

Abstract
Rationale
Alveolar epithelial cell (AEC) injury and mitochondrial dysfunction are important in the development of lung fibrosis. Our group has shown that in the asbestos exposed lung, the generation of mitochondrial reactive oxygen species (ROS) in AEC mediate mitochondrial DNA (mtDNA) damage and apoptosis which are necessary for lung fibrosis. These data suggest that mitochondrial-targeted antioxidants should ameliorate asbestos-induced lung.
Objective
To determine whether transgenic mice that express mitochondrial-targeted catalase (MCAT) have reduced lung fibrosis following exposure to asbestos or bleomycin and, if so, whether this occurs in association with reduced AEC mtDNA damage and apoptosis.
Methods
Crocidolite asbestos (100 μg/50 μL), TiO2 (negative control), bleomycin (0.025 units/50 μL), or PBS was instilled intratracheally in 8–10 week-old wild-type (WT - C57Bl/6 J) or MCAT mice. The lungs were harvested at 21 d. Lung fibrosis was quantified by collagen levels (Sircol) and lung fibrosis scores. AEC apoptosis was assessed by cleaved caspase-3 (CC-3)/Surfactant protein C (SFTPC) immunohistochemistry (IHC) and semi-quantitative analysis. AEC (primary AT2 cells from WT and MCAT mice and MLE-12 cells) mtDNA damage was assessed by a quantitative PCR-based assay, apoptosis was assessed by DNA fragmentation, and ROS production was assessed by a Mito-Sox assay.
Results
Compared to WT, crocidolite-exposed MCAT mice exhibit reduced pulmonary fibrosis as measured by lung collagen levels and lung fibrosis score. The protective effects in MCAT mice were accompanied by reduced AEC mtDNA damage and apoptosis. Similar findings were noted following bleomycin exposure. Euk-134, a mitochondrial SOD/catalase mimetic, attenuated MLE-12 cell DNA damage and apoptosis. Finally, compared to WT, asbestos-induced MCAT AT2 cell ROS production was reduced.
Conclusions
Our finding that MCAT mice have reduced pulmonary fibrosis, AEC mtDNA damage and apoptosis following exposure to asbestos or bleomycin suggests an important role for AEC mitochondrial H2O2-induced mtDNA damage in promoting lung fibrosis. We reason that strategies aimed at limiting AEC mtDNA damage arising from excess mitochondrial H2O2 production may be a novel therapeutic target for mitigating pulmonary fibrosis.
1. Introduction
Mitochondrial reactive oxygen species (ROS) are implicated in the pathogenesis of aging and lung diseases, some of which include idiopathic pulmonary fibrosis (IPF), asbestosis, chronic obstructive lung disease (COPD), and lung cancer (see for reviews: [1–7]). ROS, including H2O2, oxidize multiple cellular targets (i.e. DNA, proteins, and lipids) which activate a wide range of biological processes, such as mitochondrial dysfunction, DNA damage-response (i.e. p53 activation), apoptosis, altered cell growth, and signal transduction that can result in tissue injury, aberrant wound healing, and fibrosis [1–6]. Alveolar epithelial cell (AEC) injury from ‘exaggerated’ lung aging and mitochondrial dysfunction is prominently involved in the pathogenesis of pulmonary fibrosis [5–11]. In asbestos-exposed AEC, our group has shown that the generation of mitochondrial ROS is necessary for asbestos-induced mitochondrial DNA (mtDNA) damage and apoptosis via the mitochondrial dependent (intrinsic) pathway. In mice that were deficient in an oxidant-induced DNA repair enzyme, 8-oxoguanine DNA glycosylase (Ogg1−/−), we found there was increased alveolar type II (AT2) cell mitochondrial dysfunction, mtDNA damage, intrinsic apoptosis, and lung fibrosis [12–17]. Collectively, these data support a link between oxidant-induced AEC mtDNA damage and apoptosis in the pathophysiology of pulmonary fibrosis, however, evidence that mitochondrial ROS are necessary for the development of asbestos-mediated lung fibrosis are lacking.
Transgenic mitochondria-targeted human catalase enforced expression (MCAT) mice have a prolonged lifespan associated with reduced mitochondrial H2O2 production, mtDNA damage, and preserved mitochondrial function [18]. Compared to wild-type (WT), MCAT mice are less susceptible to/are protected against degenerative diseases involving the brain, cardiac fibrosis, pulmonary hypertension, and lung cancer [18–26]. However, the role of MCAT in preventing AEC mitochondria dysfunction and lung fibrosis has not been investigated.
We reasoned that MCAT mice would be protected from lung fibrosis in part by mechanisms involving reduced AT2 cell mitochondrial ROS production, mtDNA damage, and apoptosis. In this study, we show that MCAT mice have decreased lung fibrosis following exposure to asbestos or bleomycin as compared to WT mice. Further, we demonstrate that AT2 cells from MCAT mice have reduced AEC mtDNA damage, apoptosis, and mitochondrial ROS production following exposure to asbestos. Taken together, these data suggest an important role for AEC mitochondrial catalase in limiting mtDNA damage and apoptosis following exposure to fibrogenic agents such as asbestos or bleomycin.
2. Material and methods
2.1. Reagents
Crocidolite and amosite amphibole asbestos fibers employed were Union International Centere le Cancer (UICC) reference standards kindly supplied by Dr. Andy Ghio (U.S. Environmental Protection Agency) as characterized previously [13,14]. Euk-134 were purchased from Cayman chemical (Ann Arbor, MI). All other reagents were purchased from Sigma (St. Louis, MO) unless specified. Anti-human catalase antibody was purchased from Athens Research and Technology (Athens, GA), and anti-cytochrome oxidase IV antibody was purchased from Cell Signaling Technology (Danvers, MA).
2.2. Animals
The Institutional Animal Care and Use Committee (IACUC) at Northwestern University and the Jesse Brown VA Medical Center approved all animal studies herein. Male and female 8- to 10- week old C57Bl/6 J wild-type (WT) mice (Jackson Labs, Bar Harbor, ME) and MCAT (C57Bl/6 J background; kindly provided by Peter S. Rabinovitch; [18]) were used for lung fibrosis studies [16,27,28]. Using a Mitochondria Isolation kit (Thermo Fisher Scientific Inc., Rockford, IL) according to the manufacturer’s recommendations as we previously described [6], we obtained mitochondrial protein from lung and heart to show the mitochondrial catalase protein.
2.3. Asbestos preparation and instillation into mice
Intratracheal instillation of asbestos or TiO2 to induce pulmonary fibrosis was performed as described [16]. Stock solutions (2 mg/mL) of crocidolite asbestos was prepared in Phosphate Buffered Saline (PBS) and 15 mM HEPES (Sigma, St. Louis, MO) and sonicated at 40% power (Sonicator: Branson, Danbury, CT) for 8 min to disrupt fiber clumps. Eight to ten week-old male or female WT (C57Bl/6J) or MCAT-EE mice were anesthetized with 3% isofluorane (Butler Animal Health, Dublin, OH), intubated orally with a 20-gauge angiocatheter (BD, Sandy, UT), and 100 μg of crocidolite asbestos or TiO2 (negative control particle) suspended in 50 μL sterile PBS was instilled in 2 equal aliquots given 2 min apart. After each aliquot the mice were placed in the right and then the left decubitus position for 10–15 s.
2.4. Bleomycin preparation and instillation into mice
Intratracheal instillation of bleomycin to induce pulmonary fibrosis was performed as we have described [27,28]. Mice were anesthetized and intubated as described above. Bleomycin (0.025 units in 50 μL normal saline, APP Pharmaceuticals, Schaumberg, IL) or 50 μL normal saline (control) was administered in two equal aliquots given 2 min apart. After each aliquot, the mice were placed in the right and then the left decubitus position for 10–15 s.
2.5. Lung harvest and semi-quantitative analysis of cleaved caspase-3 (CC-3) and surfactant protein C (SFTPC) immunohistochemistry (IHC)
The lungs were harvested 21 days after instillation of asbestos, bleomycin, or control to assess the histology as well as CC-3 and SFTPC co-localization by IHC as we have previously described [16]. Briefly, a 20-gauge angiocatheter was sutured into the trachea, the right lung was ligated at the hilum and after removal of the left lung (which was saved separately for collagen determination) was inflated to 15 cm H2O with 10% formalin. The right lung was then fixed in paraffin and serial 5 μm sections were stained for hematoxylin and eosin, Masson’s trichrome, and subject to CC-3 and pro-SFTPC IHC. In serial sections, co-localization of the number of CC-3 and SFTPC positive AEC were counted per alveolar branch point, and expressed as the number of CC-3 or SFTPC positive cells per 220 AEC ± SEM from 3 or more animals in each group.
2.6. Lung collagen detection
For soluble collagen measurement, the left lung was homogenized with acetic acid using a polytron (Kinematica, Bohemia, NY) then a dounce homogenizer and cleared by centrifugation. Equal volumes of cleared homogenate were subject to the Sircol assay for soluble collagen based on a modified Picosirius Red collagen precipitation assay as previously described by our group [16,27].
2.7. Fibrosis scoring system
The lung fibrosis score, which is based upon the severity and extent of lung fibrosis and not inflammation present in the peribronchial and interstitial tissues, was assessed by one of us who is a pulmonary pathologist (AY) as previously described [16]. Lungs were assigned a severity score from 0 (no fibrosis) to 4 (severe fibrosis) while the extent of involvement was quantified on a scale of 1 (occasional alveolar duct and bronchiole involvement) to 3 (more than half of the alveolar ducts and respiratory bronchioles involved). The fibrosis score was calculated as the severity (0–4) multiplied by the extent (1–3).
2.8. Murine alveolar type II cell isolation
Murine AT2 cells were isolated as initially described [30] and modified by our group [14–17]. The AT2 cells obtained routinely display > 90% purity as assessed by pro-SFTPC (Millipore, Temecula, CA) immunofluorescence analysis and > 95% viability via trypan blue staining.
2.9. Cell culture
The MLE-12 cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cultured AECs were maintained in DMEM (Invitrogen, Grand Island, NY) with 2 mM L-glutamine supplemented with 10% fetal bovine serum, penicillin (100 units/mL) and streptomycin (100 μg/mL). Cells were plated in 6-well plates or 100-mm dishes and grown to confluence before adding asbestos or H2O2 for up to 24 h as previously described [13] and harvesting protein extracts for Western blotting or, in separate experiments, obtaining nuclear and mitochondrial DNA for DNA fragmentation and a PCR-based DNA damage assay.
2.10. Quantitative PCR-based mtDNA damage assay
Nuclear and mtDNA damage were assessed by Q-PCR exactly as previously described [15]. Briefly, genomic DNA was extracted using the Qiagen Genomic-Tip 20/G and Qiagen DNA Buffer Set (Qiagen, Gaithersberg, MD) per the manufacturer’s protocol. PCR was performed using Ex-Taq (Clontech, Mountain View, CA) with specific primers to amplify a mitochondrial genomic fragment in both short and long form and nuclear DNA (beta globin) as previously described [15]. DNA was quantified by Pico-Green (Life Technologies) using the FL600 microplate fluorescence reader with excitation and emission wavelengths of 485 nm and 530 nm, respectively. The data obtained from the mitochondria small fragment were used to normalize the results of the mitochondria long fragment. The number of mitochondrial lesions was calculated by the equation, D=(1 – 2−(Δlong-Δshort)) × 10,000 (bp)/size of the long fragment (bp).
2.11. DNA fragmentation assay
Apoptosis was evaluated using a histone-associated DNA fragmentation (mono- and oligonucleosomes) Cell Death Detection ELISAPLUS kit (Sigma-Aldrich, St Louis, MO) as previously described using the manufacturer’s protocol [12–17].
2.12. Assessment of mitochondrial ROS production
To detect mitochondrial ROS in AT2 cell from WT or MCAT-EE mice, we used a redox-sensitive MitoSOX Red O2− reagent (Molecular Probes, Eugene, OR), which detects O2− as well as other mitochondrial free radicals [29]. Briefly, the cells were exposed to asbestos or H2O2 for 3 h and incubated with 2.5 μM MitoSOX Red for the final 10 min. Cells were fixed with 4% paraformaldehyde and counterstained with Hoechst (Molecular Probes, Eugene, OR). Cells were imaged on a Zeiss Axioskiop, using identical detector settings for the red channel. Semi-quantitative analysis of relative fluorescence intensity was obtained via Image J. Identically sized regions in AT2 cells from both group of mice were drawn on ~40 cells per condition on monochrome red channel images. Scatterplots of relative fluorescence were generated using Graph Pad prism.
2.13. Statistical analysis
The results of each experimental in vitro condition were determined from the mean of duplicate or triplicate trials. The data were expressed as the means ± SEM (n=3 unless otherwise stated). For the in vivo studies, 6 animals per group were used unless noted. An independent sample two-tailed Student’s t-test was used to assess the significance between two groups. Analysis of variance was used when comparing more than two groups to a single control; differences between two groups within the set were analyzed by a Fisher’s protected least significant differences test. Probability values < 0.05 were considered significant.
3. Results
3.1. Asbestos- and bleomycin-induced pulmonary fibrosis is attenuated in MCAT mice
To determine whether MCAT overexpression reduces lung fibrosis, we studied WT and MCAT mice using both the asbestos and bleomycin model of lung fibrosis as we previously [16,27,28]. We confirmed that all of our MCAT mice overexpressed the transgene based upon PCR on genomic DNA obtained from tail samples of MCAT and WT mice to identify a 645-bp product of the MCAT gene as compared with the 324-bp product in WT mice (Fig. 1A). As previously described in MCAT mice [18,20], human catalase protein (62 kDa) and catalase activity are markedly increased in heart mitochondrial homogenates as compared to WT mice; we extended these findings to show that human catalase expression is also increased in lung homogenates in MCAT as compared to WT mice (Fig. 1B). As anticipated, crocidolite asbestos (100 μg in 50 μL PBS) induced significant bronchoalveolar duct (BAD) junction-centered lung fibrosis in WT mice at 3 weeks as compared to TiO2 (100 μg TiO2 in 50 μL PBS) (Fig. 2A and B) which was associated with increased lung fibrosis scores (Fig. 2C) and lung collagen levels (Fig. 2D). Similar to WT, MCAT mice exposed to TiO2 had normal lung architecture and negligible increases in lung fibrosis score and lung collagen (Fig. 2). Notably, asbestos-induced lung fibrosis was significantly decreased in MCAT mice as assessed by histopathology, lung fibrosis scoring and lung collagen levels (Fig. 2). To test the generalizability of our findings, we used the bleomycin model of lung fibrosis. As expected, a single IT instillation of bleomycin (0.025 IU in 50 μL PBS) significantly increased pulmonary fibrosis at 21d in WT mice as assessed histology, fibrosis score and collagen levels (Fig. 3). Compared to WT, MCAT mice were significantly protected against bleomycin-induced fibrosis in a manner similar to asbestos. These findings demonstrate that MCAT mice are less susceptible to asbestos- and bleomycin-induced lung fibrosis as compared to their WT counterparts.
Fig. 1.
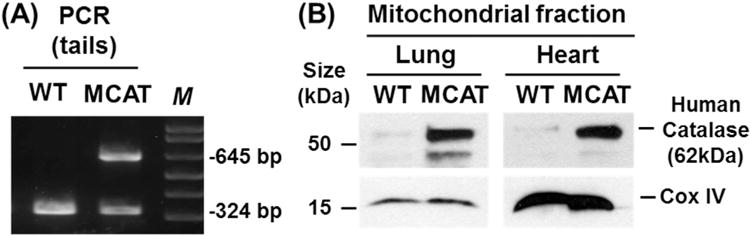
Phenotypic validation of mice expressing human mitochondrial catalase (MCAT) targeted to the mitochondrion. A. Genomic PCR of wild-type (WT) and catalase overexpressing (MCAT tail DNA: 645 bp MCAT and 324 bp (WT) amplification product. B. Lung and heart mitochondrial fractions from WT and MCAT mice showing an overabundance of human catalase protein in MCAT transgenic mice (Upper panels) as compared to WT mice. Cytochrome Oxidase IV (COX IV) was used as a loading control for mitochondrial protein (lower panels).
Fig. 2.
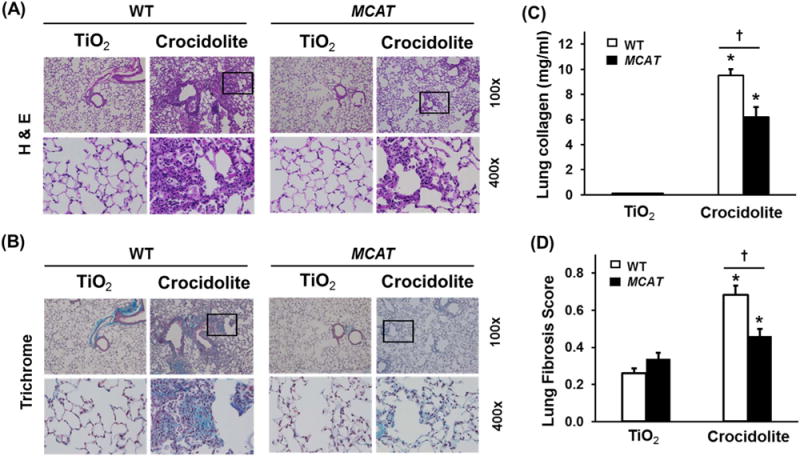
Compared to WT mice, MCAT mice are protected from pulmonary fibrosis after asbestos exposure. WT and MCAT Mice were treated with a single intra-tracheal (IT)-instillation of 100 μg in 50 μL PBS titanium dioxide (TiO2, an inert particle) or 100 μg crocidolite asbestos in 50 μL PBS and three weeks later the lungs were harvested as described in the Material and Methods and subjected to hematoxylin and eosin (H & E, A) and trichrome (B) staining, lung collagen levels (C) and lung fibrosis scores (D) as described in the Material and Methods. (A and B) Shown is representative histology from 4 to 9 mice in each group. Upper row, scale bar=0.05 mm; lower row scale bar=0.1 mm. Squares on the upper rows were enlarged in the respective lower rows. TiO2-WT or MCAT (n=4–6); crocidolite-WT or MCAT (n=7–9). The Fibrosis score =(severity: 0–4)×(extent: 1–3). (C) Lung fibrosis scores. *p < 0.05 vs. TiO2, †p < 0.001 vs. WT+crocidolite asbestos, n =4–6. (D) Collagen levels. *p < 0.05 vs. TiO2, †p < 0.05 vs. WT+crocidolite asbestos. n=7–9.
Fig. 3.
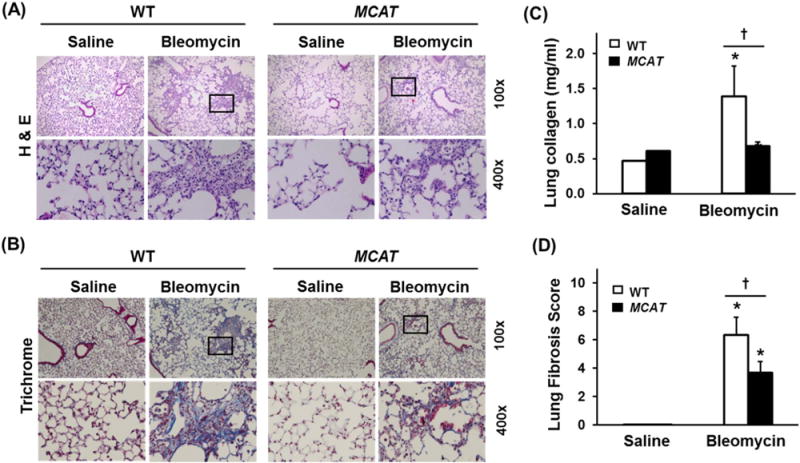
MCAT mice are protected against bleomycin-induced pulmonary fibrosis. WT and MCAT mice were treated with a single IT-instillation of 50 μL saline or 0.025 U bleomycin in 50 μL saline and three weeks later the lungs were harvested for H & E (A), Trichrome staining (B), lung collagen levels (C) and lung fibrosis scores (D) as described in the METHODS. Squares on the upper rows were enlarged in the respective lower rows. Upper row, scale bar=0.05 mm; lower row scale bar=0.1 mm. *p < 0.05 vs. saline, †p < 0.001 vs. WT+bleomycin, n =4–6.
3.2. Asbestos-induced AEC mtDNA damage and apoptosis are decreased in MCAT mice
Asbestos-induced fibrotic changes and AEC apoptosis are first evident in cells, including AT2 cells, located at the BAD junction where asbestos fiber deposition initially occurs [3,16,30]. To determine whether MCAT mice have fewer apoptotic cells at the BAD junction as compared to WT mice, we evaluated CC-3 staining using IHC and semi-quantitative analysis of serial mouse lung sections as we previously described [16]. TiO2 induced negligible increases in CC-3 positive cells at the BAD junctions in WT and MCAT mice (0.8 vs. 1.0% positive cells per 220 BAD junction cells, respectively, p= NS). However, in WT mice, asbestos significantly increased CC-3 positive cells at the BAD junction in WT mice (0.8 vs. 10% positive cells per 220 BAD junction cells, respectively, p < 0.05 vs. TiO2) and this was significantly reduced in MCAT mice (10% vs. 3% positive cells per 220 BAD junction cells, respectively, p < 0.05 vs. WT) (Fig. 4A and B). Furthermore, IHC apoptosis co-localization studies using CC-3 and SPFTC confirmed the presence of apoptotic AT2 cells in asbestos-exposed WT mice was reduced in MCAT mice though this difference did not reach statistical significance (4% vs. 3% co-positive cells per 220 BAD junction cells, respectively) (Fig. 4C).
Fig. 4.
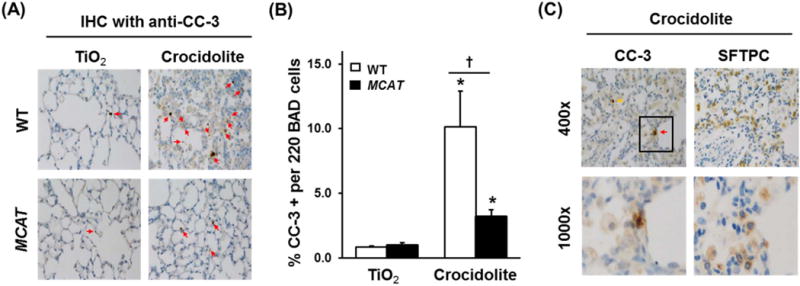
Asbestos-induced apoptosis (CC-3 activation) in cells at the BAD junction is reduced in MCAT mice versus WT mice and co-localization of CC-3- and SFTPC-(AT2)-positive cells in asbestos-exposed mice. Lungs from WT and MCAT mice were harvested 3 weeks after the IT-instillation of TiO2 or crocidolite asbestos and serial sections were subjected to IHC for CC-3 (marker of apoptosis) or SFTPC (marker for AT2 cells) IHC as detailed in the Material and Methods. (A) Apoptosis as assessed by anti CC-3 IHC in WT mice (top row) and MCAT (bottom row). (B) Semi-quantitative analysis of 220 cells at the BAD junctions. *p < 0.05 vs. TiO2, †p < 0.001 vs. WT+crocidolite asbestos, n =4–6. (C) IHC for CC-3 and SFTPC was performed on serial lung sections. Red arrows mark CC-3 and SFTPC positive cells (apoptotic AT2 cell). Yellow arrows mark CC-3 positive, SFTPC negative cells (apoptotic non-AT2 cells). Squares on the upper rows were enlarged in the respective lower rows.
We previously reported a causal role for oxidant-induced AEC mtDNA damage in mediating asbestos-induced intrinsic AEC apoptosis in vitro [14,15] and in vivo [16]. Work by others in non-lung cells suggests that MCAT overexpression reduces mtDNA damage thereby reducing mitochondrial ROS production and preserving mitochondrial function [18,24]. To directly test whether MCAT overexpression decreases asbestos-induced AT2 cell mtDNA damage, we exposed WT and MCAT mice to IT-instilled TiO2 or crocidolite asbestos for 3 weeks and then evaluated mtDNA damage in primary isolated AT2 cells. As expected, AT2 cells from WT mice had increased mtDNA damage following exposure to crocidolite asbestos as compared to TiO2 (2.6 vs. 1 ratio of lesions/10 kB frequency per fragment, p < 0.05; Fig. 5A). Remarkably, AT2 cells from asbestos-exposed MCAT mice, as compared to WT mice, had reduced mtDNA damage (1.1 vs. 2.6 lesions/10kB frequency per fragment, p < 0.05; Fig. 5A) which was comparable to our negative control particle, TiO2. As an alternative in vitro approach, we isolated primary AT2 cells from WT and MCAT mice and exposed them to amosite asbestos (25 μg/cm2) or H2O2 (250 μM) for 24 h. Asbestos- and H2O2-induced mtDNA damage in AT2 cells from WT mice were completely blocked in AT2 cells from MCAT mice (Fig. 5B). Collectively, these data show that MCAT blocks oxidant-induced AT2 cell mtDNA damage.
Fig. 5.
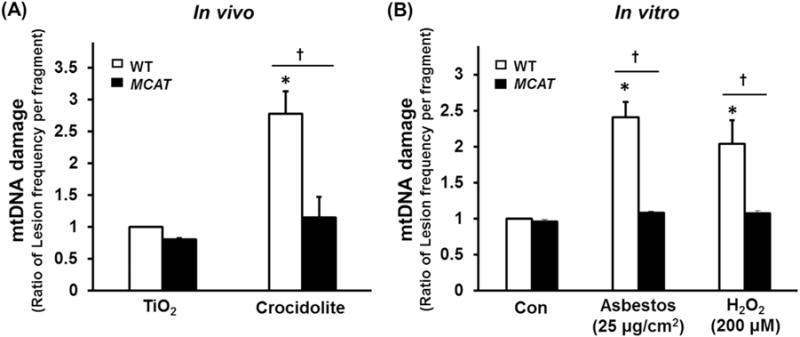
Compared to WT, AT2 cell from MCAT mice have reduced mtDNA damage after asbestos exposure in vivo (A) and in vitro (B). (A) Primary AT2 cells were isolated from the lungs of WT and MCAT mice three weeks after IT instillation of TiO2 or crocidolite asbestos and mtDNA damage were assessed by a fluorescent-based PCR mtDNA damage assay as described in the Material and Methods (expressed as the ratio of lesion frequency per fragment as compared to WT AT2 cells exposed to control – TiO2). *p < 0.05 vs. TiO2, †P < 0.05 vs. WT +crocidolite asbestos, n=4. (B) Primary AT2 cells were isolated from WT and MCAT mice, then treated with amosite asbestos or H2O2 in vitro and mtDNA damage was assessed as above (expressed as the ratio of lesion frequency per fragment as compared to WT AT2 cells exposed to control media). *p < 0.05 vs. Control of WT, †p < 0.05 vs. WT, n=4.
3.3. Euk-134 prevents oxidative stress-induced AEC mtDNA damage and apoptosis
Using highly sensitive ratiometric sensors (RoGFP) to detect ROS production in the mitochondria and cytosol, asbestos fibers and particulate matter preferentially induce mitochondrial ROS production. Euk-134, a cell-permeable combined SOD and catalase mimetic, prevents mitochondrial ROS production as well as toxin-induced endoplasmic reticulum (ER) stress response and intrinsic apoptosis in a variety of cells types including primary isolated rat AT2 cells [17,31–34]. To determine whether Euk-134 attenuates oxidant-induced AEC mtDNA damage that promotes apoptosis, we examined the protective effects of Euk-134 (20 μM) against amosite asbestos (25 μg/cm2) and H2O2 (200 μM)-induced MLE-12 cell mtDNA damage and apoptosis. Euk-134 completely blocked both asbestos- and H2O2-induced mtDNA damage (Fig. 6A) as well as apoptosis at 24 h (Fig. 6B). Taken together with the previous studies, these findings suggest an important role for mitochondria-derived ROS in mediating asbestos-induced AEC mtDNA damage that results in apoptosis.
Fig. 6.
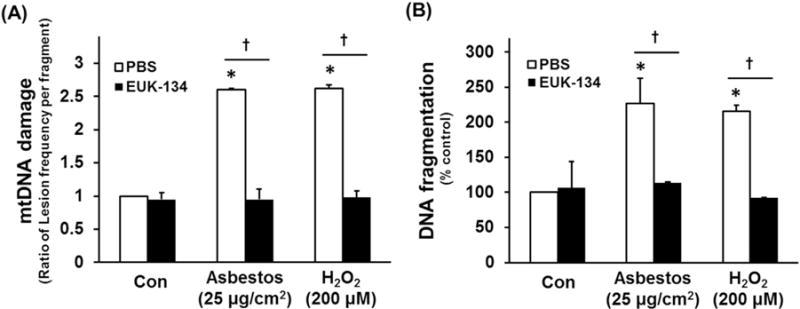
Euk-134 blocks AEC cell mtDNA damage and apoptosis. MLE 12 cells were pre-treated with vehicle (PBS) or Euk-134 (20 μL) for 4 h before exposure to oxidative stress (25 μg/ m2 amosite asbestos or 200 μM H2O2). Cells were harvested 24 h post-treatment and mtDNA damage assessed as described in the Methods, or apoptosis assessed by a DNA fragmentation ELISA assay. (A) Mitochondrial DNA damage expressed as the ratio of lesion frequency per fragment as compared to MLE-12 cells exposed to control media. (B) DNA fragmentation expressed as percent of control cells; n =3. *p < 0.05 vs. control. †p < 0.05 vs. oxidative stress (asbestos or H2O2) in PBS (vehicle control).
3.4. Asbestos-induced AT2 cell mitochondrial ROS production is decreased in MCAT as compared to WT mice
To establish whether MCAT overexpression reduces AEC mitochondrial ROS production that can result in subsequent mtDNA damage and apoptosis, we assessed mitochondrial ROS production using a MitoSox assay in primary isolated AT2 cells from both WT and MCAT mice following a 24 h exposure period to amosite asbestos (25 μg/cm2). As expected, asbestos significantly augmented WT AT2 cell mitochondrial ROS production approximately 4-fold compared to control (10,000 vs. 40,000 MitoSox fluorescence units; p < 0.05) (Fig. 7A and B). Notably, compared to WT, AT2 cells from MCAT mice have significantly reduced levels of asbestos-induced ROS production (40,000 vs. 20,000 MitoSox fluorescence units, respectively; p < 0.05) (Fig. 7A and B). These data convincingly show that AT2 cells from MCAT mice have markedly reduced levels of mitochondrial ROS production following asbestos exposure.
Fig. 7.
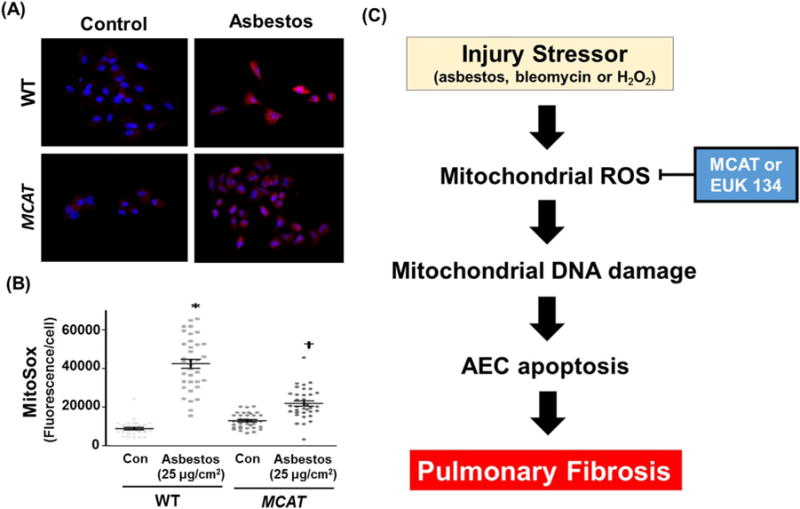
Asbestos-induced mitochondrial ROS production is reduced in AT2 cells from MCAT as compared to WT mice. (A) Primary AT2 cells were isolated from WT and MCAT mice and exposed to control media or amosite asbestos (25 μg/cm2). After 24 h, the AT2 cells were incubated with MitoSox red (90 nM) according to the manufacturer’s recommendation; shown is a representative panel from a total of 3 experiments. White punctate cytosolic staining indicative of mitochondrial ROS production as well as DAPI-stained nuclei were assessed. (B) Semi-quantitative analysis of 3 separate AT2 cell isolations from WT and MCAT mice. The graph depicts the MitoSox fluorescence intensity (mean ± SEM) in WT and MCAT murine AT2 cells; each point represents individual AT2 cell fluorescence intensities from 10 to 15 cells per experiment, lines represent mean fluorescence and brackets show standard error. *p < 0.05 vs. control/WT. †p < 0.05 vs. control/MCAT. (C) A proposed model showing that strategies aimed at reducing AEC mitochondrial ROS production (i.e. mitochondrial catalase [MCAT] or Euk-134) will attenuate AEC mitochondrial dysfunction, mtDNA damage, and apoptosis important for promoting lung fibrosis.
4. Discussion
Accumulating evidence demonstrate that AEC mitochondrial dysfunction and apoptosis are important in the pathogenesis of pulmonary fibrosis but the role of mitochondrial catalase in attenuating these effects has not been investigated. The major findings of this study are that overexpression of mitochondrial-targeted catalase reduces the severity of pulmonary fibrosis following exposure to asbestos and bleomycin. In cultured epithelial cells, mitochondrial targeted catalase reduced asbestos-induced ROS production, mtDNA damage and apoptosis. Collectively, these findings support an important role for AEC mitochondrial catalase in limiting ROS-induced AEC mtDNA damage to prevent AEC apoptosis and ameliorate lung fibrosis (see hypothetical model – Fig. 7C).
Catalase, which converts H2O2 to water, is one important regulator of ROS. There is evidence that exogenously administered catalase, either as subcutaneously polyethylene glycol-conjugated catalase or intratracheal instillation of extracellular catalase, can each attenuate asbestos-induced lung fibrosis [35,36]. However, it is unclear whether mitochondrial catalase is important in limiting pulmonary fibrosis. Because there is no ideal experimental murine model that duplicates all the features of IPF, the importance of mitochondrial catalase as a putative protective mechanistic pathway was assessed using two modes of lung fibrosis (i.e. asbestos and bleomycin) with the MCAT mice. A novel finding in this study is that MCAT mice have markedly diminished pulmonary fibrosis in both murine experimental lung fibrosis models when compared to their WT counterparts. Our data showing that MCAT affords similar levels of protection in both lung fibrosis models firmly supports the importance of mitochondrial catalase in limiting pulmonary fibrosis. The protective effects of mitochondrial catalase against lung fibrosis noted herein are in accord with studies showing that MCAT in mice attenuates other degenerative diseases involving the brain, heart, pulmonary vasculature, and lung cancer [18–26,37]. Interestingly, given the crucial role of aging in IPF and asbestosis [8,9,16], the beneficial effects of mitochondrial catalase reported here concur with the observation that MCAT mice have a prolonged survival as compared to their WT counterparts [18].
IPF and asbestosis are diseases of aging that lack highly effective treatment options [8,9,16]. Our study implicating the anti-fibrogenic effects of mitochondrial catalase enforced expression extends the work of others showing that asbestos-induced lung fibrosis is reduced by global catalase enhanced expression delivered either by infusing polyethylene glycol-conjugated catalase subcutaneously into rats [35] or by IT-instillation of catalase daily for 20 d into mice to target the alveolar space [36,38]. Our data also support studies in humans with IPF and bleomycin-induced lung fibrosis suggesting a pathogenic role for reduced alveolar and bronchiolar epithelium catalase levels and activity in association with increased oxidative stress [39–41]. An important beneficial role for mitochondrial catalase demonstrated in this study advances the work of others using genetic and pharmacologic approaches to globally modulate lung antioxidant defenses to subsequently alter bleomycin-induced lung fibrosis [42–45].
Although the mechanisms underlying the protective effects of mitochondrial catalase in our model are not entirely established, we explored two possibilities. First, we directly demonstrated that AT2 cells from MCAT as compared to WT mice had reduced levels of asbestos-induced mitochondrial ROS production as assessed by a Mito-Sox assay (Fig. 7A and B). Our finding suggesting that asbestos augments mitochondrial ROS production in AT2 cells from WT mice herein is in accord with our previous study utilizing RoGFP sensors targeted to the mitochondria and cytosol to demonstrate that asbestos induces preferential mitochondrial ROS production in AECs [17]. Recognizing the limitation of the Mito-Sox assay for precisely identifying specific free radicals as described in detail elsewhere [29], we chose this assay as a general marker of mitochondrial free radical production, as others have working with MCAT mice [20,24], because Mito-Sox localizes to the mitochondria enabling detection of mitochondrial O2− as well as other intracellular oxidants (e.g. iron and H2O2 or cytochrome c and H2O2). We also showed that Euk-134, a SOD/catalase mimetic that can attenuate toxin-induced mitochondrial ROS production as well as apoptosis [17,31,32], blocks amosite asbestos- and H2O2-induced MLE-12 cell mtDNA damage as well as apoptosis (Fig. 6). We reason that the excess lung AT2 cell mitochondrial ROS levels noted in WT mice in this study may have broader significance in promoting lung fibrosis as oxidized collagen is more resistant to degradation [46] and because asbestos-induced mitochondrial ROS can activate latent TGF-β [47–49].
A second mechanistic pathway that we explored focused on the accumulating evidence implicating ongoing AEC mitochondrial dysfunction, mtDNA damage and apoptosis in promoting lung fibrosis. Using the asbestos model, we provide two lines of evidence supporting a prominent role for these pathways. First, MCAT mice, as compared to WT, demonstrated reduced levels of apoptosis as assessed by CC-3 activation in cells at the BAD junction (Fig. 4). Co-localization studies using CC-3 and SFTPC IHC confirmed that at least some of these cells are AT2 cells. Second, using primary isolated AT2 cells from both WT and MCAT mice, we show that oxidative stress both in vivo (asbestos) and in vitro (asbestos or H2O2) each augments mtDNA damage and that mitochondrial catalase is protective (Fig. 5). Our findings showing that MCAT mice have reduced AEC mtDNA damage and apoptosis compared to their WT counterparts are in agreement with our previous study showing that Ogg1−/− mice, which are deficient in oxidative stress-induced DNA repair, have increased AEC mtDNA damage and apoptosis along with enhanced lung fibrosis following asbestos [16]. The protection afforded AT2 cells of MCAT mice following asbestos exposure noted here concur with work by others demonstrating that mitochondrial catalase reduces mtDNA damage and preserves mitochondrial function in cardiac and brain cells [18–24]. Our findings showing that mitochondrial catalase prevents lung fibrosis in part by mitigating asbestos-induced AEC mtDNA damage and apoptosis is in line with work of others as well as our group showing a direct link between the levels of mtDNA damage and intrinsic apoptosis following asbestos or bleomycin exposure that can promote lung fibrosis [15,16,50–53].
We should note some limitations with our study. Our studies have not excluded an important role for mitochondrial catalase in preventing activation of other cell types important in lung fibrosis, especially lung macrophages and fibroblasts. Carter and colleagues have shown a key role for alveolar macrophages in mediating asbestos-induced lung fibrosis that is due in part to mitochondrial electron transfer from cytochrome c to Rac1 in augmenting mitochondrial H2O2 production [36,38,54–56]. Moreover, recent flow cytometric studies of macrophage phenotypes in lung fibrosis have suggested an important role for monocyte-derived tissue macrophages [57–59]. In this regard, future studies exploring macrophage phenotypes in the MCAT mice as well as developing MCAT selectively targeted to AT2 cells will be of considerable interest. Our studies suggesting an important protective effect of mitochondrial catalase in preventing lung fibrosis do not exclude a role for other anti-oxidant lung defenses (e.g. thioredoxin-1, glutathione, Mn-SOD, Cu, Zn-SOD) or mitochondrial iron (e.g. iron-responsive element-binding protein 2), both of which have been implicated in modulating lung injury/fibrosis [54,56,60–62]. Furthermore, glutathione peroxidase-1 (GPX1), which can also reduce mitochondrial H2O2 [63], will be of interest in future studies addressing its role in mitigating lung fibrosis.
In summary, we have shown that MCAT mice, as compared to WT, have decreased lung fibrosis following exposure to asbestos or bleomycin. Furthermore, we show that the protective effects of mitochondrial catalase are in part due to decreasing AT2 cell mitochondrial ROS production, mtDNA damage and apoptosis. Given the crucial role of AEC mitochondrial dysfunction and apoptosis in promoting pulmonary fibrosis, we reason that strategies aimed at preventing AEC mtDNA damage arising from excess mitochondrial ROS levels may be a novel therapeutic target for preventing pulmonary fibrosis and possibly other degenerative diseases.
Acknowledgments
This work was supported by VA Merit 2I01BX000786-05A2 and NIH grant RO1 ES020357 to David W. Kamp, NIH/NHLBI training Grant 2T32HL076139-11A1 to Renea P. Jablonski, and VA Merit 1I01BX001910 to C. Michael Hart.
References
- 1.Schumacker PT, Gillespie MN, Nakahira K, Choi AMK, Crouser ED, Piantadosi CA, Bhattacharya J. Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiology – Lung Cell Mol Physiol. 2014;306(11):L962–L974. doi: 10.1152/ajplung.00073.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agrawal A, Mabalirajan U. Rejuvenating cellular respiration for optimizing respiratory function: targeting mitochondria. Am J Physiol – Lung Cell Mol Physiol. 2016;310(2):L103–L113. doi: 10.1152/ajplung.00320.2015. [DOI] [PubMed] [Google Scholar]
- 3.Mossman BT, Lippmann M, Hesterberg TW, Kelsey KT, Barchowsky A, Bonner JC. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J Toxicol Environ Health Part B Crit Rev. 2011;14(1–4):76–121. doi: 10.1080/10937404.2011.556047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang SX, Jaurand MC, Kamp DW, Whysner J, Hei TK. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J Toxicol Environ Health Part B Crit Rev. 2011;14(1–4):179–245. doi: 10.1080/10937404.2011.556051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. 2013;1832(7):1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SJ, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci. 2015;16(9):21486–21519. doi: 10.3390/ijms160921486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uhal BD, Nguyen H. The Witschi Hypothesis revisited after 35 years: genetic proof from SP-C BRICHOS domain mutations. Am J Physiol – Lung Cell Mol Physiol. 2013;305(12):L906–L911. doi: 10.1152/ajplung.00246.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. 2014;189(10):1161–1172. doi: 10.1164/rccm.201312-2221PP. [DOI] [PubMed] [Google Scholar]
- 9.Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, Selman M, Pardo A, White ES, Levy BD, Busse PJ, Tuder RM, Antony VB, Sznajder JI, Budinger GR. Blue journal conference. Aging and susceptibility to lung disease. Am J Respir Crit Care Med. 2015;191(3):261–269. doi: 10.1164/rccm.201410-1876PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Investig. 2015;125(2):521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, El-Chemaly S, Lee CG, Rosas IO, Elias JA, Choi AM, Morse D. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One. 2015;10(3):e0121246. doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aljandali A, Pollack H, Yeldandi A, Li Y, Weitzman SA, Kamp DW. Asbestos causes apoptosis in alveolar epithelial cells: role of iron-induced free radicals. J Lab Clin Med. 2001;137(5):330–339. doi: 10.1067/mlc.2001.114826. [DOI] [PubMed] [Google Scholar]
- 13.Panduri V, Weitzman SA, Chandel N, Kamp DW. The mitochondria-regulated death pathway mediates asbestos-induced alveolar epithelial cell apoptosis. Am J Respir Cell Mol Biol. 2003;28(2):241–248. doi: 10.1165/rcmb.4903. [DOI] [PubMed] [Google Scholar]
- 14.Panduri V, Weitzman SA, Chandel NS, Kamp DW. Mitochondrial-derived free radicals mediate asbestos-induced alveolar epithelial cell apoptosis. Am J Physiol – Lung Cell Mol Physiol. 2004;286(6):L1220–L1227. doi: 10.1152/ajplung.00371.2003. [DOI] [PubMed] [Google Scholar]
- 15.Kim SJ, Cheresh P, Williams D, Cheng Y, Ridge K, Schumacker PT, Weitzman S, Bohr VA, Kamp DW. Mitochondria-targeted Ogg1 and aconitase-2 prevent oxidant-induced mitochondrial DNA damage in alveolar epithelial cells. J Biol Chem. 2014;289(9):6165–6176. doi: 10.1074/jbc.M113.515130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheresh P, Morales-Nebreda L, Kim SJ, Yeldandi A, Williams DB, Cheng Y, Mutlu GM, Budinger GR, Ridge K, Schumacker PT, Bohr VA, Kamp DW. Asbestos-induced pulmonary fibrosis is augmented in 8-oxoguanine DNA glycosylase knockout mice. Am J Respir Cell Mol Biol. 2015;52(1):25–36. doi: 10.1165/rcmb.2014-0038OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panduri V, Liu G, Surapureddi S, Kondapalli J, Soberanes S, de Souza-Pinto NC, Bohr VA, Budinger GR, Schumacker PT, Weitzman SA, Kamp DW. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic Biol Med. 2009;47(6):750–759. doi: 10.1016/j.freeradbiomed.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 19.Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010;12(6):668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108(7):837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93(1):79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, Shirendeb U, Lo HH, Rabinovitch PS, Reddy PH. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet. 2012;21(13):2973–2990. doi: 10.1093/hmg/dds128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW., 2nd Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res. 2014;115(3):348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ Res. 2014;114(3):421–433. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adesina SE, Kang BY, Bijli KM, Ma J, Cheng J, Murphy TC, Hart C Michael, Sutliff RL. Targeting mitochondrial reactive oxygen species to modulate hypoxia-induced pulmonary hypertension. Free Radic Biol Med. 2015;87:36–47. doi: 10.1016/j.freeradbiomed.2015.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parihar VK, Allen BD, Tran KK, Chmielewski NN, Craver BM, Martirosian V, Morganti JM, Rosi S, Vlkolinsky R, Acharya MM, Nelson GA, Allen AR, Limoli CL. Targeted overexpression of mitochondrial catalase prevents radiation-induced cognitive dysfunction. Antioxid Redox Signal. 2015;22(1):78–91. doi: 10.1089/ars.2014.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morales-Nebreda LI, Rogel MR, Eisenberg JL, Hamill KJ, Soberanes S, Nigdelioglu R, Chi M, Cho T, Radigan KA, Ridge KM, Misharin AV, Woychek A, Hopkinson S, Perlman H, Mutlu GM, Pardo A, Selman M, Jones JC, Budinger GR. Lung-specific loss of alpha3 laminin worsens bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2015;52(4):503–512. doi: 10.1165/rcmb.2014-0057OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, Sennello JA, Kutuzov MA, Sitikov A, Davis JM, Lam AP, Cheresh P, Kamp D, Shumaker DK, Budinger GR, Ridge KM. Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun. 2015;6:6574. doi: 10.1038/ncomms7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic Biol Med. 2012;52(1):1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang LY, Overby LH, Brody AR, Crapo JD. Progressive lung cell reactions and extracellular matrix production after a brief exposure to asbestos. Am J Pathol. 1988;131(1):156–170. [PMC free article] [PubMed] [Google Scholar]
- 31.Soberanes S, Urich D, Baker CM, Burgess Z, Chiarella SE, Bell EL, Ghio AJ, De Vizcaya-Ruiz A, Liu J, Ridge KM, Kamp DW, Chandel NS, Schumacker PT, Mutlu GM, Budinger GR. Mitochondrial complex III-generated oxidants activate ASK1 and JNK to induce alveolar epithelial cell death following exposure to particulate matter air pollution. J Biol Chem. 2009;284(4):2176–2186. doi: 10.1074/jbc.M808844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamp DW, Liu G, Cheresh P, Kim SJ, Mueller A, Lam AP, Trejo H, Williams D, Tulasiram S, Baker M, Ridge K, Chandel NS, Beri R. Asbestos-induced alveolar epithelial cell apoptosis. The role of endoplasmic reticulum stress response. Am J Respir Cell Mol Biol. 2013;49(6):892–901. doi: 10.1165/rcmb.2013-0053OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pramanik KC, Boreddy SR, Srivastava SK. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS One. 2011;6(5):e20151. doi: 10.1371/journal.pone.0020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Na N, Chandel NS, Litvan J, Ridge KM. Mitochondrial reactive oxygen species are required for hypoxia-induced degradation of keratin intermediate filaments. FASEB J: Off Publ Fed Am Soc Exp Biol. 2010;24(3):799–809. doi: 10.1096/fj.08-128967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mossman BT, Marsh JP, Sesko A, Hill S, Shatos MA, Doherty J, Petruska J, Adler KB, Hemenway D, Mickey R, et al. Inhibition of lung injury, inflammation, and interstitial pulmonary fibrosis by polyethylene glycol-conjugated catalase in a rapid inhalation model of asbestosis. Am Rev Respir Dis. 1990;141(5 Pt 1):1266–1271. doi: 10.1164/ajrccm/141.5_Pt_1.1266. [DOI] [PubMed] [Google Scholar]
- 36.Murthy S, Adamcakova-Dodd A, Perry SS, Tephly LA, Keller RM, Metwali N, Meyerholz DK, Wang Y, Glogauer M, Thorne PS, Carter AB. Modulation of reactive oxygen species by Rac1 or catalase prevents asbestos-induced pulmonary fibrosis. Am J Physiol – Lung Cell Mol Physiol. 2009;297(5):L846–L855. doi: 10.1152/ajplung.90590.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ge X, Pettan-Brewer C, Morton J, Carter K, Fatemi S, Rabinovitch P, Ladiges WC. Mitochondrial catalase suppresses naturally occurring lung cancer in old mice. Pathobiol Aging Age Relat Dis. 2015;5:28776. doi: 10.3402/pba.v5.28776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murthy S, Ryan A, He C, Mallampalli RK, Carter AB. Rac1-mediated mitochondrial H2O2 generation regulates MMP-9 gene expression in macrophages via inhibition of SP-1 and AP-1. J Biol Chem. 2010;285(32):25062–25073. doi: 10.1074/jbc.M109.099655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Odajima N, Betsuyaku T, Nagai K, Moriyama C, Wang DH, Takigawa T, Ogino K, Nishimura M. The role of catalase in pulmonary fibrosis. Respir Res. 2010;11:183. doi: 10.1186/1465-9921-11-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inghilleri S, Morbini P, Oggionni T, Barni S, Fenoglio C. In situ assessment of oxidant and nitrogenic stress in bleomycin pulmonary fibrosis. Histochem Cell Biol. 2006;125(6):661–669. doi: 10.1007/s00418-005-0116-7. [DOI] [PubMed] [Google Scholar]
- 41.Santos-Silva MA, Pires KM, Trajano ET, Martins V, Nesi RT, Benjamin CF, Caetano MS, Sternberg C, Machado MN, Zin WA, Valenca SS, Porto LC. Redox imbalance and pulmonary function in bleomycin-induced fibrosis in C57BL/6, DBA/2, and BALB/c mice. Toxicol Pathol. 2012;40(5):731–741. doi: 10.1177/0192623312441404. [DOI] [PubMed] [Google Scholar]
- 42.Fattman CL, Chang LY, Termin TA, Petersen L, Enghild JJ, Oury TD. Enhanced bleomycin-induced pulmonary damage in mice lacking extracellular superoxide dismutase. Free Radic Biol Med. 2003;35(7):763–771. doi: 10.1016/s0891-5849(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 43.Manoury B, Nenan S, Leclerc O, Guenon I, Boichot E, Planquois JM, Bertrand CP, Lagente V. The absence of reactive oxygen species production protects mice against bleomycin-induced pulmonary fibrosis. Respir Res. 2005;6:11. doi: 10.1186/1465-9921-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teixeira KC, Soares FS, Rocha LG, Silveira PC, Silva LA, Valenca SS, Pizzol FDal, Streck EL, Pinho RA. Attenuation of bleomycin-induced lung injury and oxidative stress by N-acetylcysteine plus deferoxamine. Pulm Pharmacol Ther. 2008;21(2):309–316. doi: 10.1016/j.pupt.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Khazri O, Charradi K, Limam F, El May MV, Aouani E. Grape seed and skin extract protects against bleomycin-induced oxidative stress in rat lung. Biomed Pharmacother – Biomed Pharmacother. 2016;81:242–249. doi: 10.1016/j.biopha.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Rucklidge GJ, Milne G, McGaw BA, Milne E, Robins SP. Turnover rates of different collagen types measured by isotope ratio mass spectrometry. Biochim Biophys Acta. 1992;1156(1):57–61. doi: 10.1016/0304-4165(92)90095-c. [DOI] [PubMed] [Google Scholar]
- 47.Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. 2013;288(2):770–777. doi: 10.1074/jbc.M112.431973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pociask DA, Sime PJ, Brody AR. Asbestos-derived reactive oxygen species activate TGF-beta1. Lab Investig – J Tech Methods Pathol. 2004;84(8):1013–1023. doi: 10.1038/labinvest.3700109. [DOI] [PubMed] [Google Scholar]
- 49.Sullivan DE, Ferris M, Pociask D, Brody AR. The latent form of TGFbeta(1) is induced by TNFalpha through an ERK specific pathway and is activated by asbestos-derived reactive oxygen species in vitro and in vivo. J Immunotoxicol. 2008;5(2):145–149. doi: 10.1080/15476910802085822. [DOI] [PubMed] [Google Scholar]
- 50.Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278(3):1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- 51.Brar SS, Meyer JN, Bortner CD, Van Houten B, Martin WJ., 2nd Mitochondrial DNA-depleted A549 cells are resistant to bleomycin. Am J Physiol – Lung Cell Mol Physiol. 2012;303(5):L413–L424. doi: 10.1152/ajplung.00343.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee VY, Schroedl C, Brunelle JK, Buccellato LJ, Akinci OI, Kaneto H, Snyder C, Eisenbart J, Budinger GR, Chandel NS. Bleomycin induces alveolar epithelial cell death through JNK-dependent activation of the mitochondrial death pathway. Am J Physiol – Lung Cell Mol Physiol. 2005;289(4):L521–L528. doi: 10.1152/ajplung.00340.2004. [DOI] [PubMed] [Google Scholar]
- 53.Budinger GR, Mutlu GM, Eisenbart J, Fuller AC, Bellmeyer AA, Baker CM, Wilson M, Ridge K, Barrett TA, Lee VY, Chandel NS. Proapoptotic Bid is required for pulmonary fibrosis. Proc Natl Acad Sci USA. 2006;103(12):4604–4609. doi: 10.1073/pnas.0507604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He C, Murthy S, McCormick ML, Spitz DR, Ryan AJ, Carter AB. Mitochondrial Cu,Zn-superoxide dismutase mediates pulmonary fibrosis by augmenting H2O2 generation. J Biol Chem. 2011;286(17):15597–15607. doi: 10.1074/jbc.M110.187377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osborn-Heaford HL, Ryan AJ, Murthy S, Racila AM, He C, Sieren JC, Spitz DR, Carter AB. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J Biol Chem. 2012;287(5):3301–3312. doi: 10.1074/jbc.M111.308387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J Biol Chem. 2013;288(28):20745–20757. doi: 10.1074/jbc.M112.410720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49(4):503–510. doi: 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morales-Nebreda L, Misharin AV, Perlman H, Budinger GR. The heterogeneity of lung macrophages in the susceptibility to disease. Eur Respir Rev: Off J Eur Respir Soc. 2015;24(137):505–509. doi: 10.1183/16000617.0031-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu YR, Hotten DF, Malakhau Y, Volker E, Ghio AJ, Noble PW, Kraft M, Hollingsworth JW, Gunn MD, Tighe RM. Flow cytometric analysis of myeloid cells in human blood, bronchoalveolar lavage, and lung tissues. Am J Respir Cell Mol Biol. 2016;54(1):13–24. doi: 10.1165/rcmb.2015-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson JK, Westbom CM, MacPherson MB, Mossman BT, Heintz NH, Spiess P, Shukla A. Asbestos modulates thioredoxin-thioredoxin interacting protein interaction to regulate inflammasome activation. Part Fibre Toxicol. 2014;11:24. doi: 10.1186/1743-8977-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med. 2010;48(1):1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabon MA, Konrad C, Polverino F, Siempos II, Perez E, Mizumura K, Ghosh MC, Parameswaran H, Williams NC, Rooney KT, Chen ZH, Goldklang MP, Yuan GC, Moore SC, Demeo DL, Rouault TA, D’Armiento JM, Schon EA, Manfredi G, Quackenbush J, Mahmood A, Silverman EK, Owen CA, Choi AM. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22(2):163–174. doi: 10.1038/nm.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Handy DE, Lubos E, Yang Y, Galbraith JD, Kelly N, Zhang YY, Leopold JA, Loscalzo J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J Biol Chem. 2009;284(18):11913–11921. doi: 10.1074/jbc.M900392200. [DOI] [PMC free article] [PubMed] [Google Scholar]