

Abstract
Background
In this study, we investigated the role of artemin, a member of the glial cell‐derived neurotrophic factor of ligands, in the malignant phenotype of lung cancer.
Methods
Artemin expression was examined in various types of lung cancer and normal lung tissues, as well as in lung cancer cell lines by immunohistochemistry and semi‐quantitative PCR. Functional studies were performed using artemin overexpression or knockdown vectors in lung cancer cell lines. Methyl thiazolyl tetrazolium, flow cytometry, wound healing, and transwell assays were conducted to evaluate the contribution of artemin on tumor cell proliferation, migration, and invasion.
Results
Artemin is broadly expressed in lung cancer tissues, and is associated with tumor staging. Overexpression of artemin in NL9980 large cell lung cancer cells increased proliferating cells and enhanced migrating capability in wound healing and transwell assays, as well as demonstrating enhanced invasion capability. Silencing artemin in LTEP‐α‐2 adenocarcinoma cell lines decreased cellular proliferation, migration, and invasion capabilities.
Conclusion
Artemin could promote the proliferation and invasiveness of lung cancer cells in vitro and therefore could be a new potential target to combat lung cancer.
Keywords: Artemin, lung neoplasms, metastasis, proliferation
Introduction
Lung cancer is the top cause of cancer‐related mortality.1 Lung adenocarcinoma is one of the major pathological types of lung cancer and is predominantly driven by EGFR mutations. The discovery of EGFR‐tyrosine kinase inhibitors (TKIs), such as gefitinib or erlotinib, has contributed to the treatment of lung adenocarcinoma. However, the efficacy of EGFR‐targeted TKIs has not been satisfactory. Patients with EGFR‐mutated adenocarcinoma develop acquired resistance to TKIs, with clinical progression after a median of 10–16 months.2 Although next generation EGFR‐TKIs, such as osimertinib, have been investigated to help circumvent resistance, other mechanisms involved in EGFR TKI‐resistance, like MET pathway alteration or epithelial‐to‐mesenchymal transition, still hamper clinical efficacy.3, 4, 5, 6 Thus, the identification of novel oncogenic receptors other than EGFR may be useful to establish new TKIs and bypass the limitations in EGFR treatment.
Artemin was first discovered as a member of the glial cell‐derived neurotrophic factor (GDNF) family of ligands (GFLs) including GDNF, NRTN, and PSPN,7 which plays key roles in promoting survival, differentiation, and chemotaxis of neurons and epithelial cells.8, 9 The GFLs share a cysteine knot motif that contains seven conserved cysteine residues. Some GFLs, like NRTN or artemin, support a broad spectrum of targeted neurons, while PSPN only promotes the survival of motor and dopaminergic neurons.7 GFL receptors work in a unique multiple‐component manner, consisting of GFR α 1‐4 as a high affinity ligand binding element and the RET receptor, a receptor tyrosine kinase that functions as the signaling component.7 GDNF, NRTN, ARTN, and PSPN each use GFRα1, GFRα2, GFRα3, and GFRα4, respectively, yet cross‐talk may also occur with other combinations, such as GDNF with GFRα3 or GFRα2, artemin with GFRαα1, or NRTN and ARTN with GFRα1.7, 10 Upon interaction of GFLs with GFR receptors, the RET receptor is recruited to the ligand‐receptor complex and activated by auto‐phosphorylation to subsequently phosphorylate and activate downstream targets in the signaling transduction pathway. As a proto‐oncogene, RET is crucial for kidney and neural crust formation; knockout mice studies of Ret inactivation showed renal agenesis,11, 12 and another RET plus GFRα1 knockout mouse model exhibited neurotic death and aganglionosis of the intestinal track, mimicking Hirschsprung's disease.13
Herein, we examined the role of artemin, a GFL family member, in the malignant phenotype of lung cancer. We showed that artemin expression in lung cancer tissues was correlated with patient staging and enhanced proliferation, migration, and invasion in lung cancer cell lines.
Methods
Tissue specimens and immunohistochemical analysis
Tissue samples were obtained from 50 lung cancer patients and 10 non‐carcinoma patients from the Department of Lung Cancer Surgery, Tianjin Medical University General Hospital (Tianjin, China) between January 2010 and December 2016. All patients provided informed consent. The study was conducted in abidance with the Helsinki Declaration and approved by the institutional ethics board.
Paraffin‐embedded specimens were examined by immunohistochemical analysis. Briefly, the tissues were embedded in paraffin and sliced into 5 μm thick tissue slides. Samples were deparaffinized in xylene, rehydrated in a series of graded ethanol solutions, and heated in a LabWare microwave oven (Foshnan City, Guangdong, China) in 0.01 M sodium citrate antigen‐retrieving buffer (pH 6.0). Samples were then incubated with an anti‐artemin rabbit polyclonal antibody (AB178434, ABCAM, Cambridge, UK) in a dilution of 1:100. The staining intensity of samples was examined using a Nikon microscope (Nikon Instruments Inc., Tokyo, Japan).
Construction of artemin overexpression and short‐hairpin RNA vectors
The coding sequence of artemin messenger RNA (mRNA)‐NM_057091.2 was cloned with the following primer pairs: forward: atggaacttggacttggagg; and reverse: tcagcccaggcagccgcaggc. The sequence was digested by BglII and BamHI and then cloned into the eukaryotic overexpression vector pEGFP‐C1 (pEGFP‐artemin; in Figures, ARTN refers to the group transfected with pEGFP‐artermin). An empty vector served as the control. Three artemin short‐hairpin RNAs (shRNAs) were designed to knock down artemin: artemin‐homo‐5 (ARTN‐5): 5′‐GAACTTGGACTTGGAGGCCTCC‐3′; artemin‐homo‐434 (ARTN‐434): 5′‐GTGCGTTTCCGCTTCTGCAGCC‐3′; and artemin‐homo‐587 (ARTN‐587): 5′‐GTCTCCTTCATGGACGTCAACC‐3′. These sequences were cloned into the pGPU6/GFP/Neo‐shRNA plasmid, and the empty shRNA plasmid was used as a negative control (NC).
Cell culture
Human non‐small cell lung cancer cell lines LTEP‐α‐2 (adenocarcinoma), PC‐9 (adenocarcinoma), SPC‐A‐1 (adenocarcinoma), A549 (adenocarcinoma), NL9981 (large cell carcinoma), L9981 (large cell carcinoma), YTMLC‐9 (squamous carcinoma), and 95D (huge cell lung cancer) were cultured in RPMI‐1640 supplemented with 10% fetal bovine serum (FBS) (Gibco, Shanghai, China) at 37°C in a humidified 5% CO2 atmosphere. For cell transfection, cells were grown to 80% confluency (overexpression vector transfection) or 50% confluency (shRNA plasmid transfection) and transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and Opti‐MEM (Gibco), according to the manufacturer's instructions.
RNA isolation and PCR
Total RNA was isolated using Trizol (Invitrogen) and reverse transcribed by M‐MLV (Takara Bio Inc., Tokyo, Japan). For semi‐quantitative PCR, the RNA concentrations were determined using Nanodrop 2000 (Thermo Fisher, Scientific, Waltham, MA, USA) and standardized, and then the complementary DNA were amplified using DNA engine PCR (Bio‐Rad, Hercules, CA, USA). The PCR products were electrophoresed and captured using the ChemiDoc XRS System (Bio‐Rad). For real‐time PCR, the reverse transcribed complementary DNA was amplified using the Applied Biosystems 7900 real‐time thermal cycler (Foster City, CA, USA). GAPDH was selected as a reference gene and relative fold change was calculated using the ∆∆Ct method.14
Western blot
Cells were harvested, washed with phosphate buffered saline (PBS) twice and incubated at 98°C in radioimmunoprecipitation assay lysis buffer (Beyotime, Beijing, China). The protein extracts were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked with Quickblock Primary Antibody Dilution Buffer (Beyotime, Beijing, China) for one hour and then incubated with anti‐artemin rabbit polyclonal antibody (AB178434, ABCAM) or anti‐GAPDH antibody (KM9002T, Sungene, Tianjin, China) as a control.
Methyl thiazolyl tetrazolium proliferation assay
Cells were seeded in 96‐well plates (2 × 103 cells/well), and cells with only Lipofectamine 2000 were used as controls. After 24, 72, 96, 120, and 168 hours of incubation, methyl thiazolyl tetrazolium solution (5 mg/mL in PBS solution) was added at 20 μL per well for four hours. Formazan products were solubilized by acidic isopropanol solution, and the optic density was measured using SpectraMax M2 (Molecular Devices, San Jose, CA, USA) at 570 nm. Optic density values of each group were standardized by the values of the control group.
Flow cytometry
Cells were seeded at a density of 2 × 105 cells/well in six‐well plates. At 48 hours after seeding when cell confluence reached 75%, cells were collected and fixed with 70% ethanol at 4°C for at least one night. The cells were then washed and stained by 0.5 mL PI/RNase Staining Buffer for approximately 20 minutes and then measured using a Cell Lab Quanta SC (Beckman Coulter, Fullerton, CA, USA).
Wound healing assay
Cells were seeded in six‐well plates. After cells reached 90–100% confluence, three scratches were made with sterilized 1 mL pipette tips and the 10% FBS‐containing medium was replaced with 0.5% FBS‐containing medium. Photographs were obtained at 0 and 72 hours under an inverted microscope (TE2000, Nikon, Tokyo, Japan).
Transwell migration and invasion assays
Cell migration and invasion assays were performed using 24‐well transwell chambers (8 mm pore size, Corning Life Sciences, Tewksbury, MA, USA) without or with 1:8 diluted Matrigel (Invitrogen). Cells (1 × 104) were seeded in the upper chambers of the wells in 200 μL FBS‐free medium, and the lower chambers contained 0.6 mL 10% FBS‐containing medium. The plate was incubated for 12 hours for migration assays and 24 hours for invasion assays. The upper chambers were wiped with PBS moistened swabs and fixed with 95% alcohol for 15 to 20 minutes. Cells were stained with hematoxylin for 10 minutes, and the stained cells left in the membrane were counted using an inverted microscope (TE2000, Nikon) at a magnification of 200×.
Colony formation assay
Low melting point agarose (1.2%) was mixed with 2 × RPMI‐1640 medium into a 0.6% agarose lower layer in the six‐well plates. Then, 0.5 mL of 0.6% low melting point agarose was mixed with 0.5 mL cell suspension (1 × 103 cells per well) and seeded on the lower agarose layer. The plates were incubated for 10 days, and the colonies of each well were counted.
Statistical analysis
SPSS version 21.0 (IBM Corp., Armonk, USA) was used for data analysis. A chi‐square test was used to compare the differences in artemin levels between the two groups. Hierarchical studies and chi‐square tests were used to analyze the correlation between artemin level and clinical characteristics. P < 0.05 was considered statistically significant.
Results
ARTN expression in human lung cancer and cell lines
To explore the relationship between ARTN and lung cancer, we explored artemin expression levels in a panel of 50 human normal lung and 10 lung cancer tissues, as well as in 9 lung cancer cell lines. We found a significant difference between the artemin levels in the lung cancer and normal lung tissue groups (P = 0.001 by chi‐square test) (Fig 1). We then analyzed the correlation between artemin level and clinical characteristics. Hierarchical studies and chi‐square tests indicated that artemin levels varied in different groups of lymph node metastasis, as well as groups of tumor node metastasis staging. We used semi‐quantitative PCR to determine artemin mRNA levels in various cell lines and found that the artemin level was relatively high in LTEP‐α‐2, PC‐9, SPCA‐1, and YTMLC‐9 cells; moderate in 95D and A549 cells; and low in NL9980 and L9981 cell lines. We selected the NL9980 and LTEP‐α‐2 lines for further artemin overexpression and downregulation studies, because of their relatively low and high base artemin levels, respectively.
Figure 1.
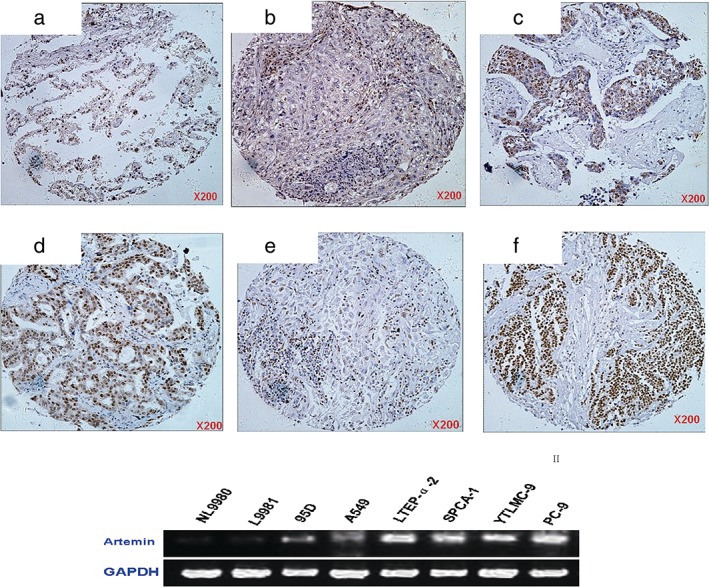
Artemin level in normal lung and lung cancer tissues, as well as in lung cancer cell lines. Immunohistochemical staining of artemin in the following samples: (a) normal lung tissue; (b) precancerous lesion; (c) lung squamous cell carcinoma; (d) lung adenocarcinoma; (e) large cell lung carcinoma; and (f) small cell lung carcinoma. Semi‐quantitative PCR of artemin in multiple cell lines, including NL9980, L9981, 95D, A549, LTEP‐α‐2, SPCA‐1, YTMLC‐9, and PC‐9.
Confirmation of effectiveness of pEGFP‐artemin and sh‐artemin vectors
We next confirmed the effectiveness of pEGFP‐artemin overexpression and artemin shRNA knockdown vectors both on RNA and protein levels by real‐time PCR and Western blot, respectively. As illustrated in Figure 2, the artemin level in NL9980 cells transfected with pEGFP‐artemin was highest compared with both controls, both in mRNA and protein levels. We examined three different shRNAs and found that the artemin‐homo‐587‐transfected group showed the most effective knockdown compared with the other shRNAs and the two control groups. These experiments confirmed the effectiveness of these constructs and our results indicate that the sh‐artemin (shARTN) vector artemin‐homo‐587 is the optimal knockdown vector for further studies.
Figure 2.
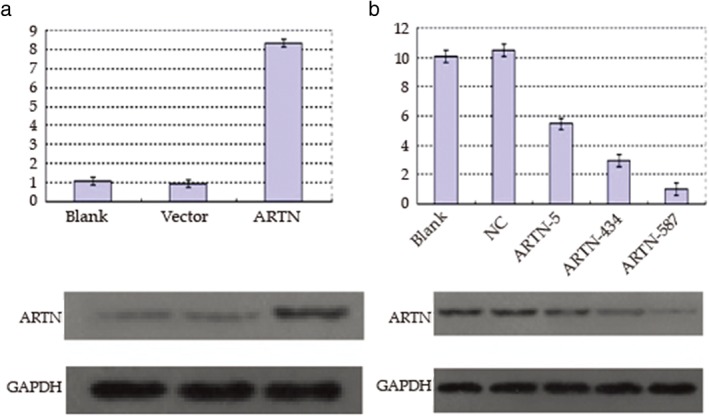
Confirmation of the artemin overexpression plasmid and silencing vectors for artemin. Relative artemin messenger RNA (mRNA) (upper) and protein level (lower) in (a) NL9980 cells and (b) LTEP‐α‐2 cells transfected as indicated and examined by real‐time PCR and Western blot, respectively.NC, negative control.
Effect of artemin on proliferation of lung cancer cell lines
We performed methyl thiazolyl tetrazolium assays to determine the potential effect of artemin on lung cancer cell proliferation. The shARTN group in LTEP‐α‐2 cells showed reduced proliferation compared with both control groups, in a time‐dependent manner (Fig 3d). In comparison, the pEGFP‐artemin group in NL9980 cells showed increased proliferation compared with both control groups. These results indicate that artemin expression promotes the proliferation of tumor cells.
Figure 3.
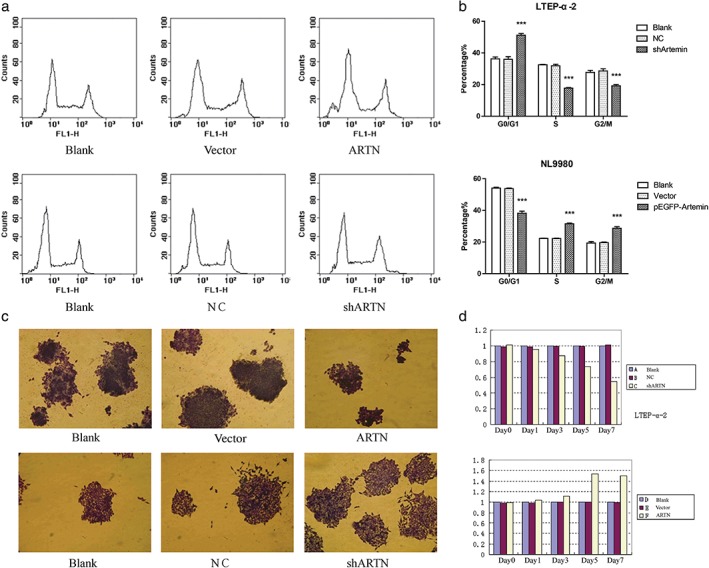
Artemin promotes proliferation of lung cancer cell lines. (a) NL9980 cells were transfected with pEGFP‐artemin or empty vector or untransfected for the indicated time. LTEP‐α‐2 cells were transfected with shARTN‐vector, negative control (NC) empty vector or untransfected. Cells were stained with propidium iodide and examined by fluorescence activated cell sorting. (b) Quantification of each cell phase is shown in histograms. Blank, NC, shARTN. Blank, Vector, pEGFP‐Artemin. (c) Effect of forced expression or knockdown of artemin on colony formation in soft agar. (d) Methyl thiazolyl tetrazolium assays in NL9980 and LTEP‐α‐2 cells transfected as indicated. A Blank, B NC, C shARTN, D Blank, E vector, F ARTN.
To further examine the growth advantage of artemin expression in lung cancer, we examined the effects of artemin on cell cycle progression. As shown in Figure 4b, the percentage of cells in the G0/G1 phase in the shARTN group was significantly higher than in the NC and blank groups, and the percentage of cells in S and G2/M phases were significantly lower in the shARTN group than in the NC and blank groups. In comparison, the portion of G0/G1 phase cells in the pEGFP‐artemin group was significantly lower than the vector and blank groups (Fig 3a,b). As the S and G2/M phases make up the overall portion of proliferation cells, this indicates that artemin level is positively correlated with proliferation phase.
Figure 4.
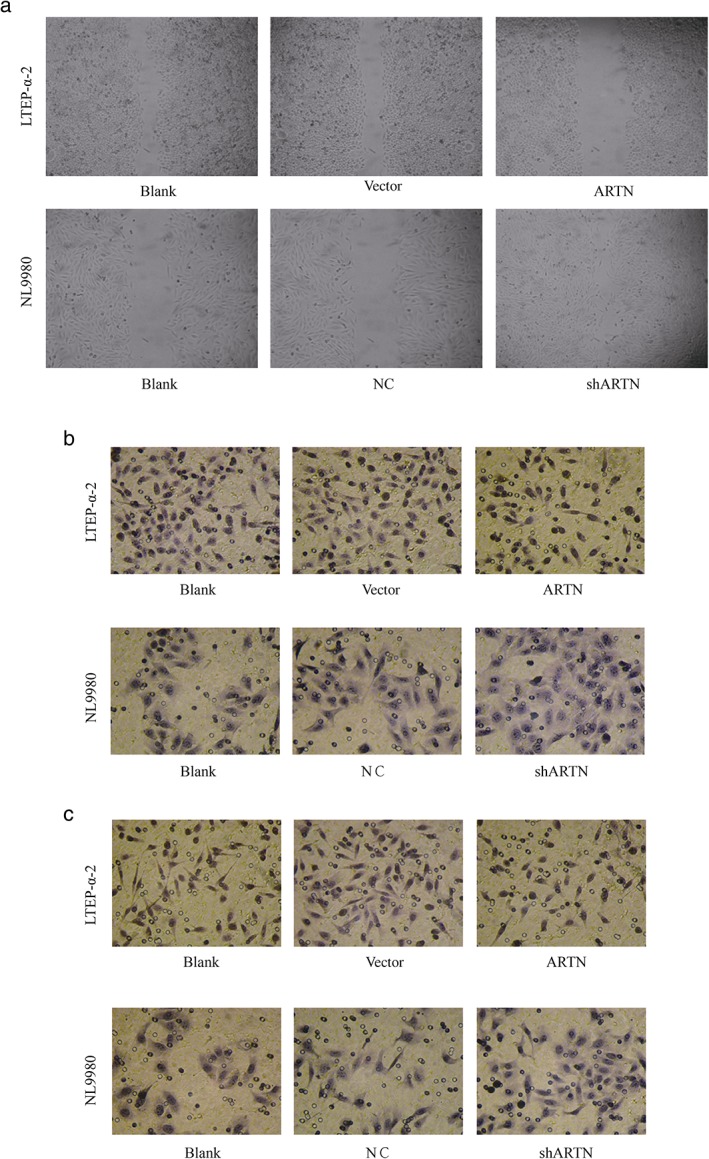
Artemin is positively correlated with metastatic capability of lung cancer cell lines. (a) Wound healing assay. NL9980 cells were transfected with pEGFP‐artemin or empty vector or untransfected, and LTEP‐α‐2 cells were transfected with shARTN‐vector or negative control (NC) empty vector. Cells were seeded in six‐well plates overnight and scratches were made to the cell layer. (b) Transwell migration and (c) Matrigel invasion assays in cells transfected as indicated.
We also examined colony formation by soft agar colony formation assay. As shown in Fig 3c, colony formation of LTEP‐α‐2 cells was reduced when artemin was knocked down, and promoted in NL9980 cells upon artemin overexpression. These results demonstrate that artemin is positively correlated with tumor cell proliferation in terms of both cell cycle and colony formation ability.
Effect of artemin on metastatic capacity of lung cancer cell lines
Wound healing and transwell migration and invasion assays were conducted to evaluate the effects of artemin on migration and invasion capabilities. The ARTN‐transfected LTEP‐α‐2 cells showed slower wound healing ability compared with the control groups, while the upregulated pEGFP‐artemin‐expressing NL9980 cells showed faster wound healing ability compared with control groups (Fig 4a). Similarly, in both transwell migration (Fig 4b) and invasion (Fig 4b) assays, fewer shARTN LTEP‐α‐2 cells reached the bottom layer of the upper chambers compared with the control groups, while more artemin‐upregulated NL9980 cells reached the bottom layer compared with the control groups. These findings indicate that artemin also positively promotes migration and invasion ability.
Discussion
Several recent studies have investigated the oncogenic role of the neurotrophic factor artemin. Artemin was detected in nearly 65% of mammary carcinoma patients, as well as in several mammary carcinoma cell lines.15 In vitro studies of mammary carcinoma confirmed that the forced expression of artemin leads to increased anchorage‐independent growth and capabilities of colony formation, migration, and invasion; similarly, in vivo xenograft models also demonstrated an association between artemin and tumor proliferation and invasion.15 Further studies have gradually uncovered the mechanism of artemin in mammary carcinoma: artemin is positively correlated with estrogen receptor (ER) and forced expression of artemin in ER‐positive mammary breast carcinoma increases the expression of ER transcription and promoted estrogen‐independent growth and tamoxifen resistance both in cell lines and xenograft models, mediated by increased BCL‐2 expression.16 Artemin is also expressed in ER‐negative mammary carcinoma, and forced expression of artemin resultes in enhanced invasion and mesenchymal characteristics, which are mediated by TWIST1 and could be abrogated by TWIST1 knockdown.17 Artemin also increases angiogenesis in ER‐negative mammary carcinoma through activation of the AKT‐TWIST‐VEGF axis.18
Artemin has been investigated in other tumors, such as pancreatic and endometrial carcinoma. In a myenteric plexus‐pancreas tissue model, nerve growth factor and artemin levels were higher in pancreatic cancer and tissues adjacent to normal pancreas tissue, and closely associated with intrapancreatic neuropathy, which clinically results in pancreatic pain.19 Artemin, as well as its receptor GFRα3, could promote pancreatic cancer cell motility and invasiveness, the mechanism of which may be through increased MMP‐2 and decreased E‐cadherin levels.20 Artemin also promotes the oncogenicity and invasiveness of endometrial carcinoma cells21 In addition, it reduces sensitivity to doxorubicin and paclitaxel, partially by enhancing CD24 expression.22
In this study, we analyzed the potential relationship between artemin and lung cancer malignancy. The artemin level was significantly higher in 50 lung cancer specimens compared with 10 normal lung cases and positively correlated with malignant characteristics after stratification by tumor node metastasis staging and lymph node metastasis. Artemin showed varied levels of expression across lung cancer cell lines. Artemin overexpression and knockdown studies were performed to evaluate the effects on proliferation‐related and metastasis‐related capabilities. Knockdown of artemin resulted in a decreased proliferation ratio and colony formation compared with the control groups. The opposite result was observed with artemin overexpression. Artemin was also positively correlated with tumor invasion and migration capacities. In wound healing and transwell migration assays, artemin was also positively correlated with faster wound healing ratio and increased cell migration. Similarly, the pEGFP‐artemin group showed increased cell invasion compared with the control group.
Our results show a positive correlation between artemin and malignant tumor characteristics, such as proliferation and metastasis, consistent with results reported by Tang et al., in which artemin stimulated BCL2 expression was confirmed to be one explanation. The BCL‐2 protein family is critical in tumor development and survival,23 and BCL‐2 functions as an oncogene23 to antagonize all major types of cell death, including apoptosis, necrosis, and autophagy.24
In brief, our study and a number of others have confirmed the oncogenic property of artemin in various kinds of tumors and shed light on the field of TKI design. More mechanism studies are needed to explore the downstream effect of artemin in the modulation of tumor biology.
Disclosure
No authors report any conflict of interest.
Acknowledgments
This work was supported by grants from the Tianjin Municipal Science and Technology Commission Fund (14JCYBJC28400), the Tianjin Municipal Education Commission Fund (20120127), and the Tianjin Municipal Health Bureau Fund (2011KZ106).
References
- 1. Jemal A, Siegel R, Ward E et al Cancer statistics, 2008. CA Cancer J Clin 2008; 58: 71–96. [DOI] [PubMed] [Google Scholar]
- 2. Mayo C, Bertran‐Alamillo J, Molina‐Vila MÁ, Gimenez‐Capitán A, Costa C, Rosell R. Pharmacogenetics of EGFR in lung cancer: Perspectives and clinical applications. Pharmacogenomics 2012; 13: 789–802. [DOI] [PubMed] [Google Scholar]
- 3. Suda K, Murakami I, Katayama T et al Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res 2010; 16: 5489–98. [DOI] [PubMed] [Google Scholar]
- 4. Onitsuka T, Uramoto H, Nose N et al Acquired resistance to gefitinib: The contribution of mechanisms other than the T790M, MET, and HGF status. Lung Cancer 2010; 68: 198–203. [DOI] [PubMed] [Google Scholar]
- 5. Miyoshi S, Kato T, Katayama H et al A case of EGFR mutant lung adenocarcinoma that acquired resistance to EGFR‐tyrosine kinase inhibitors with MET amplification and epithelial‐to‐mesenchymal transition. Onco Targets Ther 2015; 8: 783–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bean J, Brennan C, Shih JY et al MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007; 104: 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baloh RH, Tansey MG, Lampe PA et al Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3‐RET receptor complex. Neuron 1998; 21: 1291–302. [DOI] [PubMed] [Google Scholar]
- 8. Esseghir S, Todd SK, Hunt T et al A role for glial cell derived neurotrophic factor induced expression by inflammatory cytokines and RET/GFR alpha 1 receptor up‐regulation in breast cancer. Cancer Res 2007; 67: 11732–41. [DOI] [PubMed] [Google Scholar]
- 9. Plaza‐Menacho I, Mologni L, McDonald NQ. Mechanisms of RET signaling in cancer: Current and future implications for targeted therapy. Cell Signal 2014; 26: 1743–52. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev 2001; 12: 361–73. [DOI] [PubMed] [Google Scholar]
- 11. Schuchardt A, D'Agati V, Larsson‐Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994; 367: 380–3. [DOI] [PubMed] [Google Scholar]
- 12. Schuchardt A, D'Agati V, Pachnis V, Costantini F. Renal agenesis and hypodysplasia in ret‐k‐ mutant mice result from defects in ureteric bud development. Development 1996; 122: 1919–29. [DOI] [PubMed] [Google Scholar]
- 13. Uesaka T, Nagashimada M, Yonemura S, Enomoto H. Diminished Ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in mice. J Clin Investig 2008; 118: 1890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2‐△△Ct method. Methods 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- 15. Kang J, Perry JK, Pandey V et al Artemin is oncogenic for human mammary carcinoma cells. Oncogene 2009; 28: 2034–45. [DOI] [PubMed] [Google Scholar]
- 16. Kang J, Qian PX, Pandey V et al Artemin is estrogen regulated and mediates antiestrogen resistance in mammary carcinoma. Oncogene 2012; 31: 402 (Published erratum appears in ) Oncogene 2010;29: 3228–40. [DOI] [PubMed] [Google Scholar]
- 17. Banerjee A, Wu ZS, Qian P et al ARTEMIN synergizes with TWIST1 to promote metastasis and poor survival outcome in patients with ER negative mammary carcinoma. Breast Cancer Res 2011; 13: R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Banerjee A, Wu ZS, Qian PX et al ARTEMIN promotes de novo angiogenesis in ER negative mammary carcinoma through activation of TWIST1‐VEGF‐A signalling. PLoS One 2012; 7: e50098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ceyhan GO, Schäfer KH, Kerscher AG et al Nerve growth factor and artemin are paracrine mediators of pancreatic neuropathy in pancreatic adenocarcinoma. Ann Surg 2010; 251: 923–31. [DOI] [PubMed] [Google Scholar]
- 20. Meng LX, Chi YH, Wang XX et al Neurotrophic artemin promotes motility and invasiveness of MIA PaCa‐2 pancreatic cancer cells. Asian Pac J Cancer Prev 2012; 13: 1793–7. [DOI] [PubMed] [Google Scholar]
- 21. Pandey V, Qian PX, Kang J et al Artemin stimulates oncogenicity and invasiveness of human endometrial carcinoma cells. Endocrinology 2010; 151: 909–20 (Published erratum appears in Endocrinology 2012;153: 540). [DOI] [PubMed] [Google Scholar]
- 22. Pandey V, Jung Y, Kang J et al Artemin reduces sensitivity to doxorubicin and paclitaxel in endometrial carcinoma cells through specific regulation of CD24. Transl Oncol 2010; 3: 218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adams JM, Cory S. The Bcl‐2 apoptotic switch in cancer development and therapy. Oncogene 2007; 26: 1324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yip KW, Reed JC. Bcl‐2 family proteins and cancer. Oncogene 2008; 27: 6398–406. [DOI] [PubMed] [Google Scholar]