

Abstract
Pigment epithelial-derived factor (PEDF) is a multifunctional secreted glycoprotein, which exerts a variety of physiological activities. PEDF may protect against hypoxia-induced cell death associated with its antioxidative effects and p53 mitochondrial translocation in cultured cardiomyocytes and H9c2 cells. Additionally, previous studies have suggested that autophagy is an important cell survival mechanism. However, the effect of PEDF on autophagy and the associated pathway in hypoxic H9c2 cells has not been fully established. Autophagy has been reported to regulate lipid metabolism; however, little is known about whether PEDF is able to regulate lipid metabolism by promoting autophagy. In the present study, western blotting results revealed that PEDF increased the level of microtubule-associated protein 1A/1B-light chain 3 (LC3)-II. Transmission electron microscopy (TEM) and LC3 fluorescence demonstrated that PEDF increased the number of autophagosomes. PEDF also increased the viability of hypoxic H9c2 cells and decreased the level of cleaved caspase-3 protein, as evidenced by CCK-8 assays and western blotting, respectively. TEM and a triglyceride assay kit demonstrated that PEDF-induced autophagy may stimulate lipid degradation. Western blotting results revealed a novel mechanism underlying PEDF-induced H9c2 cell autophagy via the PEDF-R-mediated Atg5 pathway under hypoxic conditions. Furthermore, the results also suggest that PEDF-induced autophagy may stimulate lipid degradation. The survival function of autophagy suggests that modulation of PEDF-induced autophagy may be used as a therapeutic strategy to protect cells against lipid-associated metabolic diseases.
Keywords: pigment epithelial-derived factor, pigment epithelial-derived factor receptor, autophagy, lipid degradation, autophagy protein 5
Introduction
Lipotoxicity refers to excessive accumulation of lipid in cells, including hepatocytes, pancreatic cells and cardiomyocytes, which contributes to a loss of cellular function and cell death (1–2). A previous study suggested that excessive lipid levels may result in the death of mouse myocardiocytes, possibly causing heart failure (3). Pigment epithelial-derived factor (PEDF), a 50 kDa multifunctional glycoprotein with antiangiogenic, antitumor, anti-inflammatory, antioxidative and cardioprotective properties, protects against hypoxia-induced apoptosis and necroptosis in primary cardiomyocytes and H9c2 cells (4–6). Autophagy is an evolutionarily conserved and highly regulated process that is essential for maintaining cellular homeostasis by preventing the accumulation of damaged proteins and organelles, whilst sustaining metabolism during hypoxia (7,8). However, whether PEDF regulates lipid degradation by inducing autophagy remains to be elucidated.
The autophagy-associated-gene proteins are major regulators of the autophagy system, mediating several sequential steps of the autophagic process (7,8). Autophagy-related 5 (Atg5) is a protein required for the formation of autophagosomes (7,9) and sequesters cytoplasmic materials prior to lysosomal delivery. Atg5 serves a major role in autophagic processes; however, further investigation is required to understand whether PEDF promotes autophagy and whether this effect contributes to cytoprotection. Whether Atg5 is involved in PEDF-promoted autophagy is yet to be investigated.
Previous studies have revealed that adipose triglyceride lipase (ATGL) serves as a receptor for PEDF in retinal epithelial cells and cardiomyocytes (10,11). PEDF-receptor (PEDF-R), an 80 kDa receptor protein, was used in the present study. PEDF-R has been reported to exhibit biological effects by interacting with PEDF on cell-surface (12). PEDF-R is the key enzyme of lipid catabolism and catalyzes the lipid lipolysis cascade (13); a lack of PEDF-R in cardiac muscle causes a lipolytic defect, which leads to lipid accumulation, severe cardiac dysfunction and premature death (14). However, whether PEDF-R is associated with autophagy and whether PEDF-R-mediated autophagy is associated with lipid degradation requires further investigation.
The aim of the present study was to examine whether PEDF-promoted autophagy may protect H9c2 cells under hypoxic conditions, and whether PEDF stimulates lipid degradation by promoting autophagy. It was also investigated whether PEDF-promoted autophagy is regulated by Atg5 in H9c2 cells.
Materials and methods
Antibodies and reagents
Anti-microtubule-associated protein-light chain 3 (LC3)B (cat. no. 2775) and anti-cleaved caspase-3 (cat. no. 9662) antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Alexa Fluor 488 (cat. no. 705-546-147) was purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). Cell counting Kit-8 (CCK-8; cat. no. WO97/38985) was purchased from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). The LDH Cytotoxicity Assay kit was purchased from Nanjing KeyGen Biotech Co., Ltd (Nanjing, China). A triglyceride (TG) assay kit (GPO-POD; cat. no. E1003) was purchased from (Applygen Technologies Inc., Beijing, China). The in situ cell death detection kit (cat. no. 11684795910) was purchased from Sigma-Aldrich; (Merck KGaA, Darmstadt, Germany). Anti-Atg5 antibody (cat. no. ab227132) was purchased from Abcam (Cambridge, UK). Recombinant PEDF was synthesized by Cusabio Biotech Co., Ltd. (Hubei, China). The special inhibitor of autophagy 3-methyladenine (3-MA; cat. no. KGATGR006; Nanjing KeyGen Biotech Co., Ltd.) was donated from Dr Xiaofang Yang (Laboratory of Clinical and Experimental Pathology, Xuzhou Medical University, Jiangsu, China).
Recombinant lentivirus constructs and viral production
Recombinant lentivirus (LV) was prepared as previously described (15). The Lentivirus expressing PEDF or PEDF-R_shRNA were purchased from GENECHEM, Inc. (Daejeon, Korea). The concentrated titer of virus suspension was 2×1012 Tu/l. Transient transfection of H9c2 cells with short interfering RNA (siRNA) targeting the PEDF-R genes were performed using Lipofectamine 3000 according to the manufacturer's instructions (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Preparations of PEDF protein
Recombinant rat PEDF (GenBank™ accession no. NM_177927) was synthesized by Cusabio Biotech, Co., Ltd. (Wuhan, China).
Cell culture and hypoxia
The H9c2 cells embryonic rat heart-derived cell line was obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (both from Gibco; Thermo Fisher Scientific, Inc.) and 100 mg/ml penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Hypoxia was achieved by culturing the cells in D-Hank's liquid with glucose deprivation in a tri-gas incubator (Heal Force, Shanghai, China) saturated with 5% CO2/1% O2 at 37°C for 4 h.
TUNEL analysis
Cells were seeded in 48-well plates (Corning Inc., Corning, NY, USA). Following hypoxic incubation, cells were treated with 4% paraformaldehyde for 10 min at room temperature, and then washed three times in PBS, (pH 7.4). Next cells were incubated with 1% goat serum diluted in PBS for 1 h at room temperature. Terminal dexynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) staining was then performed using an in situ cell death detection kit (Roche Molecular Diagnostics, Pleasanton, CA, USA) according to the manufacturer's instructions. The cells were incubated with reaction buffer containing enzyme solution and label solution with an enzyme-to-label ratio of 159 at 37°C for 1 h 33 min. Cells were counterstained using Hoechst stain (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 15 min at room temperature. The cells were observed under a fluorescence microscope (Olympus, Tokyo, Japan). A total of 30 fields of view were used. Magnification, ×20.
Western blotting
Western blotting was performed following standard procedures (6). The cells were lysed in radioimmunoprecipitation assay lysis buffer: 100 mmol/l Tris-HCl, 4% SDS, 20% glycerine, 200 mmol/l dithithreitol and protease inhibitors (pH 6.8). Total cellular protein was denatured by boiling for 10 min with an equal volume of 2 3 Tris-glycine SDS buffer. Protein concentration was determined using a bicinchonic acid assay. An equal amount of protein (50 ng) for each sample was resolved via 8–15% SDS-PAGE and transferred onto a nitrocellulose membrane (EMD Millipore, Billerica, MA, USA). The membrane was blocked with 5% nonfat milk/PBS-Tween-20 for 2 h at room temperature and subsequently incubated with anti-microtubule-associated protein-light chain 3B (LC3B) (cat. no. 2775; 1:1,000) and anti-cleaved caspase-3 (cat. no. 9662; 1:1,000) antibodies overnight at 4°C, respectively. Subsequently, fluorescent-labeled secondary antibodies were added at room temperature for 2 h and then scanned by the Odyssey Infrared Imaging System with Odyssey 3.0 software (both from LI-COR Biosciences, Lincoln, NE, USA).
Immunofluorescence
H9c2 cells were cultured in 48-well plates at a density of 1×104 cells/ml. Following hypoxic treatment for 4 h (6), H9c2 cells were washed twice with PBS and fixed with freshly prepared 4% paraformaldehyde at room temperature for 15 min (6). Antigen accessibility was increased by treatment with 2% Triton X-100 at room temperature for 10 min. H9c2 cells were subsequently blocked with 3% bovine serum albumin (cat. no. VIC018; Vicmed Co., Ltd., Xuzhou, China) at room temperature for 30 min. Following incubation with anti-microtubule-associated protein-light chain 3B (LC3B) (cat. no. 2775, 1:1,000) and anti-cleaved caspase-3 (cat. no. 9662; 1:1,000) primary antibodies at 4°C overnight, respectively. Then cells were washed and incubated with anti-mouse secondary antibody (cat. no. A21207; 1:200; Thermo Fisher Scientific, Inc.) under light-protected conditions for 1 h at room temperature. H9c2 cells were then washed three times with PBS for 5 min, captured and analyzed using TCS SP8 STED 3X (Leica Microsystems GmbH, Wetzlar, Germany).
Transmission electron microscopy (TEM)
H9c2 cells were fixed with 4% paraformaldehyde and 2.5% glutaraldehyde at room temperature overnight. Subsequently, samples were incubated while protected from light with 1% osmium tetroxide at room temperature for 1 h. Following washing in distilled water, the samples were dehydrated in graded ethanol concentrations and then embedded in molds with fresh resin. Ultrathin sections (70 nm) were obtained with an EM UC7 (Leica Microsystems GmbH), the samples ultrathin sections were then incubated in 2% uranyl acetate for 0.5 h and stained with lead citrate for 10 min at room temperature. Finally, the samples were observed with a Tecnai G2 T12 at room temperature (FEI; Thermo Fisher Scientific, Inc.).
H9c2 cell viability test
H9c2 cells were seeded in 96-well plates at a density of 1×104 cells/ml. Following hypoxic treatments (6), H9c2 cells viability was analyzed using the CCK-8 kit according to the manufacturer's protocol. Absorbance was measured at 450 nm with a microplate reader (BioTek Synergy 2; BioTek Instruments, Inc., Winooski, VT, USA).
Lactate dehydrogenase (LDH) release assay
H9c2 cells were seeded in 96-well plates (1×104 cells/ml). Following hypoxic treatments the activity of LDH within H9c2 cells released into the medium was assessed as previously described (6) by a microplate reader (BioTek Synergy 2) analysis at 440 nm, using an LDH Cytotoxicity Assay kit according to the manufacturer's instructions.
TG assay
H9c2 cells were seeded in 96-well plates at a concentration of 1×104 cells/ml. The levels of intracellular TG were assessed using a TG assay kit according to the manufacturer's instructions.
Statistical analysis
Data are expressed as the mean ± standard deviation. Statistical analysis was performed using repeated-measures or one-way analysis of variance followed by the Tukey's test for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
PEDF induces autophagy via PEDF-R within hypoxic H9c2 cells
To investigate whether PEDF promotes H9c2 cell autophagy via PEDF-R under hypoxic conditions, the level of autophagy marker protein LC3 in H9c2 cells was detected under normoxic and hypoxic conditions. A total of ≤4 h of hypoxic conditions resulted in a significantly increase in the expression of LC3-II in H9c2 (P<0.05). Furthermore, overexpression of PEDF significantly increased LC3-II expression (P<0.05; Fig. 1A). In addition, LC3 staining (green) was used to further test PEDF-induced autophagy. As presented in Fig. 1B, a significant increase in LC3 puncta was detected in PEDF-treated H9c2 cells compared with cells under hypoxic conditions only (P<0.05). To further confirm that PEDF treatment induced H9c2 cell autophagy under hypoxic conditions, TEM analyses were performed. H9c2 cells treated with PEDF exhibited a marked increase in the number of autophagosomes compared with cells under hypoxic conditions only (P<0.05; Fig. 1C). Collectively, the findings of the present study indicate that PEDF is able to promote H9c2 cell autophagy via PEDF-R under hypoxic conditions.
Figure 1.

PEDF promotes autophagy via PEDF-R in hypoxic H9c2 cells. H9c2 cells were maintained in normoxic or hypoxic conditions for 4 h with or without PEDF (10 nM). RNA interference assays were used to silence PEDF-R. (A) Samples were collected for western blotting to analyze the expression of LC3-II protein (B) LC3 staining was assessed by fluorescence microscopy. Quantitative analysis of H9c2 cells containing LC3 puncta. Scale bar, 20 µm. (C) Representative images of transmission electron microscopy of H9c2 cells autophagy. Typical autophagosomes (arrows) were observed. Scale bar, 1 µm. *P<0.05. Ctrl, control; LC3, microtubule-associated protein 1A/1B-light chain 3; PEDF-R, pigment epithelial-derived factor receptor; siPEDF, short interfering PEDF RNA.
PEDF-induced autophagy contributes to lipid degradation
It has previously been reported that PEDF stimulates cardiac triglyceride degradation via ATGL (11). In addition, it has been demonstrated that autophagy may regulate lipid metabolism (16). In the present study, the association between PEDF-promoted autophagy and lipid metabolism was investigated. As presented in Figs. 2A and 4H of hypoxia led to an increase in lipid droplets (LDs), whereas PEDF-treatment resulted in decreased LDs (P<0.05). To investigate the effects of PEDF-induced autophagy on LDs, H9c2 cells were treated with 3-methyladenine (3-MA). The addition of 3-MA resulted in a marked increase in LDs (P<0.05), which suggests that PEDF may induce lipid degradation via autophagy. In addition, the TG content was significantly increased within H9c2 cells under hypoxic conditions (P<0.05). Treating H9c2 cells with PEDF significantly decreased TG levels, whereas the addition of 3-MA resulted in increased TG content (P<0.05; Fig. 2B). Collectively, these findings suggest that PEDF stimulates H9c2 cell lipid degradation by promoting autophagy.
Figure 2.

PEDF-induced autophagy contributes to lipid degradation. H9c2 cells were maintained in normoxic or hypoxic conditions for 4 h with or without PEDF (10 nM). RNA interference assays were used to silence PEDF-R expression and autophagy inhibitor 3-MA (5 mM) was added. (A) Representative images of transmission electron microscopy analysis of H9c2 cells LDs. Typical LDs (arrows) were observed. Scale bar, 1 µm. (B) TG levels were quantified with a TG assay kit. *P<0.05. Ctrl, control; LDs, lipid droplets; PEDF-R, pigment epithelial-derived factor receptor; 3-MA, 3-methyladenine; siPEDF, short interfering PEDF RNA; TG, triglyceride.
Figure 4.
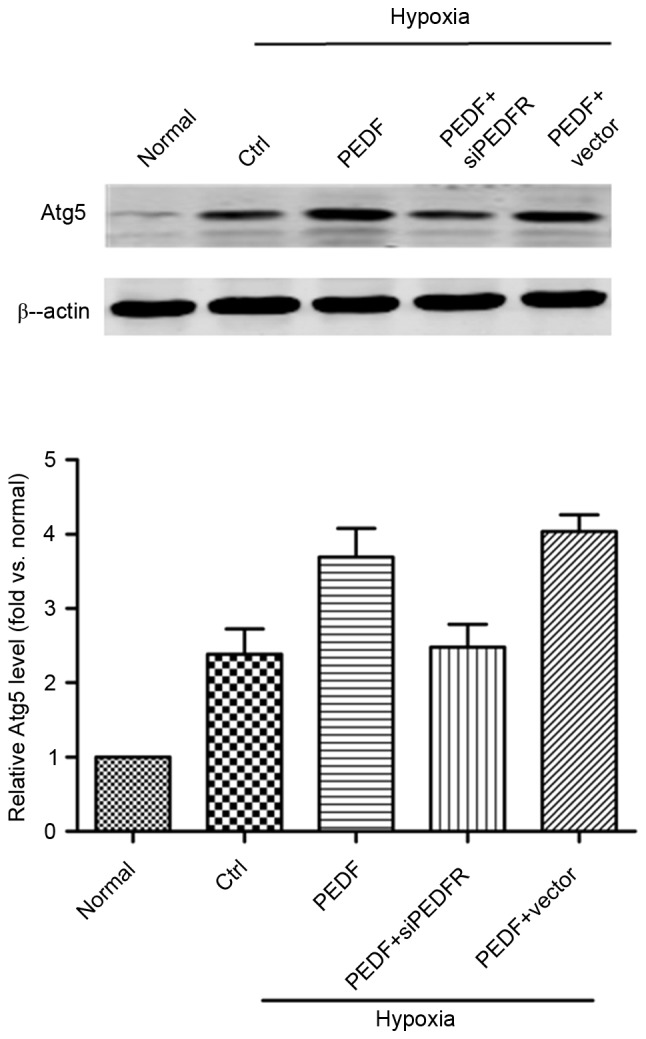
PEDF-induced autophagy is dependent of the Atg5 pathway. H9c2 cells were maintained in normoxic or hypoxic conditions for 4 h with or without PEDF (10 nM). RNA interference assays were used to silence PEDF-R. Samples were collected and analyzed for the expression levels of Atg5 protein by western blotting. n=4. Atg5, autophagy related 5; Ctrl, control; PEDF-R, pigment epithelial-derived factor receptor; siPEDF, short interfering PEDF RNA.
PEDF protects H9c2 cells against hypoxia-induced apoptosis by inducing autophagy
Following the establishment of PEDF-induced autophagy, the protective role of PEDF-induced autophagy in hypoxic H9c2 cells was investigated. CCK-8 and LDH release assays revealed similar results (P<0.05; Fig. 3A and B). Under hypoxic conditions, the viability of H9c2 cells was decreased. PEDF overexpression attenuated the hypoxia-mediated reduction in cell viability, whereas the autophagy inhibitor 3-MA attenuated this effect. The protective effects of PEDF may be achieved via reducing H9c2 cell apoptosis. TUNEL staining was used to assess H9c2 cell apoptosis and it was demonstrated that PEDF significantly reduced H9c2 cell apoptosis compared with in hypoxia only (P<0.05; Fig. 3C). Conversely, 3-MA promoted hypoxia-induced cell death (P<0.05; Fig. 3C). It was also demonstrated that PEDF significantly suppressed the levels of cleaved caspase-3, whereas the addition of 3-MA increased its expression (P<0.05; Fig. 3D). These findings indicate that PEDF-induced autophagy may be increase cell survival under hypoxic conditions by reducing hypoxia-induced apoptosis.
Figure 3.

PEDF protects H9c2 cells against hypoxia-induced apoptosis by inducing autophagy. CCK-8 and LDH release assays were employed to assess (A) cell viability and (B) the rate of cell death. (C) Effect of PEDF on H9c2 cells apoptosis. TUNEL (green) staining was performed within each group. Nuclei were stained with DAPI (blue). Scale bar, 50 µm. (D) Samples were collected and the expression of C-Casp-3 protein was analyzed by western blotting. *P<0.05. 3-MA, 3-methyladenine; C-Casp-3, cleaved caspase-3; CCK-8, Cell Counting Kit-8; Ctrl, control; LDH, lactate dehydrogenase; PEDF-R, pigment epithelial-derived factor receptor; siPEDF, short interfering PEDF RNA.
PEDF-induced autophagy is dependent on the Atg5 pathway
As presented in Fig. 4, the addition of PEDF increased Atg5 expression and the effect of PEDF treatment was inhibited by PEDF-R siRNA (P<0.05). These findings suggest that, under hypoxic conditions, PEDF may promote autophagy via the PEDF-R-induced Atg5 pathway.
Discussion
In the present study, it was confirmed that PEDF-promoted autophagy stimulates lipid degradation. In addition, PEDF was demonstrated to protect H9c2 cells against hypoxia-induced apoptosis by promoting autophagy. The results of the present study indicate a potential mechanism of PEDF promoted-autophagy that is dependent on the Atg5 pathway via PEDF-R.
Autophagy is a highly conserved and genetically programmed process, which maintains metabolic and cellular homeostasis by recycling intracellular components for use in macromolecular synthesis and inhibits the accumulation of aggregated proteins and damaged organelles that may be toxic (17–20). The results of the present study revealed that PEDF promotes autophagy via PEDF-R. This mechanism is dependent on the Atg5 pathway; the Atg5 protein serves a role in the early stage of autophagosome formation (7,9). PEDF treatment resulted in an increase in Atg5. PEDF-R, a lipase-linked cell membrane receptor for PEDF (10), binds to PEDF and stimulates triglyceride degradation (12). PEDF may therefore stimulate the activity of PEDF-R, generating intracellular signaling molecules and ultimately upregulating Atg5 under hypoxic conditions. The results of the present study suggest that PEDF upregulates the expression of Atg5 via PEDF-R to promote autophagy. However, further investigation is required to confirm the mechanisms underlying the induction of Atg5 initiated by PEDF-R in hypoxic H9c2 cells treated with PEDF.
During acute myocardial infarction, TG accumulation due to reduced oxygen availability results in H9c2 cell toxicity and serves a crucial role in the progression of cardiomyopathy and heart failure (2,21). Various diseases associated with excess lipid accumulation, including obesity and diabetes, have been reported to feature lipotoxicity (1,22). Therefore, inhibiting TG accumulation may contribute to the treatment of these diseases. In the present study, the number of LDs was significantly increased under hypoxic conditions, whereas H9c2 cells overexpressing PEDF had a markedly lower number of LDs. Conversely, the addition of 3-MA resulted in an increase in LDs. TG levels in H9c2 cells increased under hypoxic conditions and decreased in response to PEDF. Treatment with 3-MA resulted in increased TG levels. In the present study, it was demonstrated that PEDF-induced autophagy may stimulate TG degradation within hypoxic H9c2 cells. The results demonstrated a previously unreported association between PEDF-induced autophagy and lipid metabolism. However, further investigation is required to fully elucidate the molecular mechanisms of PEDF-induced autophagy in the regulation of lipid metabolism in response to hypoxia.
Autophagy is an important cell survival mechanism that eliminates aggregated proteins and unwanted organelles (23,24) and is associated with cardiovascular diseases, inflammation, hypoxia, and oxidized lipoproteins (25–27). Previous studies have demonstrated that PEDF serves a protective role in hypoxic H9c2 cells via its antioxidative effect, as well as inhibiting p53 mitochondrial translocation (5,6). To the best of the authors' knowledge, the effect of PEDF on autophagy in hypoxic H9c2 cells have not previously been investigated. In the present study, treatment with PEDF markedly increased the H9c2 cell viability compared with hypoxia alone, whereas the addition of 3-MA significantly weakened this effect. However, treatment with 3-MA could reverse the downregulation of PEDF on, the level of cleaved caspase-3. The results of the present study confirm that the autophagy facilitated by PEDF serves a protective role in H9c2 cells via inhibiting the hypoxia-induced apoptosis. The findings of the present study are generally consistent with an earlier report that autophagy may be a harmful form of procedural cell death under pathological conditions (28). Inconsistencies in the findings may be due to experimental factors, including the cell type employed, the application of the model in vivo or in vitro and treatment conditions. The results of the present study suggest that PEDF may enhance hypoxia-induced autophagy and has the potential to be a novel therapeutic approach for the treatment of H9c2 cell injury.
Acknowledgements
The present study was supported by the National Nature Science Foundation of China (grant nos. 81570242 and 81270173) and the Technology Bureau of Xuzhou of China (grant no. KC14SH106). We thank Dr. Xiaofang Yang (Laboratory of Clinical and Experimental Pathology, Xuzhou Medical University, Jiangsu, China) for providing autophagy inhibitor 3-MA.
References
- 1.Brookheart RT, Michel CI, Schaffer JE. As a matter of fat. Cell Metab. 2009;10:9–12. doi: 10.1016/j.cmet.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huss JM, Levy FH, Kelly DP. Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid X receptor gene regulatory pathway in cardiac myocytes: A mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J Biol Chem. 2001;276:27605–27612. doi: 10.1074/jbc.M100277200. [DOI] [PubMed] [Google Scholar]
- 3.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawaguchi T, Yamagishi SI, Sata M. Structure-function relationships of PEDF. Curr Mol Med. 2010;10:302–311. doi: 10.2174/156652410791065255. [DOI] [PubMed] [Google Scholar]
- 5.Gao X, Zhang H, Zhuang W, Yuan G, Sun T, Jiang X, Zhou Z, Yuan H, Zhang Z, Dong H. PEDF and PEDF-derived peptide 44mer protect cardiomyocytes against hypoxia-induced apoptosis and necroptosis via anti-oxidative effect. Sci Rep. 2014;4:5637. doi: 10.1038/srep05637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Zhang Y, Lu P, Zhang H, Li Y, Dong H, Zhang Z. PEDF attenuates hypoxia-induced apoptosis and necrosis in H9c2 cells by inhibiting p53 mitochondrial translocation via PEDF-R. Biochem Biophys Res Commun. 2015;465:394–401. doi: 10.1016/j.bbrc.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Klionsky DJ. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 8.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammond EM, Brunet CL, Johnson GD, Parkhill J, Milner AE, Brady G, Gregory CD, Grand RJ. Homology between a human apoptosis specific protein and the product of APG5, a gene involved in autophagy in yeast. FEBS Lett. 1998;425:391–395. doi: 10.1016/S0014-5793(98)00266-X. [DOI] [PubMed] [Google Scholar]
- 10.Notari L, Baladron V, Aroca-Aguilar JD, Balko N, Heredia R, Meyer C, Notario PM, Saravanamuthu S, Nueda ML, Sanchez-Sanchez F, et al. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J Biol Chem. 2006;281:38022–38037. doi: 10.1074/jbc.M600353200. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Sun T, Jiang X, Yu H, Wang M, Wei T, Cui H, Zhuang W, Liu Z, Zhang Z, Dong H. PEDF and PEDF-derived peptide 44mer stimulate cardiac triglyceride degradation via ATGL. J Transl Med. 2015;13:68. doi: 10.1186/s12967-015-0432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramanian P, Locatelli-Hoops S, Kenealey J, DesJardin J, Notari L, Becerra SP. Pigment epithelium-derived factor (PEDF) prevents retinal cell death via PEDF Receptor (PEDF-R): Identification of a functional ligand binding site. J Biol Chem. 2013;288:23928–23942. doi: 10.1074/jbc.M113.487884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 14.Haemmerle G, Moustafa T, Woelkart G, Büttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Wang Z, Feng SJ, Xu L, Shi HX, Chen LL, Yuan GD, Yan W, Zhuang W, Zhang YQ, et al. PEDF improves cardiac function in rats with acute myocardial infarction via inhibiting vascular permeability and cardiomyocyte apoptosis. Int J Mol Sci. 2015;16:5618–5634. doi: 10.3390/ijms16035618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 20.Viventi J, Blanco JA. Development of high resolution, multiplexed electrode arrays: Opportunities and challenges; Conf Proc IEEE Eng Med Biol Soc; 2012; pp. 1394–1396. [DOI] [PubMed] [Google Scholar]
- 21.van Herpen NA, Schrauwen-Hinderling VB. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol Behav. 2008;94:231–241. doi: 10.1016/j.physbeh.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 22.Chavez JA, Summers SA. Lipid oversupply, selective insulin resistance and lipotoxicity: Molecular mechanisms. Biochim Biophys Acta. 2010;1801:252–265. doi: 10.1016/j.bbalip.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan SH, Shui G, Zhou J, Li JJ, Bay BH, Wenk MR, Shen HM. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin) J Biol Chem. 2012;287:14364–14376. doi: 10.1074/jbc.M111.294157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.CIR.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 26.Xuan H, Xue W, Pan J, Sha J, Dong B, Huang Y. Downregulation of miR-221, −30d and −15a contributes to pathogenesis of prostate cancer by targeting Bmi-1. Biochemistry (Mosc) 2015;80:276–283. doi: 10.1134/S0006297915030037. [DOI] [PubMed] [Google Scholar]
- 27.Ma X, Liu H, Murphy JT, Foyil SR, Godar RJ, Abuirqeba H, Weinheimer CJ, Barger PM, Diwan A. Regulation of the transcription factor EB-PGC1α axis by beclin-1 controls mitochondrial quality and cardiomyocyte death under stress. Mol Cell Biol. 2015;35:956–976. doi: 10.1128/MCB.01091-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park M, Sabetski A, Kwan Chan Y, Turdi S, Sweeney G. Palmitate induces ER stress and autophagy in H9c2 cells: Implications for apoptosis and adiponectin resistance. J Cell Physiol. 2015;230:630–639. doi: 10.1002/jcp.24781. [DOI] [PubMed] [Google Scholar]