The activation of mitochondrial BK potassium channels contributes to the protective effects of naringenin against myocardial ischemia/ reperfusion injury. Biochem Pharmacol. 2013;85:1634–1643. doi: 10.1016/j.bcp.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Saponara S, Testai L, Iozzi D, Martinotti E, Martelli A, Chericoni S, Sgaragli G, Fusi F, Calderone V. (+/-)-Naringenin as large conductance Ca2+-activated K+ (BKCa) channel opener in vascular smooth muscle cells. Br J Pharmacol. 2006;149:1013–1021. doi: 10.1038/sj.bjp.0706951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bednarczyk P, Kicinska A, Jarmuszkiewicz W, Debowska R, Szewczyk A. Flavonoids as natural modulators of mitochondrial potassium channel. Biophys J. 2017;112:405a–406a. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Morphological and metabolic analysis of mitochondria isolated from rat cortices.
The effects of mitochondrial K+ channel inhibitors on nobiletin-induced ΔΨm changes in intact cortical neurons.