

Abstract
Cigarette smoking is one of the leading risks for lung cancer and is associated with the insensitivity of non‐small cell lung cancer (NSCLC) to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). However, it remains undetermined whether and how cigarette smoke affects the therapeutic efficacy of EGFR TKIs. In this study, our data showed that chronic exposure to cigarette smoke extract (CSE) or tobacco smoke‐derived carcinogen benzo[α]pyrene, B[α]P, but not nicotine‐derived nitrosamine ketone (NNK), reduced the sensitivity of wild‐type EGFR‐expressing NSCLC cells to EGFR TKIs. Treatment with TKIs almost abolished EGFR tyrosine kinase activity but did not show an inhibitory effect on downstream Akt and ERK pathways in B[α]P‐treated NSCLC cells. CSE and B[α]P transcriptionally upregulate c‐MET and activate its downstream Akt pathway, which is not inhibited by EGFR TKIs. Silencing of c‐MET reduces B[α]P‐induced Akt activation. The CSE‐treated NSCLC cells are sensitive to the c‐MET inhibitor crizotinib. These findings suggest that cigarette smoke augments oncogene addiction to c‐MET in NSCLC cells and that MET inhibitors may show clinical benefits for lung cancer patients with a smoking history.
Keywords: benzo[α]pyrene, cigarette smoke, c‐MET, EGFR‐TKI, lung cancer
Abbreviations
- 5‐mC
anti‐5‐methylcytosine
- B[α]P
benzo[α]pyrene
- CM
conditioned medium
- CSE
cigarette smoke extract
- DCR
disease control rate
- DEPC
diethyl pyrocarbonate
- DEPC
diethyl pyrocarbonate
- HGF
hepatocyte growth factor
- NC
nitrocellulose
- NF‐κB
nuclear factor kappa B
- NNK
nicotine‐derived nitrosamine ketone
- NNN
N′‐nitrosonornicotine
- PA
polycyclic aromatic hydrocarbon
- PFS
progression‐free survival
- PVDF
polyvinylidene difluoride
- RTK
receptor tyrosine kinase
- SCLC
small cell lung cancer
- TBST
Tris‐buffered saline Tween‐20
- TKI
tyrosine kinase inhibitor
1. Introduction
Lung cancer is one of leading cancer types in both males and females, and has a high incidence and mortality worldwide (Siegel et al., 2016). Lung cancer is grouped into two major types: non‐small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC accounts for 80–85% of lung cancer and can be further classified into three major sub‐types: adenocarcinoma, squamous cell carcinoma and large cell carcinoma. Cigarette smoke and secondhand smoke have been demonstrated as the risk factors for lung cancer (de Groot and Munden, 2012; Kenfield et al., 2008; Molina et al., 2008). The vast majority (85%) of lung cancer occurs in people aged over 50 years with a history of cigarette smoking (Siegel et al., 2016); only 10–15% of cases were non‐smokers (Thun et al., 2008).
Cigarette smoke is a mixture including more than 5000 chemicals (Adams et al., 1987; Borgerding and Klus, 2005; Talhout et al., 2011; Thielen et al., 2008). Over 60 carcinogens, including benzene, benzo[α]pyrene (B[α]P), dibenz[a,h]anthracene, catechol, nitromethane, 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone (NNK), N′‐nitrosonornicotine (NNN), were identified in the mixtures of cigarette smoke (Adams et al., 1987; Hecht, 2003; Talhout et al., 2011). These carcinogens can be classified into three groups: strong carcinogen, weak carcinogen and co‐carcinogen. Strong carcinogens, including polycyclic aromatic hydrocarbon (PAH) nitrosamines and aromatic amines, can induce tumor formation in immune‐completed laboratory animals after treatment at microgram or milligram level. Weak carcinogens, including acetaldehyde, induce carcinogenesis at a relatively high dose. Co‐carcinogens are chemicals such as nicotine that promote the effects of a carcinogen in the induction of cancer (Hecht, 2012). Among these chemicals, NNK and PAH are two major carcinogens and induce lung cancer formation in fully immune laboratory animals (Hecht, 2003; Huang and Chen, 2011). NNK, a nicotine‐derived nitrosamine ketone, can induce multiple cancer formation, including lung (major), nasal, oral, liver, pancreatic and cervical cancers (Hecht, 2003; Huang and Chen, 2011). It not only mutates or activates oncogenes and tumor suppressor, such as Adrb2, K‐Ras, p53 and TxA2 (Huang and Chen, 2011; Huang et al., 2011; Kerr et al., 2007; Matzinger et al., 1995; Zheng and Takano, 2011) but also induces hypermethylation of multiple tumor suppressor gene promoters, such as IGFBP‐3, FHIT, p16IKK4a and RARB (Harada et al., 2013; Lin et al., 2010). Some studies showed that NNK stimulated Erk signaling pathway and induced cell transformation and proliferation through the EGFR signaling pathway (Askari et al., 2005; Laag et al., 2006). Benzo[α]pyrene, a PAH, can be metabolized by cytochrome P450s (CYPs), such as CYP1A1, CYP1A2 and CYP1B1 (Chinai et al., 2015; Eling et al., 1986; Shimada, 2006). B[α]P induces carcinogenesis in mouse models through diolepoxide and radical‐cation mechanism, leading to G to T and G to A mutations in codon 12 of K‐Ras in lung cancer (Mass et al., 1993), G to T mutation at codon 13, and A to T mutation at codon 61 in H‐Ras in skin cancer (Chakravarti et al., 2008). Tp53 tumor‐suppressor gene is also mutated by B[α]P in skin and lung cancer through G to T transversion mutation in codons 157, 248 and 273 (Denissenko et al., 1996; Ruggeri et al., 1993).
In addition to genetic alternations associated with tumorigenesis, cigarette smoking behavior was also associated with the insensitivity to EGFR TKIs and poor progression‐free survival (PFS) in different types of cancer patients with EGFR overexpression (Gazdar, 2009; Lee et al., 2006; Miller et al., 2004; Takano et al., 2004). EGFR belongs to the membrane‐bound ErbB (HER) tyrosine kinase receptor family and is important for maintaining cell survival, differentiation and mitogenesis in various cancer types, including NSCLC (Scaltriti and Baselga, 2006). Upon binding with its ligands, EGFR forms homodimers or heterodimers with other ErbB family members including ErbB2, ErbB3 and ErbB4, leading to activation of its tyrosine kinase and downstream signals, such as PI3K/AKT and p42/p44 MAPK pathways (Scaltriti and Baselga, 2006). Overexpression or aberrant activation of EGFR has been demonstrated to cause tumor growth and progression of NSCLC (Dacic et al., 2006; Fontanini et al., 1995; Normanno et al., 2005). Based on this prevailing phenomenon, EGFR is therefore a rational and feasible target for suppression of tumor growth. Gefitinib (ZD1839, Iressa) and erlotinib (OSI‐774, Tarceva) are small molecule EGFR TKIs that function by binding to their ATP‐binding pocket (Noble et al., 2004) and have been approved for NSCLC patients. However, they are more effective in certain populations of NSCLC patients: Asian women, never‐smokers and adenocarcinoma patients. The good response of non‐smoker NSCLC patients to EGFR TKIs is associated with activating EGFR mutations (Lynch et al., 2004; Paez et al., 2004; Pao et al., 2004). The majority of these activating mutations are L858R mutation and exon 19 deletion of EGFR (Riely et al., 2006; Sharma et al., 2007), which showed protein structural alteration and higher binding affinity with EGFR TKIs at their ATP‐binding sites (Carey et al., 2006) and rendered this receptor more vulnerable to TKI inhibition. Therefore, these activating mutations have been viewed as a single biomarker to select patients for these TKIs (Lynch et al., 2004; Sordella et al., 2004; Zhang and Chang, 2008). However, it remains unknown whether and how cigarette smoke influences the sensitivity to EGFR TKIs in lung cancer. Moreover, even though their response rate is lower than with mutant EGFR‐expressing patients, 20–30% of NSCLC patients with amplified wtEGFR can still derive significant survival benefit from an EGFR TKI regimen (Bell et al., 2005; Cappuzzo et al., 2005; Tsao et al., 2005). Furthermore, no EGFR mutation was identified in about 10–20% gefitinib‐responsive patients (Bell et al., 2005; Cappuzzo et al., 2005; Huang et al., 2004; Kim et al., 2008; Nishimura et al., 2008; Pao et al., 2004). These observations suggest that certain NSCLC patients with wtEGFR expression still respond to EGFR TKIs. Therefore, identification of the cigarette smoke‐induced factors causing the insensitivity to these drugs may be helpful not only for the selection of wtEGFR‐expressing responders but also for the development of precise medicine to treat smoker patients.
In this study, we used a non‐biased strategy to explore whether c‐MET upregulation and downstream Akt activation can be elicited by cigarette smoke and its component B[α]P and contribute to EGFR TKI resistance. Our findings not only define the molecular mechanism accounting for the insensitivity to EGFR TKIs in NSCLC patients with a history of cigarette smoking, but also suggest that treatment with c‐MET inhibitors may benefit such patients.
2. Materials and methods
2.1. Chemicals and reagents
B[α]P was purchased from Sigma‐Aldrich (St. Louis, MO, USA). NNK was purchased from Toronto Research Chemicals (North York, Canada). Crizotinib was purchased from MedChem Express (Monmouth Junction, NJ, USA). Recombinant human hepatic growth (HGF) was purchased from PEPROTECH (Rocky Hill, NJ, USA). All chemicals were dissolved in DMSO and stored at 4 °C or −20 °C.
2.2. Cell culture
Non‐small cell lung cancer mucoepidermoid carcinoma NCI‐H292 cell line was cultured in RPMI 1640 (HyClone, Logan, UT, USA) supplemented with 10 mm HEPES, 10 mm sodium pyruvate, 10% FBS (GIBCO‐BRL, Gaithersburg, MD, USA), 100 units·mL−1 penicillin, and 100 g·mL−1 streptomycin (Thermo Scientific, Waltham, MA, USA). NSCLC adenocarcinoma HCC827 and PC9 lines were cultured in RPMI 1640 (HyClone) supplemented with 10% FBS, 100 units·mL−1 penicillin and 100 g·mL−1 streptomycin (Thermo). All cells were incubated in a humidified incubator in 5% CO2 at 37 °C. HCC827 and PC9 cell lines express EGFR exon19del E746–A750 mutant. These cell lines were gifts from Prof. Mien‐Chie Hung (MD Anderson Cancer Center, Houston, TX, USA).
2.3. Preparation of cigarette smoke extract medium
Commercial cigarettes (from Taiwan Tobacco & Liquor Corporation, Taipei, Taiwan) contain nicotine (0.8 mg per cigarette) and tar (10 mg per cigarette). Twenty‐five cigarettes were used to prepare 250 mL of culture medium (RPMI 1640 with or without HEPES serum‐free medium). Smoke from burning 25 cigarettes was inflated into 250 mL culture medium by pump and the pH of medium was adjusted to 7.4. The cigarette smoke extract (CSE) medium was filtrated with a 0.22‐μm filter to remove large particles. The CSE stock medium was stored at −30 °C until use. The stock of CSE medium is 100% (1 cigarette per 10 mL medium) and was diluted to the working concentrations with complete medium (Ishii et al., 2001; Ratovitski, 2010; Su et al., 1998; Sundar et al., 2012; Yang et al., 2006).
2.4. MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide] assay
Cells (5 × 103 cells per well) were seeded in 96‐well tissue culture plates and then treated with various EGFR TKIs including erlotinib or gefitinib at the indicated concentrations. Cell viability analysis was measured after treatment for 3 days using MTT (Sigma‐Aldrich), which was dissolved in DMSO. After treatment with TKIs, cells were washed with PBS twice and dissolved in DMSO for the measurement at absorbance 570 nm.
2.5. Analysis of phospho‐receptor tyrosine kinase array
Total lysates prepared from both control and B[α]P‐treated cells were subjected to Human Phospho‐Receptor Tyrosine Kinases Array Kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's protocol. The changes of phospho‐receptor tyrosine kinase (RTK) expressions were quantified using imagej (NIH, Bethesda, MD, USA).
2.6. Gene silencing by lentiviral small hairpin RNA
HEK293T cells (2.4 × 106) were seeded in 6‐cm culture dish in 5 mL DME/F‐12 with 10% FBS and incubated at 37 °C overnight. Three plasmids (pLKO.1‐shRNA or pLenti‐gene:pCMV‐ΔR8.91:pMD.G = 2:2:0.2 μg) were co‐transfected into HEK293T cells with 10.6 μL F2000 transfection reagent. The medium was refreshed with 10% FBS, P/S and 1% BSA after incubation for 6 h. Medium containing lentivirus was collected after 24 h of transfection and stored at −80 °C until use.
Cells (2 × 105 cells per well) were seeded in six‐well tissue culture plate and incubated overnight. Next day, cells were infected with lentivirus at MOI 125 with 8 g·mL−1 polybrene (hexadimethrine bromide, Sigma‐Aldrich). After infection for 16–18 h, culture medium was refreshed and 1 μg·mL−1puromycin added for selection for 3 days.
2.7. Genomic DNA isolation
Genomic DNA was extracted from stable clones with cell lysis buffer (10 mm pH 8.0 Tris‐HCl, 100 mm EDTA and 0.5% SDS), and incubated at 37 °C until completely dissolved, followed by treatment with RNase A at 37 °C for 15–60 min. Ammonium acetate was then added and mixed by vortex, and centrifuged at 14 000 g for 1 min. Supernatant was transferred to new tubes and 300 μL 100% isopropanol added, shaken 50 times, and centrifuged at 14 000 g for 1 min. Supernatant was removed and the pellet was washed in 300 μL 70% ethanol, and centrifuged at 14 000 g for 1 min. The pellet was dried for 15 min and re‐dissolved in TE buffer (pH 8.0). An optical density at 260 (OD260) and 280 (OD280) were determined for the concentration and purity of samples, respectively.
2.8. RNA extraction
Total RNA was extracted from stable clones with TriPure Isolation Reagent (Roche, Mannheim, Germany). First, each sample was mixed with 0.2 mL chloroform per 1 mL TriPure and then centrifuged at 12 000 g for 15 min to separate the aqueous phase, interphase and organic phases. Total RNA from the aqueous phase was mixed with 0.4–0.6 mL isopropanol at −30 °C for over 30 min. The mixtures were then centrifuged at 12 000 g for 15 min, washed in 1 mL 75% ethanol twice, and centrifuged at 12 000 g for 15 min. Finally, supernatant was removed and the RNA pellet dried, followed by re‐dissolution in diethyl pyrocarbonate (DEPC) water at 4 °C overnight.
2.9. Reverse‐transcription and polymerase chain reaction
The RT was performed with 1 μg of RNA using MMLV First‐Strand Synthesis Kit (GeneDireX, Las Vegas, NV, USA). The relative mRNA expression of c‐MET was determined using SYBR FAST qPCR kit (KAPA Biosystems, Wilmington, MA, USA). Primer sequences for c‐MET used in real‐time quantitative PCR were F’: 5′‐ CCCGAAGTGTAAGCCCAACT‐3′, R’: 5′‐AGGATACTGCACTTGTCGGC‐3′; 18s rRNA: F’: 5′‐CGGCGACGACCCATTCGAAC‐3′, R’: 5′‐GAATCGAACCCTGATTCCCCGTC‐3′; c‐MET genomic exon 2: F’: 5′‐ATAAACCTCTCATAATGAAGGCC‐3′, R’: 5′‐TTTGCTAGTGCCTCTTTACACTC‐3′.
2.10. Protein extraction and western blot analysis
Cells were lysed using RIPA lysis buffer with protease and phosphatase inhibitors and centrifuged at 12 000 g for 30 min. Samples were quantified using Braford assay (Bio‐Rad, Hercules, CA, USA). All samples were separated by 8–12% SDS/PAGE and transferred to 0.45 μm polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) or 0.22 μm nitrocellulose (NC) membranes (GE Healthcare, Amersham, UK). Non‐specific protein binding was blocked in 5% skim milk with Tris‐buffered saline Tween‐20 (TBST) for 1 h at room temperature. The membranes were hybridized with primary antibodies against phospho‐HER2 (Y1221/1222), phospho‐HER3 (Y1289), phospho‐EGFR (Y1068), phospho‐MET (Y1234/1235), c‐MET, Akt, phospho‐Erk (T202/Y204), Erk (Cell Signaling, Danvers, MA, USA), phospho‐Akt (S473), HER2, HER3 and EGFR (Santa Cruz Biotechnology, Dallas, TX, USA), tubulin, actin (Sigma‐Aldrich) and phosphotyrosine (Merck Millipore, Belmopán, Belize) at 4 °C overnight, followed by incubation with HRP‐labeled secondary antibodies at room temperature for 1 h. The expression of proteins was detected with enhanced chemiluminescence (ECL, GE Healthcare, or Millipore).
2.11. Conditioned medium treatment
Cells were cultured and plated on 100‐mm dishes. After 24 h, conditioned media from H292 parental, H292/1%CSE, H292/5%CSE, H292/DMSO and H292/B[α]P 1 μm cells was collected. Fresh conditioned media was centrifuged at 200 g for 5 min before H292 parental cells were treated with different conditioned media for 6 h. HGF treatment was used as positive control for c‐MET activation. The treated cells were lysed with RIPA lysis buffer to prepare total protein.
2.12. ChIP analysis
Cigarette smoke extract‐/B[α]P‐treated H292 cells were fixed with 1% formaldehyde at room temperature for 10 min to cross‐link protein and DNA, and the reaction was stopped by adding glycine. Cross‐linked cells were washed twice with cold PBS and resuspended in 1 mL PBS with protease inhibitor cocktail. These cells were centrifuged at 700 g for 10 min at 4 °C and the supernatant removed. DNA was digested by treating with micrococcal nuclease (MNase, Thermo Scientific) for an appropriate incubation time at 37 °C after adding nuclear lysis buffer (50 mm Tris‐Cl pH 8.0, 10 mm EDTA, 1% SDS) to break the nuclear membrane. These cell pellets were centrifuged at 900 g for 5 min at 4 °C and then the supernatant was collected and pre‐cleaned with 60 μL protein A agarose (GE Healthcare, New York, NY, USA) in 900 μL dilution buffer (0.01% SDS, 1% Triton X‐100, 2 mm EDTA, 20 mm Tris‐Cl (pH 8.0), 500 mm NaCl) with protein inhibitor cocktail at 4 °C for 1 h. The supernatants were then centrifuged at 860 g at 4 °C for 3 min and incubated with primary antibody against MeCP2 (Millipore), 5‐methylcysine (Calbiochem, San Diego, CA, USA) or control IgG at 4 °C overnight. Antibody‐protein‐DNA complex was immunoprecipitated with 60 μL salmon sperm DNA/protein A at room temperature for 2 h. Pellets were eluted by freshly prepared elution buffer (20% SDS and 1 m NaHCO3) for 15 min at 65 °C after extensive washing. Nucleic acids were extracted by adding phenol/chloroform/isoamyl alcohol (25 : 24 : 1) (Invitrogen, Carlsbad, CA, USA) and used as the template in SYBR FAST qPCR kit (KAPA Biosystems). Primer sequences for the methylation of c‐MET promoter were M1 region (+84 to +484), F′: 5′‐AGCACGTGTCTGTTCGTCCCTG‐3′, R′: 5′‐ CCTTGCCAGCTGTATCACCCTG‐3′; M2 region (−261 to +98), F′: 5′‐ GGACAAACCTAGAGCGACAGGG‐3′, R′: 5′‐ACGCGGCTGGAGTTTGTACC‐3′; M3 region (−563 to −223), F′: 5′‐GCTTTGCGCGGGTGACTTTG‐3′, R′: 5′‐AGCACGTGTCTGTTCGTCCCTG‐3′; M4 region (−710 to –409), F′: 5′‐ATCCGTCCATGCACTCCCAAC‐3′, R′: 5′‐CGGCAAGGTGAAACTTTCTAGG‐3′.
2.13. Statistical analyses
Data are presented as the mean ± standard error of the mean (SEM) of three independent results. A two‐tailed t‐test was used for most comparisons, with P < 0.05 considered significant.
3. Results
3.1. Cigarette smoke and B[α]P cause the insensitivity of wtEGFR‐expressing NSCLC to EGFR TKIs
To investigate whether and how cigarette smoke influenced the sensitivity to EGFR TKIs in NSCLC, we first established CSE‐treated clones from wtEGFR‐expressing NCI‐H292 and mutEGFR‐expressing HCC827 and PC9 cells by chronic exposure to 1% CSE or 5% CSE for at least 2 months. Since nuclear factor kappa B (NF‐κB) activation by cigarette smoke has been reported (McMillan et al., 2011; Sundar et al., 2012; Yang et al., 2006), we first examined p65 S536 phosphorylation as a positive control to monitor the proper response of NSCLC cells to CSE treatment (Fig. 1A). The viabilities of these clones under the treatment with EGFR TKIs were then examined in MTT assays. Our data showed that CSE renders H292 cells (Fig. 1B) but not HCC827 or PC9 cells (Fig. 1C,D), more insensitive to both gefitinib and erlotinib as compared with their parental cells. We also tested whether B[α]P or NNK can affect the sensitivity of lung cancer cells to EGFR TKIs. Our data showed that H292/B[α]P stable clones are more resistant to both gefitinib and erlotinib (Fig. 2A), but NNK treatment did not change the sensitivity of H292 cells to EGFR TKIs (Fig. 2B). Similar to the results in HCC827/CSE and PC9/CSE clones, HCC827/B[α]P, HCC827/NNK and PC9/B[α]P stable clones remain sensitive to EGFR TKIs (Fig. 2C–E). These results suggest that B[α]P among the CSE‐derived carcinogens may render wtEGFR‐expressing lung cancer cells more resistant to EGFR tyrosine kinase inhibitors.
Figure 1.
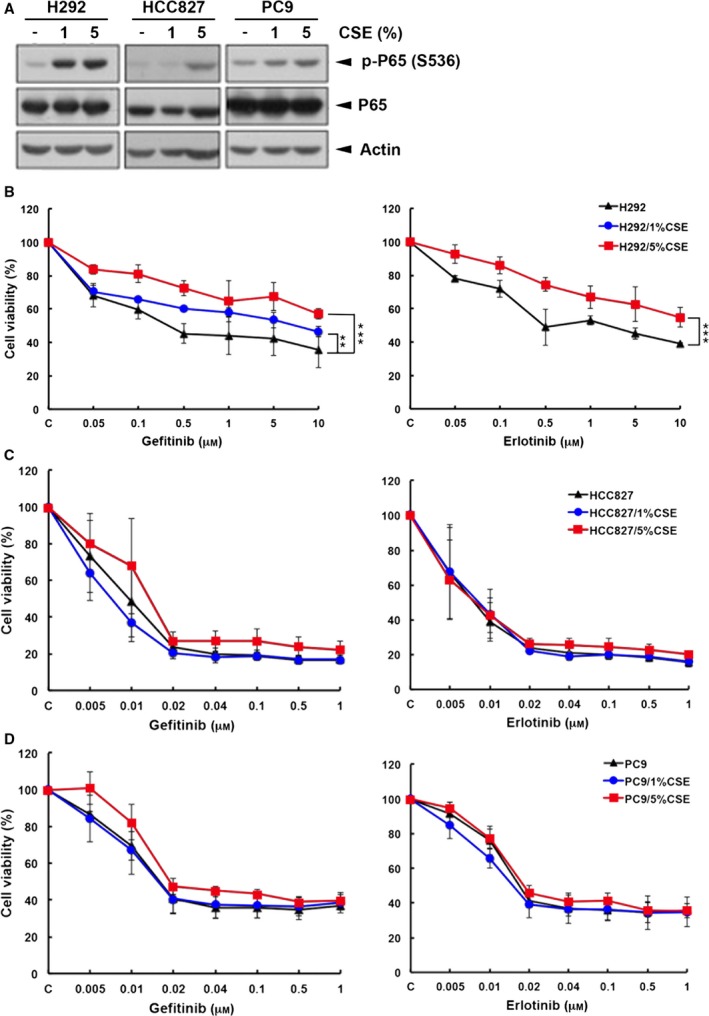
Cigarette smoke extract treatment rendered wtEGFR‐ but not EGFR mutant‐expressing NSCLC more resistant to EGFR tyrosine kinase inhibitors. Whole‐cell extracts prepared from H292/CSE, HCC827/CSE and PC9/CSE. Stable cells were subjected to western blot analysis to determine p65 phosphorylation at ser536 (A). CSE‐treated H292 (B), HCC827 (C) and PC9 (D) were treated with gefitinib (left) or erlotinib (right) for 3 days and their viabilities were determined in MTT assays.
Figure 2.
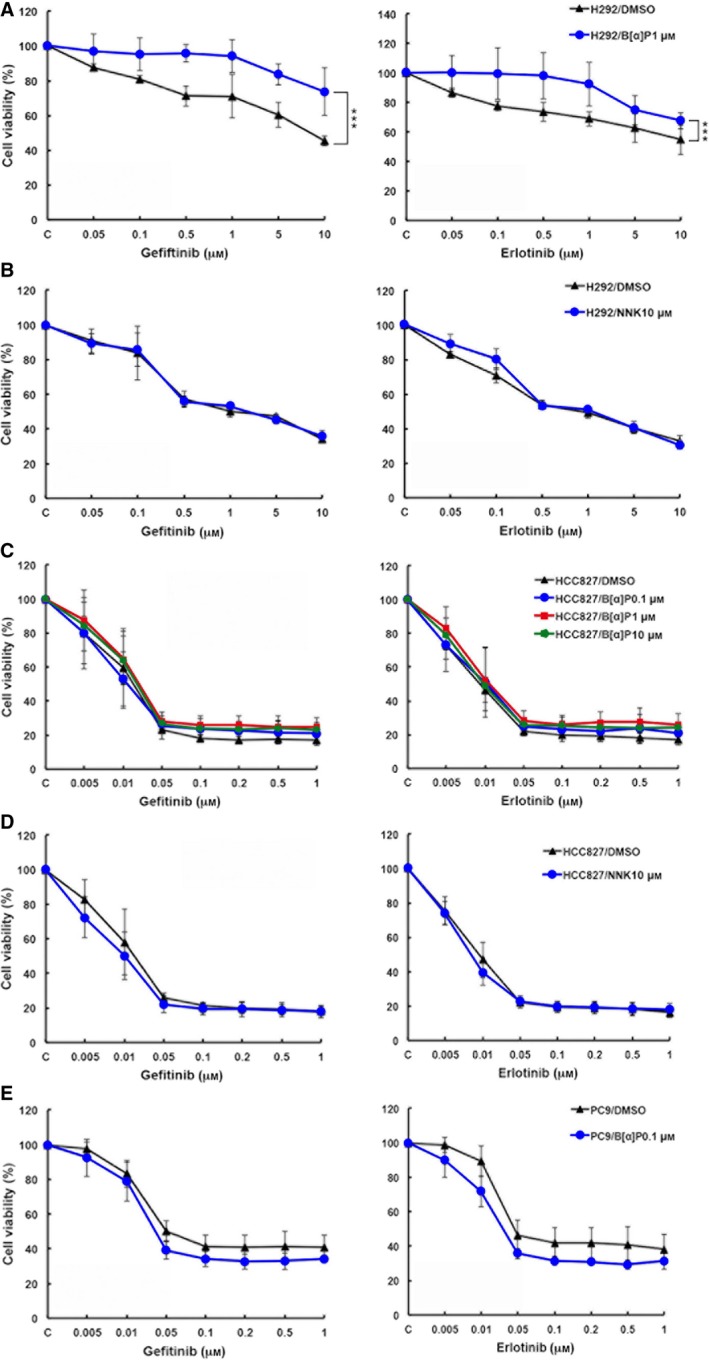
Treatment with B[α]P but not NNK reduces the sensitivity of H292 cells to EGFR tyrosine kinase inhibitors. H292 (A, B), HCC827 (C, D), and PC9 (E) selected with 1 μm B[α]P (A, C, E) or 10 μm NNK (B, D) were treated with gefitinib (left) or erlotinib (right) for 3 days, and their viabilities were determined in MTT assays.
We next examined whether EGFR and its downstream signaling pathway in H292, HCC827 and PC9 cells were affected by CSE, B[α]P or NNK. EGFR activity in all CSE‐, B[α]P‐ and NNK‐treated cells was not changed (Fig. 3A,B). However, the Akt activity in wtEGFR‐expressing H292 cells was significantly enhanced in response to CSE and B[α]P but not NNK (Fig. 3A). In EGFR mutant‐expressing HCC827 cells, Akt activation by CSE, B[α]P and NNK was barely detected (Fig. 3B). In both H292 and HCC827 cells, ERK activity was induced by CSE and B[α]P but not NNK (Fig. 3A,B). These findings suggest that the elevation of Akt activity may be critical for CSE‐ and B[α]P‐induced EGFR TKI resistance.
Figure 3.
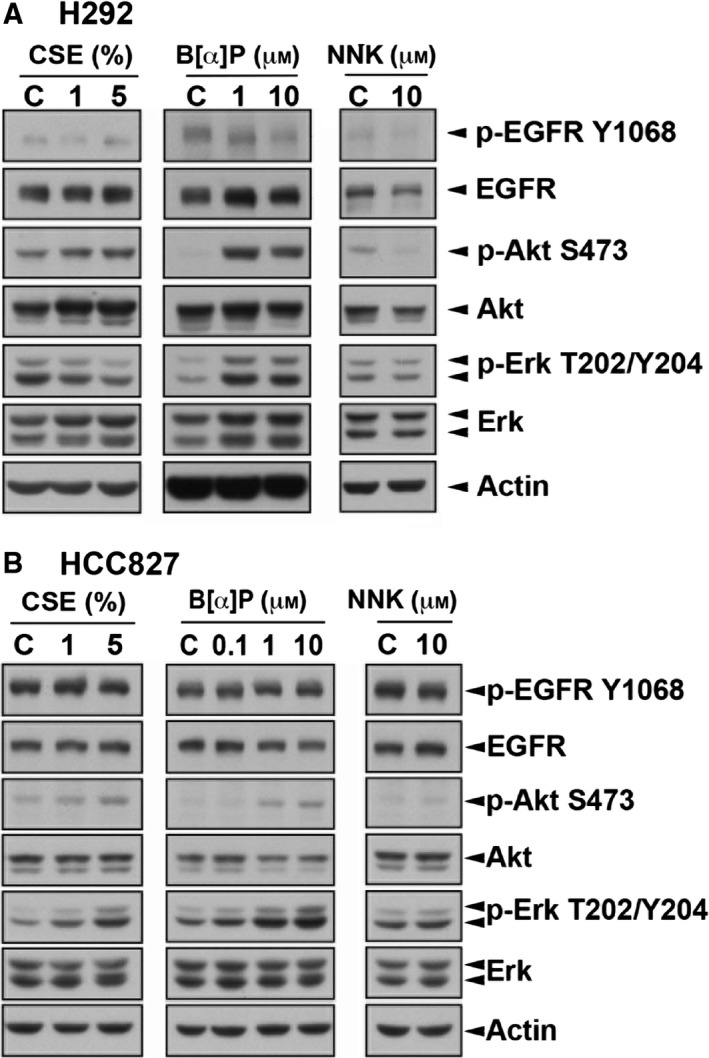
Activities of Akt and Erk were higher in CSE‐ and B[α]P‐selected H292 cells. The activities and protein levels of EGFR and its downstream signaling Akt and ERK were detected in H292 (A) and HCC827 (B) by western blot analysis with indicated antibodies.
We further found that both basal and EGF‐induced EGFR signaling including Akt and ERK activation can be completely inhibited by gefitinib and erlotinib in both parental (Fig. 4A) and NNK‐treated cells (Fig. 4B). In B[α]P‐treated H292 cells, however, the enhanced Akt and ERK kinase activities were not affected by these two EGFR TKIs; in fact, the EGFR activity was dramatically inhibited (Fig. 4A). Both Akt and ERK pathways remain sensitive to EGFR TKIs in CSE‐ and B[α]P‐treated HCC827 stable clones (Fig. 4C), probably due to the stronger oncogene addiction to the activated EGFR mutant in these cells. These data suggested that the increased Akt and ERK activities in response to CSE and B[α]P treatment may result from an alternative signaling and thereby contribute to the insensitivity of wtEGFR‐expressing smoker NSCLC patients to EGFR TKIs.
Figure 4.
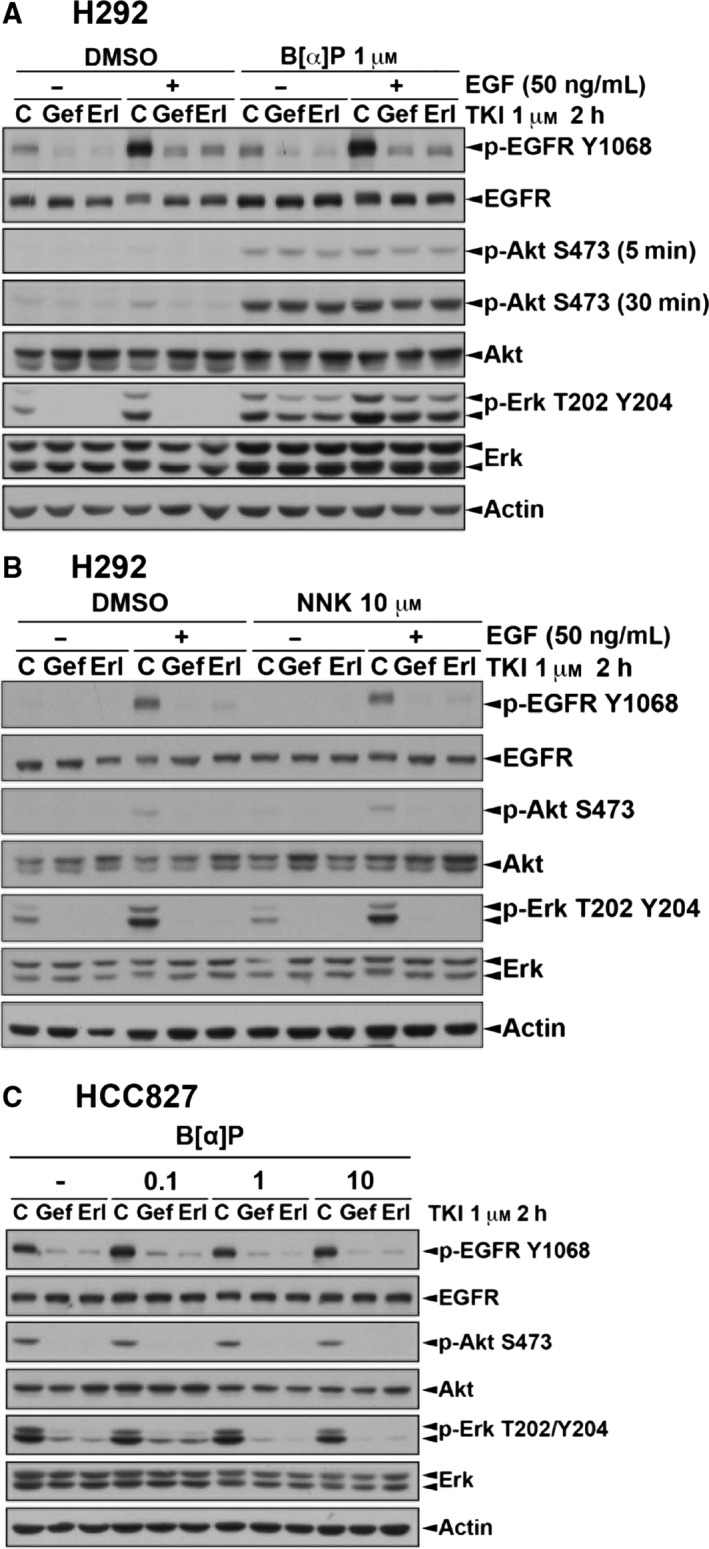
EGFR TKI failed to inhibit activation of Akt and Erk in H292/B[α]P cells but not in HCC827/ B[α]P cells. H292 (A, B) and HCC827 (C) cells selected with B[α]P or NNK were pretreated with 1 μm EGFR TKIs for 2 h followed by 50 ng·mL −1 EGF stimulation as indicated. Whole‐cell extracts were prepared and subjected to western blot analysis to determine the expression of EGFR signaling pathway with indicated antibodies.
To examine whether this resistance was caused by EGFR T790M mutation, which has been reported as a secondary mutation after treatment with EGFR TKIs (Mitsudomi and Yatabe, 2007; Sharma et al., 2007), cDNA of EGFR was extracted and prepared from these CSE‐ and B[α]P‐treated clones and sequenced. Although G to A mutation at codon 787 was found, this substitution is a silent mutation and did not alter the amino acid sequence. However, no mutation at codon 790 was found, suggesting that the resistance of CSE‐ and B[α]P‐treated H292 cells to EGFR TKI was not caused by EGFR T790M mutation (Fig. S1). We further investigated whether other receptor tyrosine kinases were involved in the elevation of Akt and ERK activities by B[α]P. We examined the expression profile of protein tyrosine phosphorylation in B[α]P‐treated cells in western blot analysis with anti‐phosphotyrosine antibody. Interestingly, an increased tyrosine phosphorylation signal around 150 kDa was detected in the B[α]P‐treated H292 cells (Fig. 5A). Therefore, we suggest that certain RTKs might be activated in response to B[α]P treatment to mediate the activations of AKT and ERK signaling. To further identify which RTK is activated by B[α]P, total lysates prepared from both parental and B[α]P‐treated cells were subjected to RTK antibody array analysis. The results showed that MET tyrosine phosphorylation is dramatically increased in the H292/B[α]P cells. However, the phosphorylation status of EGFR, HER2 and HER3 was not changed by B[α]P (Fig. 5B). As shown in Fig. 5C, the activation of c‐MET was 19‐fold higher in H292/B[α]P cells than in parental cells in the quantitated data from RTK antibody array analysis. Other RTKs, including c‐RET and EphA2, were also activated in response to B[α]P. However, IGF‐IR, Axl and Alk, which were known to play some roles in the acquired resistance to EGFR TKI (Bae et al., 2015; Guix et al., 2008; Maione et al., 2015), were not activated by B[α]P and may not be involved in the insensitivity of smoker NSCLC to EGFR TKI (Fig. 5D).
Figure 5.
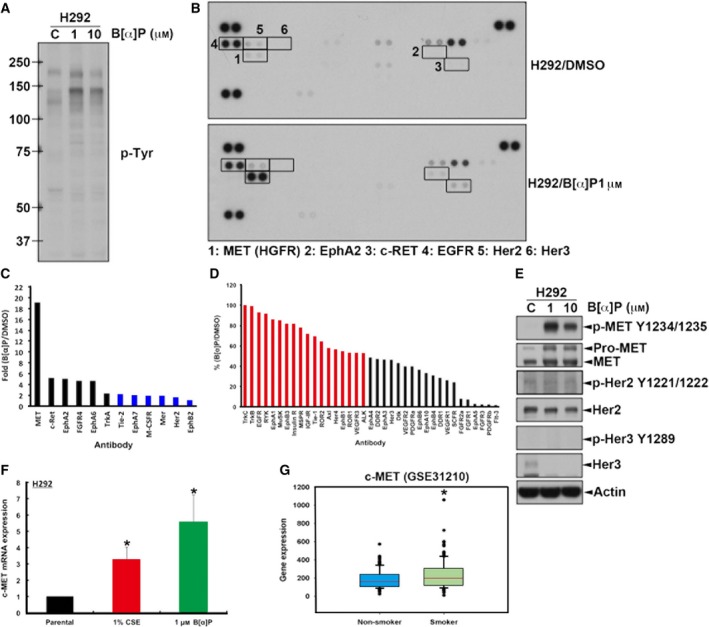
Proto‐oncogene c‐MET expression and activity were higher in H292/B[α]P cells than in control cells. (A) Whole‐cell extract from H292/B[α]P cells was subjected to western blot analysis with anti‐phosphotyrosine antibody. (B) Total lysates prepared from the H292/DMSO and H292/B[α]P were incubated with RTK antibody arrays, and phosphotyrosine was detected by anti‐phospho‐tyrosine‐HRP. The changes in tyrosine phosphorylation of RTK were labeled as indicated. The upregulation (C) and downregulation (D) of RTK tyrosine phosphorylations in H292/B[α]P cells shown in (B) were quantitated using imagej software. (E) The RTK activities and protein levels in H292/B[α]P cells determined by western blot analysis with indicated antibodies. (F) The mRNA level of c‐MET in indicated cells was detected by q‐RT‐PCR. (G) The c‐MET mRNA levels in non‐smoker and smoker NSCLC patients were analyzed from the GSE31210 GEO dataset.
3.2. CSE and B[α]P enhance the oncogene addiction of wtEGFR‐expressing NSCLC to c‐MET
To prove that cigarette smoke renders NSCLC cancer cells more resistant to EGFR TKI through induction of c‐MET signaling, we first confirmed the c‐MET activation in H292/B[α]P cells by western blot analysis. As shown in Fig. 6E, c‐MET tyrosine phosphorylation was indeed dramatically increased in B[α]P‐treated cells, and total c‐MET expression is also induced. By contrast, NNK suppressed c‐MET expression (Fig. S2). In addition, exposure to CSE, B[α]P or NNK had no dramatic effect on c‐MET phosphorylation or protein expression in PC9 and HCC827 cells (Fig. S2). The tyrosine phosphorylation of HER2 and HER3 was also determined, but no difference between B[α]P‐treated cells and control cells was detected (Fig. 5E). We further checked the mRNA level of c‐MET in CSE‐ and B[α]P‐selected cells by RT‐qPCR analysis and found an elevation of c‐MET mRNA expression in response to these treatments (Fig. 5F), suggesting that B[α]P transcriptionally upregulates c‐MET gene expression. In support of this finding, the published microarray dataset of 226 lung cancer patients (GSE31210) also showed that cMET mRNA is higher in the lung tumor tissue in smoker than in non‐smoker patients (Fig. 5G).
Figure 6.
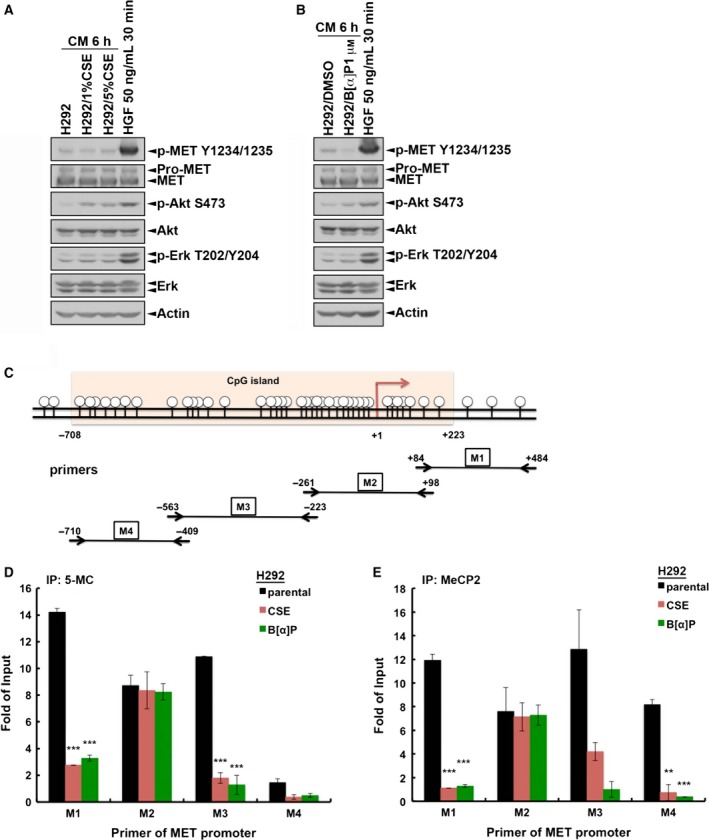
Cigarette smoke extract and B[α]P induced c‐MET expression through promoter de‐methylation. Parental H292 cells were treated with conditioned medium of (A) H292/CSE or (B) H292/B[α]P cells for 6 h. The whole‐cell lysates were then prepared and subjected to western blot analysis with indicated antibodies. (C) The CpG island on c‐MET promoter and primer sets used in ChIP analysis were illustrated. Parental H292 cells and their CSE and B[α]P clones were subjected to ChIP analysis with 5‐methylcytosine (5‐mC) (D) or anti‐MeCP2 (E) antibodies followed by quantitative PCR with primers targeting c‐MET promoter. ‘Fold of Input’ is expressed as the ratio between the amount of total immunoprecipitated DNA (bound) and the amount of input DNA. Data are expressed as means ± SD. **P < 0.005; ***P < 0.001.
In addition to c‐MET gene expression, the induction of c‐MET activity may also be due to the stimulation by autocrine ligand, which could be induced by CSE and B[α]P and released into the medium. According to Bogler's model, autocrined HGF activates c‐MET signaling to enhance HGF expression in ΔEGFR‐expressing GBM cells (Garnett et al., 2013). We therefore collected the conditioned medium (CM) from control cells or from CSE or B[α]P stable cells to treat parental H292 cells, followed by examination of c‐MET signaling. Unlike the stimulation by HGF, however, the conditioned medium from CSE‐ (Fig. 6A) or B[α]P‐treated (Fig. 6B) cells did not induce c‐MET activity in parental H292 cells. Therefore, we excluded the possibility that the CSE‐ or B[α]P‐induced c‐MET activity is due to the autocrine ligand stimulation. There is a CpG island (from –708 to +233) on the promoter of c‐MET gene, and methylation of this region suppressed c‐MET transcription through recruitment of MeCP2 (Morozov et al., 2008; Plummer et al., 2013). To examine whether CSE and B[α]P upregulated c‐MET expression by modulating the DNA methylation status on c‐MET promoter, ChIP was performed with anti‐5‐methylcytosine (5‐mC) and anti‐MeCP2 antibodies, followed by quantitative PCR with four different primer sets for c‐MET promoter (as illustrated in Fig. 6C). The results showed that all four regions of c‐MET promoter were detectable in both anti‐5mC and anti‐MeCP2 immunoprecipitates of parental H292 cells (Fig. 6D,E). More importantly, treatments with CSE and B[α]P can suppress cytosine methylation and MeCP2 recruitment on c‐MET promoter in M1, M2 and M4 regions. These results suggest that CSE and B[α]P may upregulate c‐MET expression through removal of DNA methylation on its promoter.
To further investigate whether the increased c‐MET expression and activity is responsible for Akt and Erk activation and cell viability of CSE‐ and B[α]P‐treated cells, expression and kinase activity of c‐MET were down‐regulated by shRNA and pharmacological inhibitor, respectively. As shown in Fig. 7, Akt but not ERK activity was reduced in CSE‐ and B[α]P‐treated cells but not in parental cells after silencing c‐MET expression (Fig. 7A,B) and treatment with c‐MET inhibitor crizotinib (Fig. 7C). In addition, inhibition of c‐MET by crizotinib also showed a suppressive effect on the viability of both CSE‐treated and parental cells (Fig. 7D). These results indicated that cigarette smoke increased the PI3K/Akt survival pathway for insensitization to EGFR TKIs through enhancing c‐MET expression and activity.
Figure 7.
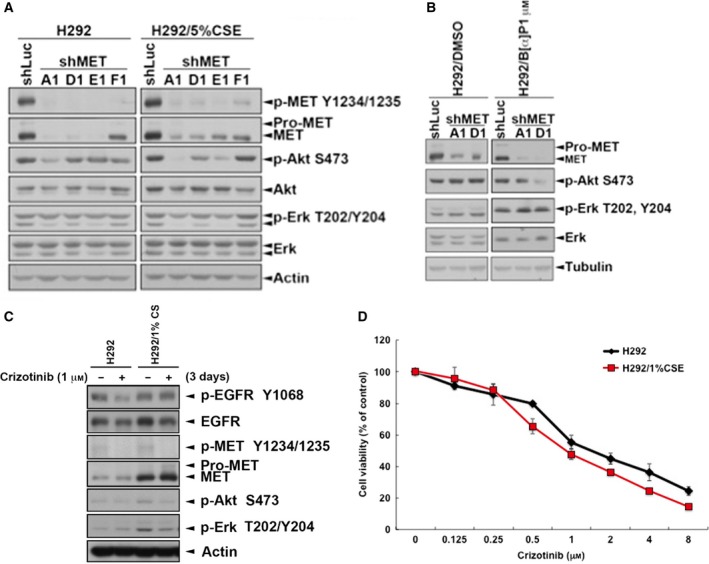
The activity of Akt was reduced by inhibition of c‐MET in H292/CSE and H292/B[α]P cells. c‐MET expression was knocked down by shRNA in H292/CSE (A) and H292/B[α]P (B) cells for 3 days. (C,D) These stable clones were treated with 1 μm crizotinib for 3 days. Total lysates were collected and subjected to western blots with indicated antibodies (C) and the cell viability was analyzed in MTT assay (D).
In conclusion, we demonstrated that cigarette smoke extract and its derived carcinogen, benzo[α]pyrene, activated proto‐oncogene MET signaling through ligand‐independent pathway to dominate Akt activation, which may thereby lead to the EGFR TKI resistance in wild‐type EGFR‐expressing NSCLC cell line but not in mutant EGFR‐expressing NSCLC cell lines (Fig. 8). Therefore, our findings not only indicate the possibility of c‐MET activation as the molecular mechanism underlying cigarette smoke‐related EGFR TKI resistance, but also suggest that c‐MET inhibitors may benefit NSCLC patients who smoke, who harbor wtEGFR‐expression.
Figure 8.
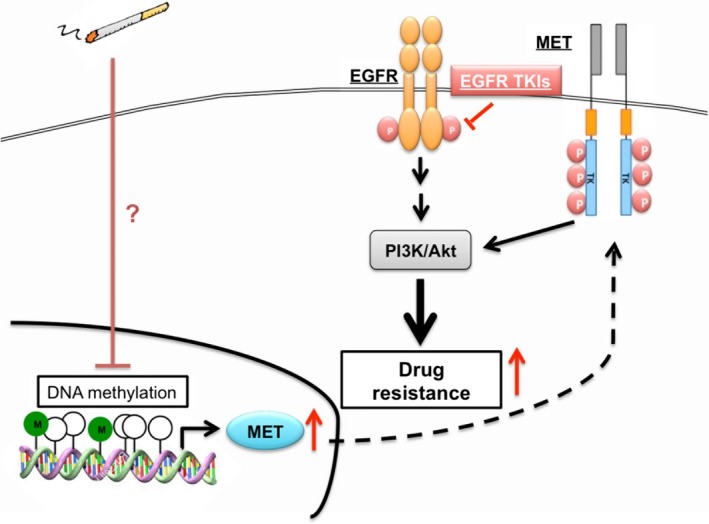
Current hypothetic model of this study. Cigarette smoke de‐represses c‐MET expression through reduction of promoter methylation. The induced c‐MET induced drug resistance to EGFR TKIs by maintaining Akt activity in wtEGFR‐expressing lung cancer cells.
4. Discussion
Cigarette smoke is an important risk factor of lung cancer progression. Clinical studies showed that lung cancer patients who are former or current smokers, are more resistant to EGFR TKIs (Organ and Tsao, 2011). Induction of cytochromes CYP1A1/1A2 by cigarette smoking has been hypothesized to alter erlotinib pharmacokinetics (Hamilton et al., 2006). However, our data showed that EGFR TKIs almost abolished the EGFR activity in CSE/B[α]P‐treated cell lines, suggesting that drug metabolism may not contribute to the EGFR TKI insensitization of NSCLC by cigarette smoke. In this study, we found that CSE and its carcinogen B[α]P may render wtEGFR‐expressing lung cancer cells more insensitive to EGFR TKI through activation of the c‐MET/Akt signaling axis. However, CSE/B[α]P‐induced c‐MET activation was not observed in HCC827 or PC9 cells and did not cause EGFR TKI resistance. Consistent to our findings, c‐MET amplification was frequently detected in wtEGFR, but not in mutant EGFR‐expressing NSCLC patients, and is associated with a poor prognosis (Song et al., 2017). Since HCC827 cells express EGFR activating mutant, which dominantly controls pro‐survival Akt signal, cigarette smoke may only switch the oncogene addition from the EGFR to MET pathway in wtEGFR‐expressing NSCLC cancer cells. Interestingly, the activation of β1‐adrenergic receptor (β1‐AR), a G protein‐coupled receptor, has been reported to mediate the NNK‐induced IGF‐1R activation and tumorigenesis of human bronchial epithelial cells (Min et al., 2016). β‐AR2 was recently demonstrated to cause EGFR TKI resistance through inactivation of LKB1 and induction of interleukin‐6 expression in NSCLC harboring EGFR activating mutation (Nilsson et al., 2017). However, our data did not show a change in TKI sensitivity of H292 and HCC827 cells caused by NNK (Fig. 2). These studies suggest that NNK‐activated β1‐AR may be involved in NSCLC carcinogenesis but not its EGFR TKI resistance.
The receptor‐tyrosine kinase c‐MET is the receptor for hepatocyte growth factor (HGF) and activates a wide range of different cellular signaling pathways, including PI3K/Akt, MAPK, JNK, PKC and FAK, which contribute to proliferation, motility, migration and invasion (Organ and Tsao, 2011). In the network enrichment analysis, c‐MET has been identified as a hub which plays key roles in cancer initiation and progression of cigarette smoking‐associated NSCLC (Pazhouhandeh et al., 2017). Activation of the HGF/c‐MET signaling pathway has also been found to be associated with a poor prognosis in various solid tumors including NSCLC (Guo et al., 2014; Zhang et al., 2015), indicating a predictive value for this disease. The critical role of crosstalk between the c‐MET and ErbB family in the development of resistance to cancer therapeutics has been well elucidated (Lai et al., 2009). The increased expression and activation of c‐MET in response to EGFR TKIs (Engelman et al., 2007) and cytotoxic anticancer agent (Ozasa et al., 2014) contribute to drug resistance in lung cancer cells through compensatory enhancing of Akt signaling. Suppression of c‐MET expression by siRNA or inhibition of c‐MET activation by pharmacological inhibitor re‐sensitized the resistant cells to these anticancer agents (Ozasa et al., 2014), indicating the importance of c‐MET overexpression in the development of acquired resistance. In support of our finding, NSCLC patients with soluble c‐Met levels > 766 ng·mL−1 have shown significant short median PFS after EGFR‐TKI treatment (Gao et al., 2016).
Mechanisms resulting in constitutive or prolonged activation of c‐MET during tumor growth or cancer progression include the occurrence of specific genetic lesions, including translocations, gene amplifications and activating mutations; and transcriptional upregulation of the c‐MET protein in the absence of gene amplification or via ligand‐dependent autocrine or paracrine mechanisms (Danilkovitch‐Miagkova and Zbar, 2002). High c‐MET gene copy number is associated with poor survival in NSCLC, ovarian clear cell adenocarcinoma and gastric cancer (Beau‐Faller et al., 2008; Cappuzzo et al., 2009; Chen et al., 2011; Go et al., 2010; Park et al., 2012; Shi et al., 2012; Yamashita et al., 2013). In oral lichen planus, the expression of c‐MET was found to be higher in smokers than non‐smokers (Kłosek et al., 2011). However, few studies reported how cigarette smoke increases c‐MET expression and activity. Previous studies suggested that mutation of c‐MET at the sema or juxtamembrane domain, including N375S, R988C, T1010I, S1058P or splicing mutation, caused c‐MET activation (Lawrence and Salgia, 2010; Onozato et al., 2009). In Soundararajan's study, most of the N375S mutation carriers were eastern Asian, male, squamous and smokers (Johnson et al., 2012; Krishnaswamy et al., 2009). Based on these studies, the exon2 (sema domain) and exon 14 (juxtamembrane domain) of c‐MET were amplified and sequenced. These mutations did not occur after treatment with CSE and B[α]P (data not shown). Gene amplification was also not observed in the PCR analysis (data not shown). Therefore, we ruled out the possibility that CSE‐ or B[α]P‐induced c‐MET activation is due to the mutation or amplification of c‐MET gene. Activation of MET classically is through binding with its ligand, HGF, inducing receptor homodimerization, and then the C‐terminal tail transphosphorylation, leading to the initiation of downstream signals (Feng et al., 2012). Chen et al. (2006) suggested that cigarette smoke could overexpress HGF in type II pneumocytes and lung cancer cells. HGF‐dependent MET activation via endocrine, paracrine or autocrine signaling had been observed in lung cancer (Feng et al., 2012). In our study, however, the condition medium from benzo[α]pyrene‐treated clone did not stimulate MET tyrosine phosphorylation, suggesting that activation of MET in these cells is not through the HGF autocrine‐dependent pathway.
Several microRNAs, including miR‐613 (Li et al., 2016b), miR‐138 (Li et al., 2016a), miR‐206 (Zheng et al., 2015), miR‐185 (Fu et al., 2014) and miR‐101 (Hu et al., 2013), have been demonstrated to target c‐MET for cancer proliferation and progression. Cigarette smoke globally downregulates microRNA expression in human alveolar macrophages (Graff et al., 2012). Further studies are required to demonstrate the possibility that CSE induces c‐MET expression through downregulation of microRNAs. In addition, physical protein interactions with other receptor tyrosine kinases have been reported to contribute to MET activation (Cassinelli et al., 2009; Ju and Zhou, 2013; Tanizaki et al., 2011). An association between integrin beta1 and c‐MET has also been reported to mediate EGFR TKI resistance through activation of the c‐MET signaling pathway in NSCLC (Ju and Zhou, 2013). Further investigations are required to examine the possibility that these potential mechanisms underlie benzo[α]pyrene‐induced MET activation.
In a meta‐analysis, targeting c‐MET therapies has been found to improve progression‐free survival (PFS) and disease control rate (DCR) in advanced or metastatic NSCLC patients. However, c‐MET inhibitors did not show the therapeutic benefits on their overall survival and objective response rate (Ye et al., 2016). It would be interesting to analyze the impact of cigarette smoke on the therapeutic efficacy of c‐MET inhibitor in NSCLC patients. In our results, when MET expression was silenced in CSE‐ and B[α]P‐treated cells, the activity of Erk is not affected by MET inhibition. This observation suggests that elevation of other unidentified driver genes may be responsible for the increased Erk signals, which may thereby become an obstacle to the treatment of NSCLC patients who are smokers, even if MET inhibitors are used. Therefore, further investigation of the underlying mechanisms of B[α]P‐induced ERK activation are indicated.
5. Conclusions
In this study, our data showed that cigarette smoke and its derivative B[α]P reduce the sensitivity of wtEGFR‐ but not EGFR mutant‐expressing NSCLC cells to EGFR TKIs. c‐MET is upregulated and activated by CSE and B[α]P for the compensatory Akt activation and resistance to EGFR inhibitors. These findings not only explain the critical role of c‐MET in the primary resistance of NSCLC patients who are smokers, to EGFR TKIs, but also suggest that targeting c‐MET may benefit such patients.
Author contributions
Study concepts: WCH and CYT. Study design: WCH and CMC. Data acquisition: FJC, SLW, YCH, BWW, YHH and YJC. Quality control of data and algorithms: CHC and TCH. Data analysis and interpretation: ISH and CHY. Statistical analysis: FJC, SLW and YLY. Manuscript preparation: WCH, FJC, SLW and CYT. Manuscript editing: CYT. Manuscript review: WCH. All authors read and approved the final manuscript.
Supporting information
Fig. S1. EGFR mutations were not found in CSE‐ or B[α]P‐treated cells. The EGFR cDNA in H292/1%CSE (A), H292/5%CSE (B) or H292/B[α]P cells (C) were prepared by RT‐qPCR, and sequenced.
Fig. S2. c‐MET phosphorylation and protein expression in NSCLC cells in response to cigarette smoke and its oncogene ingredients. Total protein lysate of H292, PC9 and HCC827 cells and their CSE, B[α]P and NNK‐treated clones were prepared and subjected to western blot analysis with indicated antibodies.
Acknowledgements
This work was supported by grants from the Ministry of Science Technology, Taiwan (MOST105‐2320‐B‐039‐056‐MY3, MOST‐105‐2314‐B‐039‐035‐MY3, MOST105‐2314‐B‐040‐011‐MY3), China Medical University Hospital (DMR‐106‐23) and the Ministry of Health and Welfare (MOHW105‐TDU‐B‐212‐134003). The grantee had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Contributor Information
Fang‐Ju Cheng, Email: fanju27@gmail.com.
Chuan‐Mu Chen, Email: chchen1@dragon.nchu.edu.tw.
Shu‐Ling Wang, Email: susan00376@gmail.com.
Wei‐Chien Huang, Email: whuang@mail.cmu.edu.tw.
References
- Adams JD, O'Mara‐Adams KJ and Hoffmann D (1987) Toxic and carcinogenic agents in undiluted mainstream smoke and sidestream smoke of different types of cigarettes. Carcinogenesis 8, 729–731. [DOI] [PubMed] [Google Scholar]
- Askari MD, Tsao MS and Schuller HM (2005) The tobacco‐specific carcinogen, 4‐ (methylnitrosamino)‐1‐ (3‐pyridyl)‐1‐butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta‐adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol 131, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SY, Hong JY, Lee HJ, Park HJ, Lee SK (2015) Targeting the degradation of AXL receptor tyrosine kinase to overcome resistance in gefitinib‐resistant non‐small cell lung cancer. Oncotarget 6, 10146–10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beau‐Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM, Massard G et al (2008) MET gene copy number in non‐small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol 3, 331–339. [DOI] [PubMed] [Google Scholar]
- Bell DW, Lynch TJ, Haserlat SM, Harris PL, Okimoto RA, Brannigan BW, Sgroi DC, Muir B, Riemenschneider MJ, Iacona RB et al (2005) Epidermal growth factor receptor mutations and gene amplification in non‐small‐cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol 23, 8081–8092. [DOI] [PubMed] [Google Scholar]
- Borgerding M and Klus H (2005) Analysis of complex mixtures – cigarette smoke. Exp Toxicol Pathol 57, 43–73. [DOI] [PubMed] [Google Scholar]
- Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I et al (2005) Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non‐small‐cell lung cancer. J Natl Cancer Inst 97, 643–655. [DOI] [PubMed] [Google Scholar]
- Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, Del Grammastro M, Sciarrotta MG, Buttitta F, Incarbone M et al (2009) Increased MET gene copy number negatively affects survival of surgically resected non‐small‐cell lung cancer patients. J Clin Oncol 27, 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N and Sliwkowski MX (2006) Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res 66, 8163–8171. [DOI] [PubMed] [Google Scholar]
- Cassinelli G, Favini E, Degl'Innocenti D, Salvi A, De Petro G, Pierotti MA, Zunino F, Borrello MG, Lanzi C (2009) RET/PTC1‐driven neoplastic transformation and proinvasive phenotype of human thyrocytes involve Met induction and beta‐catenin nuclear translocation. Neoplasia 11, 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti D, Venugopal D, Mailander PC, Meza JL, Higginbotham S, Cavalieri EL and Rogan EG (2008) The role of polycyclic aromatic hydrocarbon‐DNA adducts in inducing mutations in mouse skin. Mutat Res 649, 161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YT, Chang JW, Liu HP, Yu TF, Chiu YT, Hsieh JJ, Chen YT, Chen YR, Wu HD and Huang SF (2011) Clinical implications of high MET gene dosage in non‐small cell lung cancer patients without previous tyrosine kinase inhibitor treatment. J Thorac Oncol 6, 2027–2035. [DOI] [PubMed] [Google Scholar]
- Chen JT, Lin TS, Chow KC, Huang HH, Chiou SH, Chiang SF, Chen HC, Chuang TL, Lin TY and Chen CY (2006) Cigarette smoking induces overexpression of hepatocyte growth factor in type II pneumocytes and lung cancer cells. Am J Resp Cell Mol Biol 34, 264–273. [DOI] [PubMed] [Google Scholar]
- Chinai JM, Janakiram M, Chen F, Chen W, Kaplan M and Zang X (2015) New immunotherapies targeting the PD‐1 pathway. Trends Pharmacol Sci 36, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacic S, Flanagan M, Cieply K, Ramalingam S, Luketich J, Belani C and Yousem SA (2006) Significance of EGFR protein expression and gene amplification in non‐small cell lung carcinoma. Am J Clin Pathol 125, 860–865. [DOI] [PubMed] [Google Scholar]
- Danilkovitch‐Miagkova A and Zbar B (2002) Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Investig 109, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denissenko MF, Pao A, Tang M and Pfeifer GP (1996) Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 274, 430–432. [DOI] [PubMed] [Google Scholar]
- Eling T, Curtis J, Battista J and Marnett LJ (1986) Oxidation of (+)‐7,8‐dihydroxy‐7,8‐dihydrobenzo[a]pyrene by mouse keratinocytes: evidence for peroxyl radical‐ and monoxygenase‐dependent metabolism. Carcinogenesis 7, 1957–1963. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Feng Y, Thiagarajan PS and Ma PC (2012) MET signaling: novel targeted inhibition and its clinical development in lung cancer. J Thorac Oncol 7, 459–467. [DOI] [PubMed] [Google Scholar]
- Fontanini G, Vignati S, Bigini D, Mussi A, Lucchi H, Angeletti CA, Pingitore R, Pepe S, Basolo F and Bevilacqua G (1995) Epidermal growth factor receptor (EGFr) expression in non‐small cell lung carcinomas correlates with metastatic involvement of hilar and mediastinal lymph nodes in the squamous subtype. Eur J Cancer 31A, 178–183. [DOI] [PubMed] [Google Scholar]
- Fu P, Du F, Yao M, Lv K and Liu Y (2014) MicroRNA‐185 inhibits proliferation by targeting c‐Met in human breast cancer cells. Exp Therap Med 8, 1879–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HF, Yang JJ, Chen ZH, Zhang XC, Yan HH, Guo WB, Zhou Q, Gou LY, Dong ZY and Wu YL (2016) Plasma dynamic monitoring of soluble c‐Met level for EGFR‐TKI treatment in advanced non‐small cell lung cancer. Oncotarget 7, 39535–39543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett J, Chumbalkar V, Vaillant B, Gururaj AE, Hill KS, Latha K, Yao J, Priebe W, Colman H, Elferink LA et al (2013) Regulation of HGF expression by ΔEGFR‐mediated c‐Met activation in glioblastoma cells. Neoplasia 15, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF (2009) Activating and resistance mutations of EGFR in non‐small‐cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 28 (Suppl. 1), S24–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go H, Jeon YK, Park HJ, Sung SW, Seo JW and Chung DH (2010) High MET gene copy number leads to shorter survival in patients with non‐small cell lung cancer. J Thorac Oncol 5, 305–313. [DOI] [PubMed] [Google Scholar]
- Graff JW, Powers LS, Dickson AM, Kim J, Reisetter AC, Hassan IH, Kremens K, Gross TJ, Wilson ME and Monick MM (2012) Cigarette smoking decreases global microRNA expression in human alveolar macrophages. PLoS One 7, e44066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot P and Munden RF (2012) Lung cancer epidemiology, risk factors, and prevention. Radiol Clin North Am 50, 863–876. [DOI] [PubMed] [Google Scholar]
- Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL et al (2008) Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF‐binding proteins. J Clin Invest 118, 2609–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Cen H, Tan X, Liu W and Ke Q (2014) Prognostic value of MET gene copy number and protein expression in patients with surgically resected non‐small cell lung cancer: a meta‐analysis of published literatures. PLoS One 9, e99399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton M, Wolf JL, Rusk J, Beard SE, Clark GM, Witt K and Cagnoni PJ (2006) Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res 12, 2166–2171. [DOI] [PubMed] [Google Scholar]
- Harada A, Jogie‐Brahim S and Oh Y (2013) Tobacco specific carcinogen 4‐ (methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone suppresses a newly identified anti‐tumor IGFBP‐3/IGFBP‐3R system in lung cancer cells. Lung Cancer 80, 270–277. [DOI] [PubMed] [Google Scholar]
- Hecht SS (2003) Tobacco carcinogens, their biomarkers and tobacco‐induced cancer. Nat Rev Cancer 3, 733–744. [DOI] [PubMed] [Google Scholar]
- Hecht SS (2012) Lung carcinogenesis by tobacco smoke. Int J Cancer 131, 2724–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Lin Y, Chen H, Mao Y, Wu J, Zhu Y, Xu X, Xu X, Li S, Zheng X et al (2013) MicroRNA‐101 suppresses motility of bladder cancer cells by targeting c‐Met. Biochem Biophys Res Comm 435, 82–87. [DOI] [PubMed] [Google Scholar]
- Huang RY and Chen GG (2011) Cigarette smoking, cyclooxygenase‐2 pathway and cancer. Biochim Biophys Acta 1815, 158–169. [DOI] [PubMed] [Google Scholar]
- Huang RY, Li MY, Hsin MK, Underwood MJ, Ma LT, Mok TS, Warner TD and Chen GG (2011) 4‐Methylnitrosamino‐1‐3‐pyridyl‐1‐butanone (NNK) promotes lung cancer cell survival by stimulating thromboxane A2 and its receptor. Oncogene 30, 106–116. [DOI] [PubMed] [Google Scholar]
- Huang SF, Liu HP, Li LH, Ku YC, Fu YN, Tsai HY, Chen YT, Lin YF, Chang WC, Kuo HP et al (2004) High frequency of epidermal growth factor receptor mutations with complex patterns in non‐small cell lung cancers related to gefitinib responsiveness in Taiwan. Clin Cancer Res 10, 8195–8203. [DOI] [PubMed] [Google Scholar]
- Ishii T, Matsuse T, Igarashi H, Masuda M, Teramoto S and Ouchi Y (2001) Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S‐transferase P1. Am J Physiol Lung Cell Mol Physiol 280, L1189–L1195. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Pillai S and Chellappan SP (2012) Genetic and biochemical alterations in non‐small cell lung cancer. Biochem Res Int 2012, 940905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju L and Zhou C (2013) Association of integrin beta1 and c‐MET in mediating EGFR TKI gefitinib resistance in non‐small cell lung cancer. Cancer Cell Int 13, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenfield SA, Wei EK, Stampfer MJ, Rosner BA and Colditz GA (2008) Comparison of aspects of smoking among the four histological types of lung cancer. Tobacco Control 17, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr KM, Galler JS, Hagen JA, Laird PW and Laird‐Offringa IA (2007) The role of DNA methylation in the development and progression of lung adenocarcinoma. Dis Markers 23, 5–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, Li LY, Watkins CL, Sellers MV, Lowe ES et al (2008) Gefitinib versus docetaxel in previously treated non‐small‐cell lung cancer (INTEREST): a randomised phase III trial. Lancet 372, 1809–1818. [DOI] [PubMed] [Google Scholar]
- Kłosek SK, Sporny S, Stasikowska‐Kanicka O and Kurnatowska AJ (2011) Cigarette smoking induces overexpression of c‐Met receptor in microvessels of oral lichen planus. Arch Med Sci 7, 706–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy S, Kenteti R, Duke‐Cohan JS, Loganathan S, Liu W, Ma PC, Sattler M, Singleton PA, Ramnath N, Innocenti F et al (2009) Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res 15, 5714–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laag E, Majidi M, Cekanova M, Masi T, Takahashi T and Schuller HM (2006) NNK activates ERK1/2 and CREB/ATF‐1 via beta‐1‐AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int J Cancer 119, 1547–1552. [DOI] [PubMed] [Google Scholar]
- Lai AZ, Abella JV and Park M (2009) Crosstalk in Met receptor oncogenesis. Trends Cell Biol 19, 542–551. [DOI] [PubMed] [Google Scholar]
- Lawrence RE and Salgia R (2010) MET molecular mechanisms and therapies in lung cancer. Cell Adh Migr 4, 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Han JY , Kim HT and Lee JS (2006) Gefitinib is of more benefit in chemotherapy‐naive patients with good performance status and adenocarcinoma histology: retrospective analysis of 575 Korean patients. Lung Cancer 53, 339–345. [DOI] [PubMed] [Google Scholar]
- Li X, Sun X, Wu J and Li Z (2016b) MicroRNA‐613 suppresses proliferation, migration and invasion of osteosarcoma by targeting c‐MET. Am J Cancer Res 6, 2869–2879. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li B, Yang XX, Wang D and Ji HK (2016a) MicroRNA‐138 inhibits proliferation of cervical cancer cells by targeting c‐Met. Eur Rev Med Pharmacol Sci 20, 1109–1114. [PubMed] [Google Scholar]
- Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF and Wang YC (2010) The tobacco‐specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest 120, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG et al (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 350, 2129–2139. [DOI] [PubMed] [Google Scholar]
- Maione P, Sacco PC, Sgambato A, Casaluce F, Rossi A and Gridelli C (2015) Overcoming resistance to targeted therapies in NSCLC: current approaches and clinical application. Ther Adv Med Oncol 7, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass MJ, Jeffers AJ, Ross JA, Nelson G, Galati AJ, Stoner GD and Nesnow S (1993) Ki‐ras oncogene mutations in tumors and DNA adducts formed by benz[j]aceanthrylene and benzo[a]pyrene in the lungs of strain A/J mice. Mol Carcinog 8, 186–192. [DOI] [PubMed] [Google Scholar]
- Matzinger SA, Crist KA, Stoner GD, Anderson MW, Pereira MA, Steele VE, Kelloff GJ, Lubet RA and You M (1995) K‐ras mutations in lung tumors from A/J and A/J x TSG‐p53 F1 mice treated with 4‐ (methylnitrosamino)‐1‐ (3‐pyridyl)‐1‐butanone and phenethyl isothiocyanate. Carcinogenesis 16, 2487–2492. [DOI] [PubMed] [Google Scholar]
- McMillan DH, Baglole CJ, Thatcher TH, Maggirwar S, Sime PJ and Phipps RP (2011) Lung‐targeted overexpression of the NF‐κB member RelB inhibits cigarette smoke‐induced inflammation. Am J Pathol 279, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VA, Kris MG, Shah N, Patel J, Azzoli C, Gomez J, Krug LM, Pao W, Rizvi N, Pizzo B et al (2004) Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non‐small‐cell lung cancer. J Clin Oncol 22, 1103–1109. [DOI] [PubMed] [Google Scholar]
- Min HY, Boo HJ, Lee HJ, Jang HJ, Yun HJ, Hwang SJ, Smith JK, Lee HJ and Lee HY (2016) Smoking‐associated lung cancer prevention by blockade of the beta‐adrenergic receptor‐mediated insulin‐like growth factor receptor activation. Oncotarget 7, 70936–70947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsudomi T and Yatabe Y (2007) Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci 2007, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Yang P, Cassivi SD, Schild SE and Adjei AA (2008) Non‐small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clinic proceedings. Mayo Clinic 83, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov VM, Massoll NA, Vladimirova OV, Maul GG and Ishov AM (2008) Regulation of c‐met expression by transcription repressor Daxx. Oncogene 27, 2177–2186. [DOI] [PubMed] [Google Scholar]
- Nilsson MB, Sun H, Diao L, Tong P, Liu D, Li L, Fan Y, Poteete A, Lim SO, Howells K et al 2017. Stress hormones promote EGFR inhibitor resistance in NSCLC: implications for combinations with beta‐blockers. Sci Transl Med 9, eaao4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Yoshioka K, Bereczky B and Itoh K (2008) Evidence for efficient phosphorylation of EGFR and rapid endocytosis of phosphorylated EGFR via the early/late endocytic pathway in a gefitinib‐sensitive non‐small cell lung cancer cell line. Mol Cancer 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble ME, Endicott JA and Johnson LN (2004) Protein kinase inhibitors: insights into drug design from structure. Science 303, 1800–1805. [DOI] [PubMed] [Google Scholar]
- Normanno N, Bianco C, Strizzi L, Mancino M, Maiello MR, De Luca A, Caponigro F and Salomon DS (2005) The ErbB receptors and their ligands in cancer: an overview. Curr Drug Targets 6, 243–257. [DOI] [PubMed] [Google Scholar]
- Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y and Mitsudomi T (2009) Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol 4, 5–11. [DOI] [PubMed] [Google Scholar]
- Organ SL and Tsao MS (2011) An overview of the c‐MET signaling pathway. Ther Advan Med Oncol 3, S7–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozasa H, Oguri T, Maeno K, Takakuwa O, Kunii E, Yagi Y, Uemura T, Kasai D, Miyazaki M and Niimi A (2014) Significance of c‐MET overexpression in cytotoxic anticancer drug‐resistant small‐cell lung cancer cells. Cancer Sci 105, 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ et al (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L et al (2004) EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 101, 13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Choi YL, Sung CO, An J, Seo J, Ahn MJ, Ahn JS, Park K, Shin YK, Erkin OC et al (2012) High MET copy number and MET overexpression: poor outcome in non‐small cell lung cancer patients. Histol Histopathol 27, 197–207. [DOI] [PubMed] [Google Scholar]
- Pazhouhandeh M, Samiee F, Boniadi T, Khedmat AF, Vahedi E, Mirdamadi M, Sigari N, Siadat SD, Vaziri F, Fateh A et al (2017) Comparative network analysis of patients with non‐small cell lung cancer and smokers for representing potential therapeutic targets. Sci Rep 7, 13812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer JT, Evgrafov OV, Bergman MY, Friez M, Haiman CA, Levitt P and Aldinger KA (2013) Transcriptional regulation of the MET receptor tyrosine kinase gene by MeCP2 and sex‐specific expression in autism and Rett syndrome. Transl Psychiat 3, e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratovitski EA (2010) LKB1/PEA3/ΔNp63 pathway regulates PTGS‐2 (COX‐2) transcription in lung cancer cells upon cigarette smoke exposure. Oxid Med Cell Longev 3, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riely GJ, Politi KA, Miller VA and Pao W (2006) Update on epidermal growth factor receptor mutations in non‐small cell lung cancer. Clin Cancer Res 12, 7232–7241. [DOI] [PubMed] [Google Scholar]
- Ruggeri B, DiRado M, Zhang SY, Bauer B, Goodrow T and Klein‐Szanto AJ (1993) Benzo[a]pyrene‐induced murine skin tumors exhibit frequent and characteristic G to T mutations in the p53 gene. Proc Natl Acad Sci USA 90, 1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti M and Baselga J (2006) The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 12, 5268–5272. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J and Haber DA (2007) Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 7, 169–181. [DOI] [PubMed] [Google Scholar]
- Shi J, Yao D, Liu W, Wang N, Lv H, He N, Shi B, Hou P, Ji M (2012) Frequent gene amplification predicts poor prognosis in gastric cancer. Int J Mol Sci. 13, 4714–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T (2006) Xenobiotic‐metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab Pharmacokinet 21, 257–276. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD and Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66, 7–30. [DOI] [PubMed] [Google Scholar]
- Song Z, Wang X, Zheng Y, Su H and Zhang Y (2017) MET gene amplification and overexpression in Chinese Non‐small‐cell lung cancer patients without EGFR mutations. Clin Lung Cancer 18(213–219), e212. [DOI] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA and Settleman J (2004) Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science 305, 1163–1167. [DOI] [PubMed] [Google Scholar]
- Su Y, Han W, Giraldo C, De Li Y and Block ER (1998) Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 19, 819–825. [DOI] [PubMed] [Google Scholar]
- Sundar IK, Chung S, Hwang JW, Lapek JD Jr, Bulger M, Friedman AE, Yao H, Davie JR and Rahman I (2012) Mitogen‐ and stress‐activated kinase 1 (MSK1) regulates cigarette smoke‐induced histone modifications on NF‐κB‐dependent genes. PLoS One 7, e31378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Ohe Y, Kusumoto M, Tateishi U, Yamamoto S, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Tamura T et al (2004) Risk factors for interstitial lung disease and predictive factors for tumor response in patients with advanced non‐small cell lung cancer treated with gefitinib. Lung Cancer 45, 93–104. [DOI] [PubMed] [Google Scholar]
- Talhout R, Schulz T, Florek E, van Benthem J, Wester P and Opperhuizen A (2011) Hazardous compounds in tobacco smoke. Int J Environment Res Public Health 8, 613–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanizaki J, Okamoto I, Sakai K and Nakagawa K (2011) Differential roles of trans‐phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br J Cancer 105, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielen A, Klus H and Muller L (2008) Tobacco smoke: unraveling a controversial subject. Exp Toxicol Pathol 60, 141–156. [DOI] [PubMed] [Google Scholar]
- Thun MJ, Hannan LM, Adams‐Campbell LL, Boffetta P, Buring JE, Feskanich D, Flanders WD, Jee SH, Katanoda K, Kolonel LN et al (2008) Lung Cancer occurrence in never‐smokers: an analysis of 13 cohorts and 22 cancer registry studies. PLoS Med 5, e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel‐Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M et al (2005) Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med 353, 133–144. [DOI] [PubMed] [Google Scholar]
- Yamashita Y, Akatsuka S, Shinjo K, Yatabe Y, Kobayashi H, Seko H, Kajiyama H, Kikkawa F, Takahashi T and Toyokuni S (2013) Met is the most frequently amplified gene in endometriosis‐associated ovarian clear cell adenocarcinoma and correlates with worsened prognosis. PLoS One 8, e57724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I (2006) Cigarette smoke induces proinflammatory cytokine release by activation of NF‐kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 291, L46–L57. [DOI] [PubMed] [Google Scholar]
- Ye S, Li J, Hao K, Yan J and Zhou H (2016) The efficacy and risk profile of c‐Met inhibitors in non‐small cell lung cancer: a meta‐analysis. Sci Rep 6, 35770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X and Chang A (2008) Molecular predictors of EGFR‐TKI sensitivity in advanced non‐small cell lung cancer. Int J Med Sci 5, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Xie F, Gao W, Yu S, Qiu L, Lin W, Sun Y and Jia T (2015) Expression of hepatocyte growth factor and c‐Met in non‐small‐cell lung cancer and association with lymphangiogenesis. Mol Med Rep 11, 2797–2804. [DOI] [PubMed] [Google Scholar]
- Zheng HC and Takano Y (2011) NNK‐induced lung tumors: a review of animal model. J Oncol 2011, 635379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Yan D, Chen X, Huang H, Chen K, Li G, Zhou L, Zheng D, Tu L and Dong XD (2015) MicroRNA‐206: effective inhibition of gastric cancer progression through the c‐Met pathway. PLoS One 10, e0128751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. EGFR mutations were not found in CSE‐ or B[α]P‐treated cells. The EGFR cDNA in H292/1%CSE (A), H292/5%CSE (B) or H292/B[α]P cells (C) were prepared by RT‐qPCR, and sequenced.
Fig. S2. c‐MET phosphorylation and protein expression in NSCLC cells in response to cigarette smoke and its oncogene ingredients. Total protein lysate of H292, PC9 and HCC827 cells and their CSE, B[α]P and NNK‐treated clones were prepared and subjected to western blot analysis with indicated antibodies.