

Abstract
Dexmedetomidine (DEX), an α2 adrenoceptor agonist, has sedative and analgesic properties and myocardial protective effects. However, the mechanism underlying the protective effects of DEX on the myocardium remain unclear. The present study aimed to determine whether DEX serves an important role on cardioprotection through the endoplasmic reticulum (ER)- and mitochondria-mediated apoptosis signaling pathways. Neonatal rat cardiomyocytes (NRCMs) were cultured and divided four groups: i) Normal culture medium with 10% fetal bovine serum (control group); ii) H2O2 at 500 µM (H2O2 group); iii) DEX at 5 µM (DEX group); and iv) H2O2 plus DEX (H2O2 + DEX group). The levels of apoptosis and oxidative stress of NRCMs were investigated by ELISA, western blotting, flow cytometry and cell immunofluorescence. DEX significantly suppressed H2O2-induced apoptosis, and increased activity of caspases 3, 8 and 9 of NRCMs. DEX inhibited mitochondria-mediated oxidative stress and apoptosis, as evidenced by decreased levels of reactive oxygen species and lactic dehydrogenase, alleviated mitochondrial membrane potential depolarization, and increased Bcl-2-associated X protein/B-cell lymphoma 2 ratio. In addition, DEX decreased the activity of caspase 12, and the expression levels of glucose-regulated protein 78 kDa and serine/threonine-protein kinase/endoribonuclease IRE1, three major signaling molecules involved in the ER stress-mediated apoptosis pathway. Preventive treatment with DEX alleviates cardiomyocyte against H2O2-induced oxidative stress injury through attenuating the mitochondria- and ER-mediated apoptosis pathways.
Keywords: dexmedetomidine, oxidative stress, reactive oxygen species, apoptosis, cardiomyocyte
Introduction
Dexmedetomidine (DEX) has been widely used in intensive care units (ICU) and clinical anesthesia due to its anxiolytic and sedative effects without respiratory depression and with minor adverse effects (1). Studies have suggested that perioperative use of DEX reduced the incidence of complications and mortality after cardiac surgery (2). Oxidative stress plays an important role in the development of many diseases, and previous studies suggest that pretreatment with DEX protects against oxidative stress injury and/or cell apoptosis of many organs including lung, kidney, intestine and hippocampus. (3–5). Yoshitomi et al (6) reported that DEX exerted a direct protective effect against ischemia/reperfusion (I/R) injury on the myocardium. However, it is still unclear whether oxidative stress signaling pathway was involved in the cardioprotective effect of DEX.
Reactive oxygen species (ROS) are generated in the ischemic myocardium, especially after reperfusion, and the major source of ROS in the I/R myocardium are mitochondria (7,8). Organ I/R injury has been associated with significant increase in ROS and depolarization of mitochondrial transmembrane potential (ΔΨm), these factors play a key role in mitochondrial oxidative stress-induced cell injury (9,10). Fu et al (3) reported that DEX attenuated lipopolysaccharide (LPS)-induced acute lung injury through inhibiting oxidative stress and ROS. These studies suggest that oxidative stress may be involved in the cardioprotective effect of DEX. In addition, endoplasmic reticulum (ER) stress is also one of the important pathways involved in oxidative stress, ischemia, and other physiological and pathological conditions (11,12). Unfolded protein response (UPR), the key step in ER stress-mediated apoptosis, restored ER environment in the early stage of ER stress or leads to changes in cell function if UPR continues (13).
Based on literature review, this study aimed to raise and investigate the hypothesis that DEX exerts a cardioprotective effect through attenuating mitochondria and ER stress-mediated injury and apoptosis in NRCMs.
Materials and methods
Reagents
Dexmedetomidine hydrochloride (DEX) (C13H16N2·HCl, MW 236.7 g/mol) was purchased from Singch Pharm (Jiangsu, China). DEX was added to DMEM to obtain the desired concentration. H2O2 solution and collagenase type II were obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Cell culture and treatment
All protocols in this study were approved by the Ethics Committee of Southwest Medical University. Neonatal rats (2–3 days) was used for the isolation and culture of neonatal rat cardiomyocytes (NRCMs) as we reported previously (14). Cultured NRCMs in DMEM with 10% FBS were further divided into four groups by different treatments: i) Control group: normal DMEM adding with 10% FBS, ii) H2O2 group [with H2O2 for 6 h at 500 µM (15)], iii) DEX group [with DEX at 5 µM (16)], and iv) H2O2 + DEX group (with 5 µM DEX for 2 h before exposing to 500 µM H2O2 for 6 h). Cells with different treatments then underwent serial analyses including enzyme-linked immunosorbent assay (ELISA), western blotting, flow cytometry (FCM) and fluorescent microscopy.
ELISA
ELISA was used in this study to detect the levels of lactic dehydrogenase (LDH), glutathione (GSH) and the activities of caspase 3, 8, 9 and 12 with ELISA kits according to the instructions and our previous research (14).
FCM
The level of apoptosis, intracellular ROS level and mitochondrial membrane potential (ΔΨm) in NRCMs were detected with FCM (BD Biosciences, Franklin Lakes, NJ, USA) according to the instructions and our previous research (14). The level of apoptosis in NRCMs was analyzed with FITC-Annexin V Apoptosis Detection kit I (BD Biosciences). Briefly, cells were harvested with cold PBS and resuspended at a density of 1×106 cells/ml. Five µl of FITC Annexin V and 10 µl propidium iodide (PI) were added in 100 µl cell suspension and incubated at room temperature (25°C) for 15 min. Four hundred µl of 1X binding buffer was added to each tube and apoptosis was tested with FCM within 1 h. To measure the intracellular ROS level, NRCMs were washed with PBS and incubated with the probe for ROS, membrane permeable dichloro-dihydro-fluorescein diacetate (DCFH-DA) (final concentration 10 µM) in PBS at 37°C for 20 min. At the end of incubation, cells were washed with PBS and the level of ROS was analyzed by flow cytometry. ΔΨm was also measured with FCM using the JC-1 MitoScreen kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The ratio of monomeric JC-1 represented ΔΨm. The increased ratio represented the depolarization of ΔΨm, while a decreased ratio indicated the hyperpolarization of ΔΨm.
Immunofluorescence microscopy
Immunofluorescence microscopy of NRCMs was conducted as previously described (14). In brief, NRCMs were fixed in cold 4% paraformaldehyde, then blocked with 5% BSA and treated with 0.1% Triton-X 100. NRCMs were further incubated with the monoclonal primary antibody against α-actin produced in mouse (1:100; Wuhan Boster Biological Technology, Ltd., Wuhan, China) overnight at 4°C and then incubated with DyLight 488-conjugated anti-mouse IgG secondary antibody (1:200; Abcam, Cambridge, UK) for 1 h at room temperature. In addition, for the staining of F-actin, NRCMs were treated with rhodamine phalloidin (with final concentration, 100 nM) for 30 min at room temperature. Images were acquired with a fluorescence microscope (Olympus IX-81; Olympus Corp., Tokyo, Japan).
Tunnel asay
Tunnel assay was also used to investigate the effect of DEX on H2O2 induced cell apoptosis. The protocol was manipulated according to the instructions of tunnel assay kit (Beyotime Institute of Biotechnology, Haimen, China). In brief, NRCMs with different treatment was fixed with cold 4% paraformaldehyde for 30 min and 0.3% Triton X-100 was treated for 5 min. The Tunnel detecting solution [including Terminal Deoxynucleotidyl Transferase (TdT) and fluorescence solution] was added and incubated for 60 min at 37°C. Images were acquired with a fluorescence microscope (Olympus IX-81).
Western blot analysis
Western blotting was performed to determine the protein expression level under various conditions. Lysates with 50 µg protein were separated by SDS-PAGE. The proteins were transferred to PVDF membrane and blocked with 5% BSA in TBS-T solution. The PVDF membrane was incubated with primary antibodies against Grp78 (1:1,000; Abcam), IRE1α (1:1,000), Bcl-2 (1:1,000), Bax (1:1,000; all Cell Signaling Technology, Inc., Danvers, MA, USA) and GAPDH (1:10,000) from Sigma-Aldrich; Merck KGaA as house-keeping protein overnight at 4°C, respectively. The membrane was then incubated with HRP-conjugated IgG as a secondary antibody (1:2,000; BBI, China) for 1 h at room temperature and the immunoreactive bands were developed using an enhanced chemiluminescent substrate (Engreen Biosystem, Ltd., Beijing, China). Images of blotting bands and statistical analysis of the gels were used Bio-Rad QuantityOne® software (Bio-Rad Laboratories, Richmond, CA, USA).
Statistical analysis
The results obtained were presented as the mean ± standard error of mean (SEM). Analysis of variance (ANOVA) was used to statistical analysis for differences. Least Significant Difference (LSD) was used for further multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
DEX protects H2O2-induced structural impairment of NRCMs
We detected the effects of DEX on H2O2-induced injury of NRCMs. As shown in Fig. 1, green colors indicated the T-tube tagged with α-actin and red colors represented the cytoskeleton tagged with F-actin in NRCMs. NRCMs without H2O2 treatment exhibited clear and neatly arranged myocardial stripes (green) and cytoskeleton (red) (the first row in Fig. 1). Treatment with DEX alone did not significantly affect the structure of NRCMs (the second row in Fig. 1). Treatment of H2O2 significantly changed the shape of NRCMs from typical fusiform cell to irregularly shaped cell and H2O2 impaired the structure of NRCMs with disordered a-actin and F-actin as shown in Fig. 1 (the third row). Pre-treatment with DEX (5 µM) and H2O2 significantly attenuated the impairment of cell structure induced by H2O2 (the fourth row in Fig. 1). The right column in Fig. 1 showed that the effect of DEX on the structure of NRCMs with more details.
Figure 1.
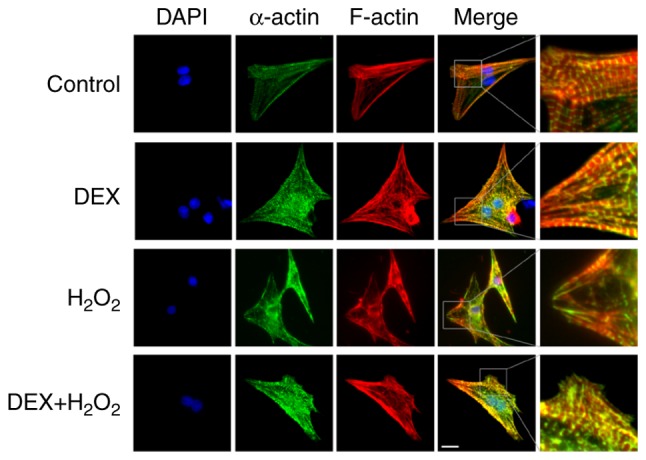
Images of NRCMs with immunofluorescence microscopy showing the attenuating effects of DEX on H2O2-induced structural impairment of NRCMs. α-actin (green) indicated T-tubule marker and F-actin (red) indicated cytoskeleton marker. Nuclei were tagged with DAPI (blue). The right column showing the effect of DEX on the structure of NRCMs with more details. Scale bar, 20 µm. DEX, dexmedetomidine; NRCMs, neonatal rat cardiomyocytes.
DEX alleviates H2O2-induced apoptosis of NRCMs
We investigated the effect of DEX on apoptosis of NRCMs induced by H2O2 with FCM, and results were shown in Fig. 2. We first tested the dose-dependent effect of H2O2 (0–1250 µM) on apoptosis of NRCMs and results showed that H2O2 concentration-dependently increased the cell apoptosis ratio with an EC50 of 574.95 µM (Fig. 2A). Therefore, H2O2 at 500 µM was used in this study in the later study. We further found that DEX (0.01–100 µM) reduced H2O2 induced apoptosis of NRCMs with an IC50 of 2.08 µM, and thus 5 µM DEX was used in later study (Fig. 2B). The concentration of DEX was similar with that used in a previous report (16).
Figure 2.

The effect of DEX on H2O2-induced apoptosis of NRCMs. (A) The dose-dependence of H2O2 on apoptosis of NRCMs. (B) The dose-dependence of DEX on apoptosis of NRCMs induced by 500 µM H2O2. (C) Typical original FCM plots showing the effect of DEX on apoptosis of NRCMs induced by H2O2. Cells with both FITC Annexin V and PI negative were viable and not underwent apoptosis. Cells with FITC Annexin V positive and PI negative were in early stage apoptosis, while cells with both FITC Annexin V and PI positive were in the late stage of apoptosis. (D) Statistical bar graph showing the effect of DEX on the apoptosis ratio (n=4). (E) Statistical bar graph showing the activities of caspases 3, 8, and 9 in NRCMs underwent different treatment. (F) Tunnel assay showing the effect of DEX on the cell apoptosis. Scale bar, 50 µm. **P<0.01 vs. control. #P<0.05 vs. H2O2. DEX, dexmedetomidine; NRCMs, neonatal rat cardiomyocytes; FCM, flow cytometry; PI, propidium iodide.
H2O2 increased the ratio of apoptosis-positive cells located in the first (Q2 area in Fig. 2C) and fourth quadrants (Q3 area in Fig. 2C) of the FCM plots from the control group 19.36±6.38 to 57.49±10.52% (Fig. 2C and D), while DEX replenishment decreased the ratio to 39.86±9.35%. Treatment with DEX alone did not affect the apoptosis of NRCMs. Consistent with the FCM results, pretreatment with DEX also significantly attenuated the H2O2-induced activity increase of caspases 3, 8, and 9, the important apoptosis-inducing molecules. Treatment of DEX alone did not change the baseline activities of caspases 3, 8, and 9 (Fig. 2E). Tunnel assay shown the similar results of DEX on cell apoptosis as that with FCM (Fig. 2F). These results indicate that DEX attenuates H2O2-induced apoptosis of NRCMs.
DEX suppresses H2O2-induced mitochondrial oxidative stress injury in NRCMs
To evaluate the effect of DEX on oxidative stress induced by H2O2, we further determined the levels of ROS, GSH and LDH, the important indices of oxidative stress. Fig. 3A showed that ROS level was evaluated with FCM in NRCMs with different treatments. As shown in Fig. 3B and C, H2O2 treatment increased the levels of ROS and LDH and reduced GSH release. Pretreatment with DEX alleviated H2O2-induced increment of ROS and LDH and refreshed H2O2-induced reductions of GSH significantly.
Figure 3.

Effects of DEX on H2O2-induced mitochondrial oxidative stress injury and apoptosis of NRCMs. (A) The original flow cytometry histogram showing the evaluation the ROS level in NRCMs with different treatments. (B) Effects of DEX on ROS in H2O2-challenged NRCMs. n=5. (C) Effects of DEX on LDH and GSH in H2O2-challenged NRCMs. n=5. (D) The original flow cytometry plot showing the evaluation the ΔΨm level in NRCMs. The percentage of JC-1 monomer in P3 area indicating the level of ΔΨm. (E) Flow cytometry showing that DEX inhibited H2O2-induced ΔΨm depolarization monitored by JC-1 dye and indicated by JC-1 monomer (n=5). CCCP, a mitochondrial uncoupler, was used as a positive control drug to induce depolarization of ΔΨm. (F) Western blotting showing the protein levels of Bax and Bcl-2 (upper) and the ratio of Bax/Bcl2 (lower) (n=5). *P<0.05, **P<0.01 vs. control. #P<0.05 vs. H2O2. DEX, dexmedetomidine; NRCMs, neonatal rat cardiomyocytes ROS, reactive oxygen species; LDH, lactic dehydrogenase; GSH, glutathione; ΔΨm, mitochondrial transmembrane potential; CCCP, carbonyl cyanide 3 chlorophenylhydrazone.
ΔΨm was also an important oxidative stress index. We investigated whether DEX affects the ΔΨm monitored with JC-1 dye. The percentage of monomeric JC-1 represented ΔΨm and typical recording with FCM was shown in Fig. 3D. As shown in Fig. 3E, H2O2 significantly depolarized the ΔΨm, while pretreatment with DEX suppressed the ΔΨm depolarizing effect of H2O2.
We further checked the Bax/Bcl2 ratio which reflects the status of mitochondrial-mediated apoptosis. As shown in Fig. 3F, compared with that in control group, H2O2 significantly increased the ratio of Bax/Bcl2, while pretreatment of DEX reduced the ratio compared with H2O2 challenge.
DEX attenuates H2O2-induced ER oxidative stress injury in NRCMs
In addition, to determine whether ER-mediated apoptosis pathway was also involved in the effect of DEX on NRCMs, the expression of Grp78 and IRE1α and activity of caspase 12 were measured in NRCMs with different treatment. As shown in Fig. 4A and B, treatment of H2O2 significantly increased the expression levels of Grp78 and IRE1α and the activity of caspase 12, while these effects of H2O2 were abolished by pretreatment of DEX. Treatment of DEX alone did not change the expression of Grp78 and IRE1α and the activity of caspase 12 compared with the control group significantly (Fig. 4A and B).
Figure 4.

Effects of DEX on the signaling molecules involved in ER-mediated oxidative stress pathway of NRCMs. (A) Western blotting showing the attenuating effects of DEX on the expressions of IRE1α and Grp78 in NRCMs (n=4). (B) ELISA showing the suppressing effects of DEX on elevated activities of caspase 12 induced by H2O2 in NRCMs (n=6). (C) Schematic representation of the signaling pathways underlying the protective effect of DEX on H2O2-induced injury and apoptosis of NRCMs. *P<0.05, **P<0.01 vs. control. #P<0.05 vs. H2O2. DEX, dexmedetomidine; NRCMs, neonatal rat cardiomyocytes; ER, endoplasmic reticulum; ELISA, enzyme-linked immunosorbent assay.
The above results showed that DEX alleviated H2O2-induced oxidative stress injury through both mitochondria and ER stress-mediated apoptosis pathways and a putative signaling network was shown in Fig. 4C. For the mitochondria mediated oxidative stress pathway, treatment of H2O2 first induced oxidative stress injury with increment of intracellular ROS level. Mitochondria oxidative stress injury and increased ROS induced a series of cascade reactions, including ΔΨm depolarization, increased Bax/Bcl-2 ratio, increment of LDH release and decrement of GSH. Pretreatment of DEX reversed the H2O2 induced mitochondrial oxidative stress injury. In addition, H2O2 also induced ER stress with increment of Grp78 and IRE1α, the important molecular indexes for reflexing ER stress with increment of UPR, while pretreatment of DEX also attenuated H2O2 induced ER stress. Both mitochondria and ER stress induced cell apoptosis through downstream reaction including activation of caspase 9 and caspase 12 then activation of caspase 3, which are the important markers for cell apoptosis. Pretreatment of DEX attenuated H2O2 induced cell apoptosis through decreasing the alleviating the mitochondria and ER mediated oxidative stress.
Discussion
It is well-documented that increased oxidative stress (OS) leads to dysfunction of relative signaling pathways and is involved in many cardiovascular diseases, such as myocardial I/R injury (8,17). Therefore, the oxidative stress signaling pathway may provide therapeutic targets against cardiac I/R injury. In the present study, we found that DEX, a drug widely used in clinical anesthesia, protects NRCMs against oxidative stress injury and apoptosis through modulating the mitochondria and ER stress-mediated apoptosis signaling pathways.
Mitochondria are the important cellular components for biological oxidation in eukaryotic cells (9). In the present study, DEX attenuated H2O2-induced increment of intracellular ROS level and LDH release, and restored the reduction of GSH induced by H2O2 in NRCMs and inhibited H2O2-induced depolarization of ΔΨm, which are the important molecular for reflexing the mitochondria oxidative stress status. These results support that mitochondria are the important target organelle of DEX for its cardioprotective action. Fu et al (3) reported that DEX attenuates LPS-induced acute lung injury of rats by inhibiting oxidative stress, mitochondrial dysfunction and apoptosis. These data suggested that oxidative stress signaling pathway is an important target for the organ protection of DEX. Therefore, the present study supports our hypothesis that mitochondria-mediated signaling pathway is involved in the protective effect of DEX on cardiomyocyte.
ER stress-mediated apoptosis is an alternative important signaling pathway in oxidative stress injury and Grp78, caspase 12, CHOP and IRE1α are the important molecules in this signaling pathway (18–20). Wang et al (21) reported that DEX produced cardioprotection against myocardial apoptosis injury through modulating expression of Grp78, CHOP and caspase 12 in grave scalding rat model. In this study, we also showed that DEX produced myocardial protection through inhibiting H2O2 induced increment of expression or activity of Grp78 and caspase 12. In addition, UPR signals are transmitted across the ER membrane to the nucleus via the proximal sensor IRE1α-a transmembrane serine/threonine kinase in the ER membrane (22). This study further demonstrated that pretreatment of DEX reduced the increased expression of IRE1α, suggesting that ER was another target of DEX on cardiac protection.
In summary, the present study supports our hypothesis that preventive treatment with DEX protects NRCMs against H2O2-induced injury and apoptosis through both mitochondria and ER stress-mediated signaling pathways. Oxidative stress (including mitochondria and ER stress) is the important target for the cardioprotective effect of DEX. The present study may encourage the widespread use of DEX in clinical anesthesia and ICU especially in high risk cardiovascular patients undergoing surgery operation such as cardiopulmonary bypass (CPB) or patients with cardiovascular diseases undergoing noncardiac surgery.
Acknowledgements
The authors would like to thank Professor Ji-Min Cao from the Chinese Academy of Medical Sciences (Shanghai, China) for critical reading of the manuscript.
Funding
The research was supported by the National Natural Science Foundation of China (grant no. 81670310) and Southwest Medical University Scientific Research Fund (grant no. 2014QN-003).
Availability of data and materials
The datasets and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
XT, XL, TL and LC designed the experiments; XL, TL, LC, YY, LLC, XF and BY performed in the experiments; XT, XL, TL and LC analyzed the data; XT, XL and TL wrote the paper.
Ethics approval and consent to participate
All protocols in the present study were approved by the Ethics Committee of Southwest Medical University (no. 20160063).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Arcangeli A, D'Alò C, Gaspari R. Dexmedetomidine use in general anaesthesia. Curr Drug Targets. 2009;10:687–695. doi: 10.2174/138945009788982423. [DOI] [PubMed] [Google Scholar]
- 2.Ji F, Li Z, Nguyen H, Young N, Shi P, Fleming N, Liu H. Perioperative dexmedetomidine improves outcomes of cardiac surgery. Circulation. 2013;127:1576–1584. doi: 10.1161/CIRCULATIONAHA.112.000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fu C, Dai X, Yang Y, Lin M, Cai Y, Cai S. Dexmedetomidine attenuates lipopolysaccharide-induced acute lung injury by inhibiting oxidative stress, mitochondrial dysfunction and apoptosis in rats. Mol Med Rep. 2017;15:131–138. doi: 10.3892/mmr.2016.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang H, Liu HZ, Wang HB, Zhong JY, Yang CX, Zhang B. Dexmedetomidine protects against cisplatin-induced acute kidney injury in mice through regulating apoptosis and inflammation. Inflamm Res. 2017;66:399–411. doi: 10.1007/s00011-017-1023-9. [DOI] [PubMed] [Google Scholar]
- 5.Xie C, Li Y, Liang J, Xiao J, Zhao Z, Li T. The effect of dexmedetomidine on autophagy and apoptosis in intestinal ischemia reperfusion-induced lung injury. Zhonghua Jie He He Hu Xi Za Zhi. 2015;38:761–764. (In Chinese) [PubMed] [Google Scholar]
- 6.Yoshitomi O, Cho S, Hara T, Shibata I, Maekawa T, Ureshino H, Sumikawa K. Direct protective effects of dexmedetomidine against myocardial ischemia-reperfusion injury in anesthetized pigs. Shock. 2012;38:92–97. doi: 10.1097/SHK.0b013e318254d3fb. [DOI] [PubMed] [Google Scholar]
- 7.Rajendran P, Nandakumar N, Rengarajan T, Palaniswami R, Gnanadhas EN, Lakshminarasaiah U, Gopas J, Nishigaki I. Antioxidants and human diseases. Clin Chim Acta. 2014;436:332–347. doi: 10.1016/j.cca.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 9.Addabbo F, Montagnani M, Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension. 2009;53:885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 12.Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol. 2012;75:49–67. doi: 10.1146/annurev-physiol-030212-183707. [DOI] [PubMed] [Google Scholar]
- 13.Li N, Zoubeidi A, Beraldi E, Gleave ME. GRP78 regulates clusterin stability, retrotranslocation and mitochondrial localization under ER stress in prostate cancer. Oncogene. 2013;32:1933–1942. doi: 10.1038/onc.2012.212. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Cao L, Li T, Chen LL, Yu YY, Huang WJ, Liu L, Tan XQ. Propofol attenuates H2O2-induced oxidative stress and apoptosis via the mitochondria- and ER-medicated pathways in neonatal rat cardiomyocytes. Apoptosis. 2017;22:639–646. doi: 10.1007/s10495-017-1349-3. [DOI] [PubMed] [Google Scholar]
- 15.Ichinose M, Yonemochi H, Sato T, Saikawa T. Diazoxide triggers cardioprotection against apoptosis induced by oxidative stress. Am J Physiol Heart Circ Physiol. 2003;284:H2235–H2241. doi: 10.1152/ajpheart.01073.2002. [DOI] [PubMed] [Google Scholar]
- 16.Yan M, Dai H, Ding T, Dai A, Zhang F, Yu L, Chen G, Chen Z. Effects of dexmedetomidine on the release of glial cell line-derived neurotrophic factor from rat astrocyte cells. Neurochem Int. 2011;58:549–557. doi: 10.1016/j.neuint.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 17.Von Harsdorf R, Li PF, Dietz R. Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation. 1999;99:2934–2941. doi: 10.1161/01.CIR.99.22.2934. [DOI] [PubMed] [Google Scholar]
- 18.Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol. 2015;308:C415–C425. doi: 10.1152/ajpcell.00334.2014. [DOI] [PubMed] [Google Scholar]
- 19.Zhang LH, Zhang X. Roles of GRP78 in physiology and cancer. J Cell Biochem. 2010;110:1299–1305. doi: 10.1002/jcb.22679. [DOI] [PubMed] [Google Scholar]
- 20.Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann N Y Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Zhang S, Xu S, Zhang L. The efficacy and mechanism of dexmedetomidine in myocardial apoptosis via the renin-angiotensin-aldosterone system. J Renin Angiotensin Aldosterone Syst. 2015;16:1274–1280. doi: 10.1177/1470320314546941. [DOI] [PubMed] [Google Scholar]
- 22.Hetz C, Martinon F, Rodriguez D, Glimcher LH. The unfolded protein response: Integrating stress signals through the stress sensor IRE1α. Physiol Rev. 2011;91:1219–1243. doi: 10.1152/physrev.00001.2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.