

Abstract
Castration-resistant prostate cancer (CRPC), also known as androgen-independent prostate cancer, frequently develops local and distant metastases, the underlying mechanisms of which remain undetermined. In the present study, surgical specimens obtained from patients with clinical prostate cancer were investigated, and it was revealed that the expression levels of ataxia telangiectasia mutated kinase (ATM) were significantly enhanced in prostate cancer tissues isolated from patients with CRPC compared with from patients with hormone-dependent prostate cancer. CRPC C4-2 and CWR22Rv1 cells lines were subsequently selected to establish prostate cancer models, and ATM knockout cells were established via lentivirus infection. The results of the present study demonstrated that the migration and epithelial-mesenchymal transition (EMT) of ATM knockout cells were significantly decreased, which suggested that ATM is closely associated with CRPC cell migration and EMT. To further investigate the mechanisms underlying this process, programmed cell death 1 ligand 1 (PD-L1) expression was investigated in ATM knockout cells. In addition, inhibitors of Janus kinase (JAK) and signal transducer and activator of transcription 3 (STAT3; Stattic) were added to C4-2-Sc and CWR22Rv1-Sc cells, and the results demonstrated that PD-L1 expression was significantly decreased following the addition of JAK inhibitor 1; however, no significant change was observed following the addition of Stattic. Furthermore, a PD-L1 antibody and JAK inhibitor 1 were added to C4-2-Sc and CWR22Rv1-Sc cells, and it was revealed that cell migration ability was significantly decreased and the expression of EMT-associated markers was effectively reversed. The results of the present study suggested that via inhibition of the ATM-JAK-PD-L1 signaling pathway, EMT, metastasis and progression of CRPC may be effectively suppressed, which may represent a novel therapeutic approach for targeted therapy for patients with CRPC.
Keywords: castration-resistant prostate cancer, metastasis, programmed cell death 1 ligand 1, ataxia telangiectasia mutated kinase, Janus kinase
Introduction
Cases of prostate cancer-associated morbidity and mortality are rapidly increasing in China (1), and prostate cancer is the most prevalent malignant tumor in the United States (2), thus affecting life expectancy and the quality of life of patients with prostate cancer. Androgen deprivation and radical resection surgery remain the primary methods of treatment, with androgen blockade having an important role in current therapeutic methods. However, during treatment, many patients with prostate cancer rapidly develop hormone resistance and enter the castration-insensitive phase of the disease [castration-resistant prostate cancer (CRPC)], in which the incidence of local and distant metastases markedly increases and patients' prognoses are seriously affected.
Investigations to determine the underlying mechanisms of prostate cancer metastasis have become an important area of research. Epithelial-mesenchymal transition (EMT) is a well-established mechanism of malignant tumor invasion and metastasis (3–7). Clinical and pathological studies have demonstrated that as the Gleason score of prostate cancer increases, the structure and basement membranes of prostate cancer epithelial cells gradually disappear, the appearance and growth of cell clusters that rapidly become more invasive increases, and the levels of epithelial cell marker E-cadherin (E-Cad) decrease, whereas the mesenchymal cell markers N-cadherin (N-Cad) and vimentin increase (8–10). Aberrant expression of these EMT markers in numerous malignant tumors, including prostate cancer, has an important role in cancer metastasis (8–13).
The mechanisms underlying EMT in prostate cancer invasion and metastasis have not yet been fully determined. Programmed cell death 1 ligand 1 (PD-L1) is a cell surface glycoprotein belonging to the B7 family of co-stimulatory molecules, which is widely expressed in numerous malignant tumor types (14–16). PD-L1 is not only involved in tumor immune evasion, but also in EMT, thus suggesting that PD-L1 is involved in tumor metastasis and prognosis (14–16). Studies have demonstrated that the Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) signaling pathway has an important role in tumor progression and metastasis (17–19). The JAK/STAT3 pathway is not only involved in prostate cancer progression, but is also closely associated with the transition from hormone dependence to hormone independence in prostate cancer (20–22).
A previous study revealed that increased ataxia telangiectasia mutated kinase (ATM) expression in lymph node metastases of breast cancer is closely associated with tumor invasion and metastasis (23). It has also been demonstrated that reduced expression of ATM can inhibit ovarian cancer metastasis (24). Considering these findings, the present study aimed to investigate the clinical findings in samples obtained from patients with prostate cancer; the results revealed that ATM expression levels in prostate tissue samples from patients with CRPC were significantly enhanced compared with prostate tissue samples from patients with hormone-dependent prostate cancer (HDPC). In the present study, to investigate the mechanisms underlying the elevated expression of ATM, the expression of the ATM gene in tumor cells was inhibited via gene knockout and the addition of inhibitors, after which cell migration and the expression levels of EMT-associated markers were investigated. The results of the present study revealed that, as determined by inhibition of PD-L1 and the JAK/STAT3 signaling pathway, the ATM-JAK-PD-L1 signaling pathway has an important role in the metastatic progression of CRPC, and therefore may represent a novel therapeutic approach for CRPC-targeted therapy.
Materials and methods
Clinical species
To investigate whether ATM is elevated in prostate cancer tissues from patients with CRPC who are prone to develop metastasis. We collected surgical specimens of prostate cancer from April 2006 to April 2016 in our hospital. All patients were male, aged 67–84 years, of whom 62 were HDPC and 50 were CRPC. 112 surgical specimens isolated from patients with prostate cancer were obtained. All patients provided informed consent. The present study was approved by the Ethics Committee of the Second Affiliated Hospital of Soochow University (Suzhou, China). Among the total patients included in the present study, 62 patients with HDPC but without local and distant metastases were included, all of which underwent radical prostatectomy. The remaining 50 patients with CRPC had developed varying local or distant metastases and underwent palliative surgery to alleviate urinary tract symptoms.
Cell culture
CRPC cell lines C4-2 and CWR22Rv1 were used in the present study. C4-2 cells were obtained from the China Center for Type Culture Collection (Wuhan, China). CWR22Rv1 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in RPMI-1640 (Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% charcoal stripped fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.). CP466722 (Selleck Chemicals, Houston, TX, USA), JAK inhibitor 1 (EMD Millipore, Billerica, MA, USA) and Stattic (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) inhibitors were added to cultured tumor cells (1:1,000 for all inhibitors) at 37°C, at 6, 12 and 24 h time intervals to inhibit the expression of ATM, JAK and STAT3, respectively. RT-qPCR, western blot and cell migration assay were performed at 6, 12 and 24 h respectively.
Immunohistochemistry (IHC)
Tumor tissues were fixed in 10% (v/v) formaldehyde in PBS, embedded in paraffin, and cut into 5-µm thick sections. The sections were air-dried, then cleared and dehydrated. Antigen retrieval was performed by boiling sections in sodium citrate buffer. Subsequently, sections were incubated for 1 h at room temperature in blocking solution 5% BSA (AR0004, Boster Biological Technology, Pleasanton, CA, USA). Incubation with primary antibody Phosphorylated (p)-ATM (MA1-46069, Thermo Fisher Scientific, Inc. USA) was at 4°C overnight. Following washes with PBS three times, incubation with secondary rabbit anti-mouse antibody (61–0200, Thermo Fisher Scientific, Inc. USA) at room temperature for 20 min followed by three additional washes with PBS. Following staining, tissues were counterstained with hematoxylin. Error bars and significance of values were determined by counting positively stained cells in three randomly selected areas of each slide. Tumor tissue sections were immunostained using a mouse and rabbit specific HRP/DAB detection IHC kit (ab80436, Abcam, Cambridge, UK). Following this, tissues were counterstained by hematoxylin. A light microscope was used (Olympus Corporation, Tokyo, Japan), at magnification, ×40. Error bars and significance values were obtained by manually counting positively stained cells from one randomly chosen area of slides of three different stains.
ATM knockout cell construction
ATM knockout cells and corresponding control cells were established via lentivirus infection. The lentiviral pLenti-II vector (Addgene, Inc., Cambridge, MA, USA) carried ATM small interfering (si)RNA (ATMSi) or scrambled (Sc) gene sequences as follows: ATM scramble forward, 5′-UUCUCCGAACGUGUCACGUdTdT-3′ and reverse, 5′-ACGUGACACGUUCGGAGAAdTdT-3′ and ATM siRNA forward, 5′-CAUACUACUCAAAGACAUUdTdT-3′ and reverse, 5′-AAUGUCUUUGAGUAGUAUGdTdT-3′. pLent-II-ATMSi or Sc, psPAX2 (virus-packaging plasmid) and pMD2G (envelope plasmid; All from Qiagen, Inc., Valencia, CA, USA) were transfected into 293T cells at a ratio of 4:3:2 using PolyFect transfection reagent (Qiagen, Inc.). Following C4-2 and CWR22Rv1 cells being virally infected overnight, the culture media containing the virus were replaced with normal culture media and maintained under normal cell culture conditions. After subculturing cells, the ATM-knockdown cells were selected by culturing cells in the presence of puromycin (2 µgxml−1; 540411; Millipore, Billerica, MA, USA) and maintained in media containing 0.1 µgxml−1 puromycin.
Migration assay
Tumor cells (1×104) were seeded into the upper chamber of a Transwell support (Corning Incorporated, Corning, NY, USA) with serum-free media, whereas medium containing 10% FBS was added to the lower chamber. Following incubation for 24 h at 37°C, cells were fixed in methanol and then stained with crystal violet. Positively stained cells in three randomly selected visual fields were counted under a light microscope (Olympus Corporation), magnification, ×20 (inlet, ×100). Each assay was repeated three times. Antibodies used were: JAK inhibitor 1 (420099; EMD Millipore), CP466722 (S2245; Selleck Chemicals), PD-L1 Antibody (62-5982-80, Thermo Fisher Scientific, Inc.).
Cell proliferation assay
Tumor cells (1×103) were seeded in a 96-well plate, the ATM inhibitor CP466722 was added and a control group was set up without CP466722. The growth of cells in MTT (5 mg/ml; Sigma-Aldrich; Merck KGaA) was assessed daily, DMSO was used to dissolve formazan in cells and the optical density of each well was determined at 570 nm relative to the control group.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA from cells (1 µg) was subjected to reverse transcription using Superscript III transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.) as follows: 25°C for 10 min, 42°C for 50 min, 70°C for 15 min and 4°C hold. Quantitative PCR was conducted using the appropriate primers and a Bio-Rad CFX96 system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with SYBR green to determine the mRNA expression levels of genes of interest. Enzyme activation and DNA initial denaturation occurred at 95°C for 3 min, followed by for 40 cycles of: DNA denaturation at 95°C for 15 sec and annealing/extension at 60°C for 45 sec. A melting curve analysis was performed. GraphPad Prism 5 was used to analyze the results. The 2−ΔΔCq method was used to calculate the relative expression level of each gene. Expression levels were normalized to GAPDH.
Western blotting
Tumor cells were collected following centrifugation at 300 × g for 5 min at 4°C) and supernatant removal. Cells were lysed in RIPA buffer (50 mM Tris-Cl at pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 µg/ml leupeptin, 1 µg/ml aprotinin, 0.2 mM PMSF). Proteins (20–40 µg) were separated on 8–10% SDS/PAGE gel and then transferred onto PVDF membranes (EMD Millipore). The PVDF membrane was placed in blocking buffer and put on shaker for 30 min at room temperature and wash three times with PBS for 10 min. Subsequently, membranes were incubated with primary antibodies (1:1,000), for 1 h at room temperature on a shaker. Following 3 washes for 10 min each, with PBS with Tween 20, HRP-conjugated secondary antibodies (1:5,000; ab97030; Abcam) incubated for 1 h at room temperature on a shaker. Finally, band intensities were visualized in Imager (Bio-Rad Laboratories) using ECL system (34095; Thermo Fisher Scientific, Inc.). Primary antibodies used in the assay included: Phosphorylated (p)-ATM (MA1-46069; Thermo Fisher Scientific, Inc.), PD-L1, p-JAK1, p-JAK2 and p-STAT3 and GAPDH (14-5983-82, 44–422G, 710928, 44–384G, MA5-15738; Thermo Fisher Scientific, Inc.).
Statistical analysis
All statistical analyzes were performed using SPSS 19.0 (IBM Corp., Armonk, NY, USA) statistical software. All experiments were performed three times which are presented as the mean ± standard deviation and differences between two groups were analyzed using a two-tailed Student's t-test. One-way analysis of variance followed by Fisher's least significant difference post hoc test was used for comparisons among multiple groups. P<0.05 was considered to indicate a statistically significant difference.
Results
ATM expression in prostate cancer tissues is higher in patients with CRPC compared with patients with HDPC
ATM expression in prostate cancer specimens was investigated via RT-qPCR and IHC. IHC was performed to determine the levels of p-ATM, since ATM is predominantly activated by phosphorylation. Based on clinical pathological findings of prostate cancer specimens, the RT-qPCR results revealed that the mRNA expression levels of p-ATM were significantly enhanced in prostate cancer tissues obtained from patients with CRPC compared with from patients with HDPC (P<0.01; Fig. 1A). The results of IHC analysis demonstrated that the expression levels of p-ATM were significantly enhanced in prostate cancer tissues obtained from patients with CRPC compared with from patients with HDPC (P<0.01; Fig. 1B and C).
Figure 1.

(A) Reverse transcription-quantitative polymerase chain reaction and (B and C) immunohistochemistry analyses revealed enhanced p-ATM expression in prostate cancer tissues from patients with CRPC compared with those with HDPC. ***P<0.01 vs. HDPC. Magnification, ×40. ATM, ataxia telangiectasia mutated kinase; CRPC, castration-resistant prostate cancer; HDPC, hormone-dependent prostate cancer; p-, phosphorylated.
Migration and EMT of ATM knockout CRPC cells, C4-2-ATMSi and CWR22Rv1-ATMSi, are suppressed
Using the CRPC cell lines C4-2 and CWR22Rv1 as model systems, the successful establishment of ATM knockout cell lines C4-2-ATMSi and CWR22Rv1-ATMSi, and control lines C4-2-Sc and CWR22Rv1-Sc, was revealed (Fig. 2A). The migration of ATM knockout cells was significantly decreased 24 h post-transfection in C4-2-ATMSi and CWR22Rv1-ATMSi cells compared with in the control cells (Fig. 2B). The expression levels of EMT-associated markers were investigated in ATM knockout cells by RT-qPCR; the results demonstrated that the expression levels of N-Cad, vimentin and vascular endothelial growth factor were significantly decreased compared with in the control cells, whereas E-Cad expression was significantly increased in ATM knockout cells compared with in the control cells (Fig. 2C). These results suggested that ATM knockout suppresses EMT in tumors.
Figure 2.

In C4-2-ATMSi and CWR22Rv1-ATMSi knockout cell lines, migration and EMT are significantly suppressed compared with in the control group. (A) C4-2-ATMSi and CWR22Rv1-ATMSi cells were established by lentivirus infection, and decreased levels of p-ATM were demonstrated by reverse transcription-quantitative polymerase chain reaction and western blotting. (B) Migration ability of ATM knockout cells was significantly decreased compared with in the Sc cells, as revealed by a cell migration assay. Magnification, ×40. (C) EMT of the ATM knockout cells was significantly decreased, as revealed by reverse transcription-quantitative polymerase chain reaction. **P<0.01; ***P<0.001 vs. the Sc group. Each experiment was performed in triplicate. ATM, ataxia telangiectasia mutated kinase; E-Cad, E-cadherin; EMT, epithelial-mesenchymal transition; N-Cad, cadherin-N; p-, phosphorylated; Sc, scramble; si, small interfering RNA; VEGF, vascular endothelial growth factor.
Expression of ATM is downregulated in cells treated with an ATM inhibitor, and the proliferation and migration of such cells are decreased
To validate the aforementioned experimental results, an ATM inhibitor (CP466722) was incubated with C4-2, CWR22Rv1, C4-2-Sc and CWR22Rv1-Sc cells. Following 6 h of incubation, a significant decrease in p-ATM expression was revealed by RT-qPCR analysis (Fig. 3A). Furthermore, the proliferative ability of the cells was significantly decreased 3 and 4 days post incubation with CP466722 compared with in the control group (Fig. 3B). In addition, the migratory ability of ATM knockout cells and cells incubated with CP466722 were significantly decreased, thus suggesting that ATM inhibition may suppress the growth and metastasis of CRPC cells (Fig. 3C).
Figure 3.


Following inhibition of ATM expression in C4-2, CWR22Rv1, C4-2-Sc and CWR22Rv1-Sc cells, the proliferation and migration of cells is significantly decreased compared with in the control cells. Similar to ATMSi, addition of the ATM inhibitor CP466722 significantly inhibits the migration and proliferation of prostate cancer cells. (A) Decreased p-ATM expression was revealed by reverse transcription-quantitative polymerase chain reaction analysis. Compared with ATMSi, the ability of ATM inhibitor CP466722 to inhibit ATM was not significantly different. (B) MTT assay demonstrated a significant decrease in proliferation of the cells on days 3 and 4 compared with in the control group. (C) Following addition of CP466722, the migration of cells was significantly suppressed compared with in the control group. There was no significant difference in the migration of prostate cancer cells between the Sc, CP466722 and ATMSi groups. Magnification, ×40. These results further demonstrated that downregulation of ATM suppresses the migration and epithelial-mesenchymal transition of CRPC cells. **P<0.01; ***P<0.001. ATM, ataxia telangiectasia mutated kinase; OD, optical density; P, primary cells; Sc, scramble; si, small interfering RNA.
Expression of PD-L1 in ATM knockout cells is suppressed, and downregulation of the JAK signaling pathway can inhibit the expression of PD-L1
The aforementioned results demonstrated that ATM has an important role in CRPC invasion and metastasis via mechanisms that are not yet fully understood. Therefore, the present study aimed to investigate the expression of PD-L1, which is closely associated with EMT (14–16,25). Firstly, the expression of PD-L1 in C4-2-ATMSi and CWR22Rv1-ATMSi cells was investigated, and the results demonstrated that compared with in the control group, the expression of PD-L1 was significantly suppressed in ATM knockout cells (Fig. 4A). In order to determine the mechanism underlying the alteration of PD-L1 levels, ATM downstream regulatory factors were examined, particularly the extensively studied JAK/STAT3 signaling pathway, which is involved in PD-L1 regulation (26,27). The expression levels of p-JAK1, p-JAK2 and p-STAT3 in ATM knockout cells were decreased compared with in the control cells (Fig. 4B). Following 6 h of co-culture with JAK inhibitor 1 or Stattic, changes in PD-L1 mRNA expression were investigated by RT-qPCR, the results of which revealed that incubation with JAK inhibitor 1 resulted in a significant decrease in the expression levels of PD-L1, whereas incubation with Stattic did not exert a significant effect (Fig. 4C). These results suggested that downregulation of the JAK signaling pathway may inhibit the expression of PD-L1.
Figure 4.
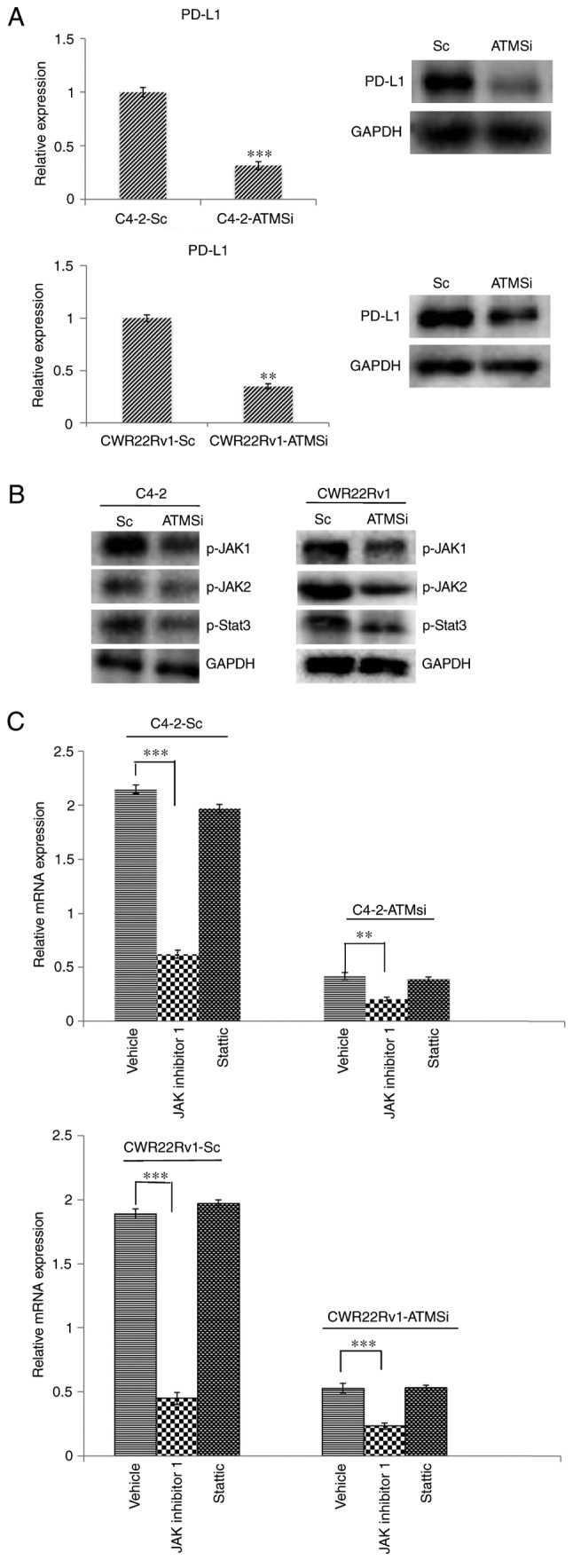
Levels of PD-L1, p-JAK1, p-JAK2 and p-STAT3 are suppressed in C4-2-ATMSi and CWR22Rv1-ATMSi cells compared with in the control cells, and JAK inhibitor 1 significantly suppresses the expression of PD-L1 in ATM knockout groups and control groups. (A) A significant decrease in PD-L1 expression was revealed in experimental groups by reverse transcription-quantitative polymerase chain reaction and western blotting. (B) Decreased levels of p-JAK1, p-JAK2 and p-STAT3 were revealed in the experimental groups by western blotting. (C) JAK inhibitor 1 and Stattic were used to treat all cell groups. Downregulation of JAK significantly reduced PD-L1 expression, whereas Stattic had no significant effect on PD-L1 expression. **P<0.01; ***P<0.001. ATM, ataxia telangiectasia mutated kinase; JAK, Janus kinase; p-, phosphorylated; PD-L1, programmed cell death 1 ligand 1; Sc, scramble; si, small interfering RNA; STAT3, signal transducer and activator of transcription 3.
Suppressed expression of PD-L1 and the JAK signaling pathway inhibits migration and EMT of CRPC cells, and thereby inhibits metastasis
To investigate the roles of PD-L1 expression and the JAK signaling pathway in ATM-associated regulation of CRPC metastasis, PD-L1 antibodies and JAK inhibitor 1 were incubated with C4-2-Sc and CWR22Rv1-Sc cells. The results demonstrated that PD-L1 antibodies and JAK inhibitor 1 significantly decreased the migration of cells (Fig. 5A). Furthermore, the results demonstrated that following incubation with either PD-L1 antibodies or JAK inhibitor 1, the overexpression of EMT-associated marker genes was effectively normalized (Fig. 5B). These findings suggested that downregulation of the JAK-PD-L1 signaling pathway may inhibit metastasis and EMT in CRPC.
Figure 5.

Downregulation of PD-L1 and JAK expression inhibits migration and EMT of C4-2-Sc and CWR22Rv1-Sc cells. C4-2-Sc and CWR22Rv1-Sc cells were treated with PD-L1 antibodies and JAK inhibitor 1. (A) Migration of tumor cells was significantly decreased following incubation with PD-L1 antibodies and JAK inhibitor 1. Magnification, ×40. (B) Expression of EMT-associated markers was reversed, and EMT of the tumor cells was inhibited. One-way analysis of variance followed by Fisher's least significant difference post hoc test was used for comparison among groups. ***P<0.001. ATM, ataxia telangiectasia mutated kinase; E-Cad, E-cadherin; EMT, epithelial-mesenchymal transition; JAK, Janus kinase; N-Cad, N-cadherin; PD-L1, programmed cell death 1 ligand 1; Sc, scramble; si, small interfering RNA; VEGF, vascular endothelial growth factor.
Discussion
Numerous studies have revealed that ATM has an important role in cell growth and DNA damage (28–31); however, the role of ATM in tumor EMT has not been extensively studied. Suppression of the ATM signaling pathway has been demonstrated to inhibit ovarian cancer metastasis (24), and in a study of macrophage paracrine signaling in breast cancer metastasis, downregulation of ATM reduced the expression of EMT-associated markers in tumor cells, and thus inhibited metastasis (32). In the present study, it was revealed that the expression levels of EMT-associated genes were decreased in ATM gene knockout CRPC cells and the migratory ability of these cells was suppressed. To further investigate this, ATM inhibitors were added to primary cells and to Sc cells, and similar effects were observed. Furthermore, it was revealed that the proliferative ability of cells was reduced in ATM knockout CRPC cells on days 3 and 4. Therefore, the results of the present study suggested that ATM may accelerate the progression of CRPC EMT and metastasis.
The mechanisms underlying EMT and tumor metastasis have been extensively studied in recent years. PD-L1 is closely associated with EMT, invasion and metastasis of breast cancer, lung cancer, kidney cancer, head and neck tumors, and other malignant tumors. Furthermore, suppression of PD-L1 expression can inhibit tumor metastasis (14–16,25). PD-L1 is highly expressed in invasive prostate cancer and enzalutamide-resistant prostate cancer (33), and can induce prostate cancer immune evasion (34). Whether PD-L1 is a downstream regulatory factor of ATM and whether its regulation affects the EMT and metastasis of CRPC has not yet been determined. Therefore, the present study aimed to investigate PD-L1 expression in ATM knockout cells. ATM knockout cells were revealed to exhibit low PD-L1 expression, thus suggesting that ATM signaling pathway inhibition suppresses PD-L1 expression.
Considering the aforementioned results, the mechanisms by which ATM regulates PD-L1 expression and affects the EMT and metastasis of CRPC cells will be investigated in future studies. The roles of the JAK/STAT3 signaling pathway in tumor EMT have been widely studied with regards to prostate cancer, lung cancer, pancreatic cancer, neck tumors and other solid tumors (35–38). JAK1/2 inhibitors have also been revealed to modulate prostate cancer metastasis by regulating interleukin (IL)-6 in animal experiments and in vitro; therefore, IL-6 has been suggested to represent a therapeutic target for the treatment of metastatic prostate cancer (39). Furthermore, previous studies have demonstrated that the JAK/STAT3 signaling pathway is involved in PD-L1 regulation (40,41). To investigate whether the JAK/STAT3 signaling pathway is associated with ATM-associated regulation of PD-L1 expression, and thus whether the JAK/STAT3 signaling pathway affects the EMT and metastasis of CRPC, the expression levels of p-JAK1, p-JAK2 and PD-L1 were investigated, and the results revealed that expression levels of p-JAK1, p-JAK2 and PD-L1were suppressed in ATM knockout CRPC cells. In addition, inhibition of the JAK signaling pathway by JAK inhibitor 1 was revealed to significantly suppress the expression of PD-L1, whereas STAT3 inhibition did not affect the expression of PD-L1. Therefore, it may be suggested that the JAK signaling pathway is involved in the regulation of PD-L1 via ATM.
To investigate the effects of PD-L1 and JAK expression on CRPC cell EMT and migration, PD-L1 antibodies and JAK inhibitor 1 were used to treatC4-2-Sc and CWR22Rv1-Sc cells; the results demonstrated that post incubation, the migratory ability of CRPC cells was significantly decreased and the expression of EMT-associated markers was effectively normalized. Therefore, these results revealed that the ATM-JAK-PD-L1 signaling pathway may serve an important role in the EMT and metastatic progression in CRPC.
In conclusion, with continuous advances in prostate cancer androgen deprivation therapy, CRPC has become difficult to treat clinically and has been extensively studied (42,43). Targeted therapy is considered a promising therapeutic approach for numerous tumor types, including prostate cancer (44–46). The present study aimed to investigate the mechanisms underlying CRPC cell EMT and metastasis, and it was revealed that inhibition of the ATM-JAK-PD-L1 signaling pathway may suppress EMT and metastatic progression of CRPC cells. This may represent a novel therapeutic approach for targeted therapy for the improvement of prognosis and quality of life for patients with CRPC.
Acknowledgements
Authors thank Soo Ok Lee for assistance with manuscript preparation.
Funding
The present study was supported by the National Natural Science Foundation of China (grant no. 81472776) for the work of Professor Dr. Yang, Dr. Xu and Professor Shan; as well as the Second Affiliated Hospital of Soochow University preponderant clinic discipline group project funding (grant no. XKQ2015008).
Availability of data and materials
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
Authors' contributions
LZ, LJX and JZ performed the in vitro and in vivo experiments and statistical analyses, and made the figures. JL and B-XX contributed to the generation of knockdown cell lines. JG and CYS provided and performed staining of human tissues. YCZ and YBZ helped with the interpretation of data and reviewed the manuscript. DRY and YXS conceived the idea and wrote the manuscript. All authors reviewed and agreed to the information in this manuscript.
Ethics approval and consent to participate
All patients provided informed consent. The present study was approved by the Ethics Committee of the Second Affiliated Hospital of Soochow University (Suzhou, China).
Consent for publication
All patients provided written informed consent for the publication of any associated data and accompanying images.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 3.Meng F, Wu G. The rejuvenated scenario of epithelial-mesenchymal transition (EMT) and cancer metastasis. Cancer Metastasis Rev. 2012;31:455–467. doi: 10.1007/s10555-012-9379-3. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Liu D, Feng Z, Mao J, Zhang C, Lu Y, Li J, Zhang Q, Li Q, Li L. MicroRNA-138 modulates metastasis and EMT in breast cancer cells by targeting vimentin. Biomed Pharmacother. 2016;77:135–141. doi: 10.1016/j.biopha.2015.12.018. [DOI] [PubMed] [Google Scholar]
- 5.Li C, Wang J, Kong J, Tang J, Wu Y, Xu E, Zhang H, Lai M. GDF15 promotes EMT and metastasis in colorectal cancer. Oncotarget. 2016;7:860–872. doi: 10.18632/oncotarget.6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suh SS, Yoo JY, Cui R, Kaur B, Huebner K, Lee TK, Aqeilan RI, Croce CM. FHIT suppresses epithelial-mesenchymal transition (EMT) and metastasis in lung cancer through modulation of microRNAs. PLoS Genet. 2014;10:e1004652. doi: 10.1371/journal.pgen.1004652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ombrato L, Malanchi I. The EMT universe: Space between cancer cell dissemination and metastasis initiation. Crit Rev Oncog. 2014;19:349–361. doi: 10.1615/CritRevOncog.2014011802. [DOI] [PubMed] [Google Scholar]
- 8.Liu CH, Tang WC, Sia P, Huang CC, Yang PM, Wu MH, Lai IL, Lee KH. Berberine inhibits the metastatic ability of prostate cancer cells by suppressing epithelial-to-mesenchymal transition (EMT)-associated genes with predictive and prognostic relevance. Int J Med Sci. 2015;12:63–71. doi: 10.7150/ijms.9982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang J, Li Y, Daniels G, Sfanos K, De Marzo A, Wei J, Li X, Chen W, Wang J, Zhong X, et al. LEF1 targeting EMT in prostate cancer invasion is regulated by miR-34a. Mol Cancer Res. 2015;13:681–688. doi: 10.1158/1541-7786.MCR-14-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong D, Sethi S, Li Y, Chen W, Sakr WA, Heath E, Sarkar FH. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. Prostate. 2015;75:161–174. doi: 10.1002/pros.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Zhang J, Ma Y, Chen J, Dong B, Zhao W, Wang X, Zheng Q, Fang F, Yang Y. Testicular orphan receptor 4 (TR4) is a marker for metastasis and poor prognosis in non-small cell lung cancer that drives the EMT phenotype. Lung Cancer. 2015;89:320–328. doi: 10.1016/j.lungcan.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 12.Ma X, Yan W, Dai Z, Gao X, Ma Y, Xu Q, Jiang J, Zhang S. Baicalein suppresses metastasis of breast cancer cells by inhibiting EMT via downregulation of SATB1 and Wnt/β-catenin pathway. Drug Des Devel Ther. 2016;10:1419–1441. doi: 10.2147/DDDT.S102541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huan H, Wen X, Chen X, Wu L, Liu W, Habib NA, Bie P, Xia F. C/EBPα short-activating RNA suppresses metastasis of hepatocellular carcinoma through inhibiting EGFR/β-catenin signaling mediated EMT. PLoS One. 2016;11:e0153117. doi: 10.1371/journal.pone.0153117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alsuliman A, Colak D, Al-Harazi O, Fitwi H, Tulbah A, Al-Tweigeri T, Al-Alwan M, Ghebeh H. Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol Cancer. 2015;14:149. doi: 10.1186/s12943-015-0421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Wang H, Zhao Q, Xia Y, Hu X, Guo J. PD-L1 induces epithelial-to-mesenchymal transition via activating SREBP-1c in renal cell carcinoma. Med Oncol. 2015;32:212. doi: 10.1007/s12032-015-0655-2. [DOI] [PubMed] [Google Scholar]
- 16.Ock CY, Kim S, Keam B, Kim M, Kim TM, Kim JH, Jeon YK, Lee JS, Kwon SK, Hah JH, et al. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget. 2016;7:15901–15914. doi: 10.18632/oncotarget.7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen W, Liang W, Wu J, Kowolik CM, Buettner R, Scuto A, Hsieh MY, Hong H, Brown CE, Forman SJ, et al. Targeting JAK1/STAT3 signaling suppresses tumor progression and metastasis in a peritoneal model of human ovarian cancer. Mol Cancer Ther. 2014;13:3037–3048. doi: 10.1158/1535-7163.MCT-14-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, Wels J, Theilen T, Granitto S, Zhang X, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. 2013;15:848–862. doi: 10.1593/neo.13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012;30:1005–1014. doi: 10.1200/JCO.2010.31.8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao B, Shen X, Kunos G, Meng Q, Goldberg ID, Rosen EM, Fan S. Constitutive activation of JAK-STAT3 signaling by BRCA1 in human prostate cancer cells. FEBS Lett. 2001;488:179–184. doi: 10.1016/S0014-5793(00)02430-3. [DOI] [PubMed] [Google Scholar]
- 21.Teng Y, Ghoshal P, Ngoka L, Mei Y, Cowell JK. Critical role of the WASF3 gene in JAK2/STAT3 regulation of cancer cell motility. Carcinogenesis. 2013;34:1994–1999. doi: 10.1093/carcin/bgt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tam L, McGlynn LM, Traynor P, Mukherjee R, Bartlett JM, Edwards J. Expression levels of the JAK/STAT pathway in the transition from hormone-sensitive to hormone-refractory prostate cancer. Br J Cancer. 2007;97:378–383. doi: 10.1038/sj.bjc.6603871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun M, Guo X, Qian X, Wang H, Yang C, Brinkman KL, Serrano-Gonzalez M, Jope RS, Zhou B, Engler DA, et al. Activation of the ATM-Snail pathway promotes breast cancer metastasis. J Mol Cell Biol. 2012;4:304–315. doi: 10.1093/jmcb/mjs048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin S, Wang P, Yang L, Liu Y, Wang Y, Liu M, Qi Z, Meng J, Shi TY, Yang G, Zang R. Wip1 suppresses ovarian cancer metastasis through the ATM/AKT/Snail mediated signaling. Oncotarget. 2016;7:29359–29370. doi: 10.18632/oncotarget.8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S, Koh J, Kim MY, Kwon D, Go H, Kim YA, Jeon YK, Chung DH. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum Pathol. 2016;58:7–14. doi: 10.1016/j.humpath.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Doi T, Ishikawa T, Okayama T, Oka K, Mizushima K, Yasuda T, Sakamoto N, Katada K, Kamada K, Uchiyama K, et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol Rep. 2017;37:1545–1554. doi: 10.3892/or.2017.5399. [DOI] [PubMed] [Google Scholar]
- 27.Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, Ritz J. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4:e1008824. doi: 10.1080/2162402X.2015.1008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subhash VV, Tan SH, Yeo MS, Yan FL, Peethala PC, Liem N, Krishnan V, Yong WP. ATM expression predicts veliparib and irinotecan sensitivity in gastric cancer by mediating P53-independent regulation of cell cycle and apoptosis. Mol Cancer Ther. 2016;15:3087–3096. doi: 10.1158/1535-7163.MCT-15-1002. [DOI] [PubMed] [Google Scholar]
- 29.Khoronenkova SV, Dianov GL. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression; Proc Natl Acad Sci USA; 2015; pp. 3997–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojewoda M, Walczak J, Duszyński J, Szczepanowska J. Selenite activates the ATM kinase-dependent DNA repair pathway in human osteosarcoma cells with mitochondrial dysfunction. Biochem Pharmacol. 2015;95:170–176. doi: 10.1016/j.bcp.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 31.McCabe N, Hanna C, Walker SM, Gonda D, Li J, Wikstrom K, Savage KI, Butterworth KT, Chen C, Harkin DP, et al. Mechanistic rationale to target PTEN-deficient tumor cells with inhibitors of the DNA damage response kinase ATM. Cancer Res. 2015;75:2159–2165. doi: 10.1158/0008-5472.CAN-14-3502. [DOI] [PubMed] [Google Scholar]
- 32.Singh R, Shankar BS, Sainis KB. TGF-β1-ROS-ATM-CREB signaling axis in macrophage mediated migration of human breast cancer MCF7 cells. Cell Signal. 2014;26:1604–1615. doi: 10.1016/j.cellsig.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 33.Bishop JL, Sio A, Angeles A, Roberts ME, Azad AA, Chi KN, Zoubeidi A. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget. 2015;6:234–242. doi: 10.18632/oncotarget.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM, van Bokhoven A, Meeker AK, Lucia MS, Anders RA, DeMarzo AM, Drake CG. Paucity of PD-L1 expression in prostate cancer: Innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015;18:325–332. doi: 10.1038/pcan.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aalinkeel R, Hu Z, Nair BB, Sykes DE, Reynolds JL, Mahajan SD, Schwartz SA. Genomic analysis highlights the role of the JAK-STAT signaling in the anti-proliferative effects of dietary flavonoid-‘Ashwagandha’ in prostate cancer cells. Evid Based Complement Alternat Med. 2010;7:177–187. doi: 10.1093/ecam/nem184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu Z, Zhao J, Zhang HT. JAK/STAT3 signaling is required for TGF-β-induced epithelial-mesenchymal transition in lung cancer cells. Int J Oncol. 2014;44:1643–1651. doi: 10.3892/ijo.2014.2310. [DOI] [PubMed] [Google Scholar]
- 37.Macha MA, Rachagani S, Gupta S, Pai P, Ponnusamy MP, Batra SK, Jain M. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT3 and Src/FAK signaling. Cancer Lett. 2013;341:166–177. doi: 10.1016/j.canlet.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9:1658–1667. doi: 10.1158/1541-7786.MCR-11-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu L, Talati P, Vogiatzi P, Romero-Weaver AL, Abdulghani J, Liao Z, Leiby B, Hoang DT, Mirtti T, Alanen K, et al. Pharmacologic suppression of JAK1/2 by JAK1/2 inhibitor AZD1480 potently inhibits IL-6-induced experimental prostate cancer metastases formation. Mol Cancer Ther. 2014;13:1246–1258. doi: 10.1158/1535-7163.MCT-13-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu L, Shen M, Chen X, Zhu R, Yang DR, Tsai Y, Keng PC, Chen Y, Lee SO. Adipocytes affect castration-resistant prostate cancer cells to develop the resistance to cytotoxic action of NK cells with alterations of PD-L1/NKG2D ligand levels in tumor cells. Prostate. 2018;78:353–364. doi: 10.1002/pros.23479. [DOI] [PubMed] [Google Scholar]
- 41.Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, Huang JA. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49:1360–1368. doi: 10.3892/ijo.2016.3632. [DOI] [PubMed] [Google Scholar]
- 42.Lee DJ, Cha EK, Dubin JM, Beltran H, Chromecki TF, Fajkovic H, Scherr DS, Tagawa ST, Shariat SF. Novel therapeutics for the management of castration-resistant prostate cancer (CRPC) BJU Int. 2012;109:968–985. doi: 10.1111/j.1464-410X.2011.10643.x. [DOI] [PubMed] [Google Scholar]
- 43.Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC) Transl Androl Urol. 2015;4:365–380. doi: 10.3978/j.issn.2223-4683.2015.05.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kübler H, Miller K. New therapy concepts for castration-resistant prostate cancer: Between hormone manipulation, targeted therapy and chemotherapy. Urologe A. 2013;52(1517–1518):1520–1521. doi: 10.1007/s00120-013-3247-9. 1524–1526. (In German) [DOI] [PubMed] [Google Scholar]
- 45.Qi WX, Fu S, Zhang Q, Guo XM. Efficacy and toxicity of molecular targeted therapies in combination with docetaxel for metastatic castration-resistant prostate cancer: A meta-analysis of phase III randomized controlled trials. J Chemother. 2015;27:181–187. doi: 10.1179/1973947814Y.0000000230. [DOI] [PubMed] [Google Scholar]
- 46.Loriot Y, Eymard JC, Patrikidou A, Ileana E, Massard C, Albiges L, Di Palma M, Escudier B, Fizazi K. Prior long response to androgen deprivation predicts response to next-generation androgen receptor axis targeted drugs in castration resistant prostate cancer. Eur J Cancer. 2015;51:1946–1952. doi: 10.1016/j.ejca.2015.06.128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.