

Abstract
Human mesenchymal stem cells (hMSCs) dynamically remodel their microenvironment during basic processes, such as migration and differentiation. Migration requires extracellular matrix invasion, necessitating dynamic cell-material interactions. Understanding these interactions is critical to advancing materials designs that harness and manipulate these processes for applications including wound healing and tissue regeneration. In this work, we encapsulate hMSCs in a cell-degradable poly(ethylene glycol)-peptide hydrogel to determine how cell-secreted enzymes, specifically matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), create unique pericellular microenvironments. Using multiple particle tracking microrheology (MPT), we characterize spatio-temporal rheological properties in the pericellular region during cell-mediated remodeling. In MPT, the thermal motion of probes embedded in the network is measured. A newly designed sample chamber that limits probe drift during degradation and minimizes high value antibody volumes required for cell treatments enables MPT characterization. Previous MPT measurements around hMSCs show that directly around the cell the scaffold remains intact with the cross-link density decreasing as distance from the cell increases. This degradation profile suggests t hat hMSCs are simultaneously secreting TIMPs, which are inactivating MMPs through MMP–TIMP complexes. By neutralizing TIMPs using antibodies, we characterize the changes in matrix degradation. TIMP inhibited hMSCs create a reaction-diffusion type degradation profile where MMPs are actively degrading the matrix immediately after secretion. In this profile, the cross-link density increases with increasing distance from the c ell. This change in material properties also increases the speed of migration. This simple treatment could increase delivery of hMSCs to injuries to aid wound healing and tissue regeneration.
Table of content image and novelty of work
We characterize dynamic pericellular re-engineering by human mesenchymal stem cell-secreted enzymes in well-defined hydrogels using multiple particle tracking microrheology.
1 Introduction
Cellularly responsive hydrogels are commonly used as scaffolds for 3D encapsulation to study and direct basic cellular functions and outside-in signaling while providing cues designed into the microenvironment1–13. Controlling basic cellular processes, such as migration and differentiation, will enable materials to instruct cells to migrate to wounds and begin the wound healing process or change lineage specification and begin building new tissue during tissue regeneration14–19. These synthetic hydrogels recapitulate aspects of the native extracellular matrix (ECM) and are also designed to allow cells to adhere to and degrade the scaffold during basic processes20. The native ECM is composed of various fibrous proteins and proteoglycans which act as a barrier for cell migration forcing complex cell-material interactions. Similarly, chemically cross-linked synthetic scaffolds necessitate cell-mediated degradation of the network, especially during motility1,18,21. To overcome these physical barriers, cells secrete protease to degrade the ECM and create channels during motility1–3,14,18,22. Due to this, the initially well-defined synthetic scaffold microenvironment continually presents new physical and chemical cues in the pericellular region6,10,20,23.
In this work, we determine how inhibition of selected cell-secreted enzymes change scaffold degradation in the pericellular region and, in turn, change motility. Our work focuses on characterization of the pericellular region around encapsulated human mesenchymal stem cells (hMSCs) and the role of tissue inhibitors of metalloproteinases (TIMPs) in matrix degradation using a novel sample chamber and microrheological characterization. hMSCs are chosen because they are key players in wound healing, migrating to wounds and regulating inflammation and tissue regeneration14,18,19,24,25. TIMPs are chosen because they are cell-secreted molecules that inhibit the activity of matrix metalloproteinases (MMPs), which degrade the hydrogel scaffold. Understanding the changes in the material microenvironment and the chemical strategies that hMSCs use to degrade the pericellular region will inform the design of new materials that mimic these microenvironments to enhance motility. This will potentially increase delivery of cells when the cell-laden hydrogels are used as implantable materials to enhance wound healing and regeneration of tissue.
hMSCs controllably degrade their microenvironment through secretion of a variety of proteases that degrade the native ECM. MMPs are one of the main family of calcium and zinc dependent endopeptidases that have the ability to degrade ECM components. More than 25 MMPs have been identified and hMSCs secrete mainly MMP–1, –2, –9 and –1322,26,27. In the extracellular space, MMP activity (activation or inhibition) is regulated by TIMPs18,22,27–29. Four TIMPs have been identified, TIMP –1, –2, –3 and –4, which are all composed of two main domains: N–terminal and C–terminal domains. These domains bind to the catalytic parts of MMPs to create MMP–TIMP complexes21,27,28,30,31. The cells used in this work are hMSCs derived from bone marrow, which only secrete TIMP–1 and –218,27. Olson et al. analyzed MMP–TIMP binding kinetics and found that TIMPs bind to MMPs quickly and unbind relatively slowly31. This creates a tight MMP–TIMP binding mechanism which results in effective MMP inhibition immediately after secretion31. This binding also severely limits scaffold degradation. In this work, we focus on the change in the cell-mediated degradation profile in the pericellular region around an encapsulated hMSC and hMSC motility as a function of inhibition of cell-secreted TIMPs.
To reduce the complexity inherent in cell-material interactions with the native ECM, we use a well-defined synthetic hydrogel scaffold for cell encapsulation. This synthetic material enables control over the initial physical and chemical cues presented to the encapsulated hMSC3,6,9,10,32. The scaffold used in this work is a poly(ethylene glycol) (PEG)-peptide hydrogel. PEG is chosen as the backbone because it has no biological activity and is resistant to protein adsorption. Additionally, PEG can be easily functionalized to change the cross-linking reaction and to tether chemical cues to the network4,5,8,33–35. The cross-linker for this scaffold is an MMP degradable peptide sequence, KCGPQG↓IWGQCK, which allows cells to remodel the pericellular region prior to and during migration2,3,8–10,36,37. Additionally, cells also adhere to the network during the migration process. An adhesion ligand (CRGDS) is tethered to the network to enable hMSC interactions with the surroundings via integrin binding3,38.
Although this hydrogel presents carefully engineered physical and chemical cues to encapsulated hMSCs, it remains unclear how hMSCs remodel the pericellular region during motility. Spatial and temporal changes in the scaffold have recently started to be characterized necessitating the adaptation and development of techniques.
Several characterization methods have combined the use of highly engineered synthetic hydrogel scaffolds with spatial measurement techniques to quantify cellular remodeling and degradation during motility. Zaman et al. have shown that migration of cells in 3D depends on density of adhesion ligand, stiffness of the hydrogel and integrin binding39. This work illustrates the importance of the physical microenvironment on cell motility. Incorporation of degradable cross-linkers in a hydrogel with fluorescence resonance energy transfer (FRET)-based dye has been used to observe cell migration and track material degradation40,41. Another method for monitoring force exerted on the pericellular region is traction force microscopy (TFM). TFM measures force exerted by the cell on the material using embedded probe particles. This method cannot be used in our hydrogel since TFM assumes that material properties are not changing over time. This assumption is violated in our degradable scaffold42,43. In addition, our previous work has characterized the microenvironment around untreated hMSCs during motility using MPT10,23.
To characterize the changes in the pericellular region spatially and temporally during hMSC-mediated degradation we use multiple particle tracking microrheology (MPT). MPT is a passive microrheological technique that measures the Brownian motion of embedded probe particles. Probe particle Brownian motion is related to rheological properties using the Generalized Stokes-Einstein Relation44–50. MPT has several unique characteristics that make it ideal for characterizing the pericellular region during cell-mediated degradation. MPT is sensitive in the low moduli (10−3 −4 Pa) and frequency (0.01–10 Hz) regimes45,48,49,51,52. This measurement sensitivity enables characterization of changes in material properties, especially of the weak incipient gel during gel-sol phase transitions. MPT also has fast acquisition times, on the order of 30 s, enabling measurements of dynamically evolving microenvironments at a quasi steady-state. Finally, MPT uses video microscopy to capture data. This enables spatial characterization of the microenvironment around an encapsulated hMSC.
Schultz et. al used MPT to study remodeling and degradation around an encapsulated hMSC in this well-defined PEG-peptide hydrogel prior to and during migration10. This work characterized the spatial and temporal degradation profile hMSCs create in the pericellular region. Scaffold degradation, caused by cell-secreted MMPs and cytoskeletal tension, is measured to be greatest in the furthest region from the cell center (at the edge of the field of view) and the material directly around the cell remains in a gel state and is not degraded. This degradation profile is the opposite of expectations, since cells are secreting MMPs the profile of degradation was expected to follow a reaction-diffusion type profile. A reaction-diffusion type profile would have the greatest degradation around the cell with cross-link density increasing as distance from the cell center is increased.10,23. To further determine the mechanism hMSCs use to create this degradation profile, we also characterized the role of cytoskeletal tension and cell-secreted MMPs in matrix degradation in this hydrogel23. We inhibited cytoskeletal tension exerted by hMSCs (using a myosin II inhibitor) and measured the degradation profile in the pericellular region. We measured the same degradation profile as untreated cells. This suggests that matrix degradation is due to cell-secreted MMPs and cytoskeletal tension has a negligible effect on scaffold degradation23. From this previous work, we hypothesize that MMPs are being inactivated by TIMPs to inhibit degradation around the hMSC and create this unique microenvironment that enables spreading and attachment prior to motility. Therefore, we investigate the role of TIMPs in cellular remodeling of the pericellular region during the migration process.
In this work, we encapsulate hMSCs in PEG-peptide hydrogels to determine the role of TIMPs in the remodeling and degradation of the pericellular region during motility. We develop a novel sample chamber that enables TIMP neutralization and MPT measurements. We use MPT to measure spatial and temporal changes in the pericellular region during hMSC degradation. These highly sensitive spatio-temporal measurements enable the real-time characterization of the pericellular region during cellular remodeling. We repeat experiments measuring untreated hMSCs and present them for comparison. We then inhibit both TIMP–1 and –2 and characterize the dynamic evolution of the pericellular region. Our work measures a reaction-diffusion type profile around TIMP inhibited hMSCs. The increase in MMP activity in the absence of TIMPs creates a profile where the scaffold is the softest directly around the hMSC. We also measure an increase in hMSC motility. These results determine that by inhibiting TIMPs, hMSC speed is increased due to increased degradation directly around the encapsulated cell. This information gives new insight into the mechanism hMSCs use to re-engineer their microenvironment. Understanding the mechanism of degradation and the hMSC response to changes in the physical properties of the scaffold, provide vital information that will enable the design of new materials. These materials can be designed to mimic cellularly-engineered pericellular regions to harness and enhance motility for more effective cell delivery to wounded areas and accelerated regeneration of tissue.
2 Experimental
2.1 Hydrogel scaffold fabrication and cell encapsulation
The PEG-peptide hydrogel used in this work was cross-linked using a thiol:ene photopolymerization reaction. This hydrogel scaffold was developed by the Anseth group and has been widely used for cell encapsulation3,6,9,53. This hydrogel consists of 3 mM four-arm star PEG end-functionalized with norbornene (Mn= 20,000 g mol−1, f = 4, where f is the number of functional groups, purchased from Sigma-Aldrich), which is chemically cross-linked with 3.9 mM of an MMP degradable peptide sequence, KCGPQG↓IWGQCK (Mn= 1,305 g mol−1, f = 2, American Peptide, Inc.)3,10,51. This peptide sequence is highly degradable by cell-secreted MMPs, therefore, this scaffold provides an environment that a cell can easily remodel to enable extending, attachment and motility3,10,23,36,54. All hydrogels characterized in this work have a thiol:ene ratio of 0.65, resulting in a soft hydrogel scaffold. 1.7 mM lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), a highly water-soluble photoinitiator, was added to the solution to initiate a step-growth photopolymerization mechanism (LAP is synthesized in the Anseth group following published protocols53).
Briefly, dimethyl phenylphosphonite was reacted with 2,4,6-trimethylbenzoyl chloride under Argon at room temperature. After mixing for 18 hrs, lithium bromide was mixed in 2-butanone and added to the reaction mixture. This mixture was then heated to 50°C. After 10 mins a solid was formed. After cooling, the mixture was filtered with 2-butanone to remove excess lithium bromide.53.
To facilitate cellular attachment and motility 1 mM CRGDS (Mn= 594 g mol−1, f = 1, American Peptide, Inc), an adhesion ligand, was covalently tethered to the scaffold. 1 µm fluorescently labeled carboxylated probe particles (2a = 1.00±0.02 µm, where a is the probe particle radius, Polysciences, Inc) were added to the precursor solution to enable MPT measurements. Finally, hMSCs were suspended in a 1× phosphate buffered saline (1× PBS, Life Technologies) and added to the hydrogel precursor solution to be encapsulated at a final concentration of 2 × 105 cells mL−1.
The precursor solution was mixed and added to the sample chamber described below. The gelation reaction was initiated by exposure to UV light (365 nm light, 10 mW cm−2, UVP, LLC) for 3 mins. Since hMSCs were in the precursor solution, upon gelation they were suspended in three-dimensions within the hydrogel scaffold. After gelation, the scaffold was immediately incubated in 4 mL of growth media. The sample chamber wast hen placed in the incubator (37°C, 5% CO2, Eppendorf, Inc.) and incubated overnight before data were collected. All data were collected 18 – 48 hrs after hMSC encapsulation. This enables cells to adjust to their environment and begin to migrate through the scaffold after encapsulation.
2.2 Device fabrication
Glass-bottomed petri dishes (D = 35 mm, MatTek Corporation) were used to make sample chambers to measure cell-laden hydrogel scaffolds. The glass-bottom of the petri dish was functionalized with thiol groups using 3-mercaptopropyl triethoxysilane (Sigma-Aldrich). This attaches the hydrogel to the glass during polymerization. To functionalize the glass, 30 mL of reagent alcohol (Fisher Scientific) was poured into a beaker while being stirred constantly. Glacial acetic acid (Fisher Scientific) was used to adjust the pH between 4.5 – 5.5. 170 µL of 3-mercaptopropyl triethoxysilane was added to the solution. After mixing, the solution was added to the glass-bottom of the petri dishes. After reacting with the glass, 2 – 3 mins, petri dishes were rinsed with reagent alcohol and placed in the oven at 80°C for 30 mins.
After glass functionalization, a tube of polydimethylsiloxane (PDMS, Dow Corning) was attached to the glass-bottom of the petri dish10,51. These chambers were used to reduce vibrations in hydrogel scaffolds and probe particle drift (or directed motion) after cell-mediated scaffold degradation. To make the PDMS tubes, the shape was cut out of a flat PDMS sheet. PDMS sheets were made by mixing silicone elastomer base with curing agent at a ratio of 10:1 (ratio recommended by manufacturer, Dow Corning). The pre-cured PDMS mixture was degassed under vacuum and cured at 65°C overnight. PDMS tubes were then cut from the PDMS sheet using two different diameter biopsy punches (6 mm and 10 mm disposable biopsy punches, Acuderm Inc.) creating a tube with an inner diameter of 6 mm and an outer diameter of 10 mm. The PDMS tube was attached to the glass-bottom of the petri dish using uncured PDMS and incubating the petri dish overnight at 65°C.
The hydrogel precursor solution was added to the PDMS tube, using a volume that does not completely fill the diameter of the tube, 17 µL. This allows hydrogels to swell when growth media was added to the petri dish. After sterilizing the petri dishes with 70% ethanol, gel solutions were added into the PDMS chambers and photopolymerized as described above.
For TIMP inhibition experiments, a small inner sample chamber was constructed inside the petri dish using O-rings (D = 15.5 mm, Small Part Inc). TIMPs were inhibited by incubating the cell-laden hydrogel in TIMP–1 and –2 antibodies, described in detail below. The smaller sample chamber minimizes the volume (and amount of antibodies) needed to treat the hydrogels, therefore, greatly reducing the cost of each experiment. O-rings were used because they are non-reactive and not porous, therefore, they effectively trap the incubation liquid within the smaller sample chamber. To make this chamber, three O-rings were stacked and fastened together with UV curable glue (NOA-81, Norland Products, Inc.). The stacked O-rings were then attached to the bottom of the petri dish using the same UV adhesive. This creates a 1 mL inner chamber where the hydrogel can be incubated with antibodies in media, Figure 1. After TIMP inhibition the entire volume of the petri dish was used for incubation of the hydrogel in media (4 mL).
Fig. 1.

Schematic of the hydrogel sample chamber enabling incubation in a reduced volume of media (1 mL).
2.3 Cell culture
hMSCs were obtained in passage 2 from Lonza. All experiments were done with cells in passages 2 – 5. Cells were cultured in a 150 cm2 tissue culture petri dish with growth media. Growth media consists of 10% fetal bovine serum (FBS, Life Technologies), 50 U mL−1 Penicillin/streptomycin (Life Technologies), 0.5 µg mL−1 Fungizone (Life Technologies) and 1 ng mL−1 recombinant human fibroblast growth factor (hFGF, PeproTech) in a low-glucose Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies). All components were used as received. For TIMP neutralization experiments, cells are cultured in serum-free (SF) and Phenol Red-free (PF) conditions27,55. In the SF condition, FBS was replaced with 1% insulin-transferrin-selenium (Life Technologies). In the PF condition, DMEM was replaced with Medium 199 (Life Technologies).
For each condition (untreated and TIMP–1 and –2 inhibition), at least three biological replicates were characterized. Within each biological replicate, two gels were made per stock solution. Cell-mediated degradation and motility were measured in both hydrogels. For each experiment, data was collected around 4 – 5 different cells per hydrogel. For untreated hMSCs data were collected around 25 different cells and for TIMP inhibited hMSCs data were collected around 44 different cells. All measurements were taken 18 – 48 hrs after encapsulation. This allows the cells to adjust to their environment after encapsulation and begin to migrate. The experiment was ended at 48 hrs, which ensures that hMSCs are not differentiating. In all of the experiments, selection of the cells measured was arbitrary. After encapsulation hMSCs have rounded morphology. After secretion of enzymes, the cells will spread in the scaffold and extend and have an elongated morphology. Data was taken around cells with different morphologies in the hydrogel. MPT measures no scaffold degradation around rounded cells.
In addition to MPT measurements of cell-laden hydrogels, three control experiments were done. Each of the control experiments were repeated three times and in each repeat at least two hydrogels were measured. The control experiments are available in the Electronic Supplementary Material† (ESI) and are as follows: (1) hydrogels were made without cells and incubated in growth, SF and PF media, (2) hydrogels were made in the absence of hMSCs and incubated with TIMP–1 and –2 antibodies and (3) hMSCs were encapsulated in hydrogels and MMP activity was inhibited using 10 µM InSolution GM 6001 (MMP inhibitor, Millipore Sigma). In all experiments no degradation was observed after incubation. The results of MMP inhibition have been previously described23. The logarithmic slope of the mean-squared displacement versus time was calculated and plotted for each control experiment separately. These are Figures 1S–3S in the ESI†.
2.4 Cell encapsulation
hMSCs were cultured in a 150 cm2 tissue culture plastic petri dish with growth media. Cell media was changed after 3 – 4 days. Cells were passaged when they reached 90% confluence, ≈ 7 days. For encapsulation, hMSCs were suspended in 1× PBS and added to the hydrogel precursor solution at a final concentration of 2 × 105 cells mL−1. The precursor solution was mixed and added to the PDMS tube in the sample chamber. The solution was exposed to the UV light for 3 mins, encapsulating the hMSCs in the hydrogel scaffold. This results in hMSCs evenly distributed throughout the hydrogel scaffold in 3D.
2.5 TIMP–1 and –2 inhibition
To determine whether TIMP–1 and TIMP–2 were effectively inhibited we used Western blots. To inhibit TIMPs, hMSCs were cultured in 2D in SF and PF media in a 12 well tissue culture plate (3.8 cm2, Sigma-Aldrich) at a concentration of 3.7 × 105 cells mL−1. After total cell attachment, approximately 5 hrs, media was aspirated and cells were washed twice with 1× PBS. Cells were treated with TIMP–1 and –2 antibodies (Polyclonal Goat IgG, Research And Diagnostic Systems, Inc.) using the concentrations of 45 µg mL−1 and 30 µg mL−1, respectively, and incubated at room temperature for 1.5 hrs. All experiments presented here neutralize both TIMP–1 and –2 simultaneously. After incubation, antibodies were removed and cells were rinsed again with 1× PBS to wash away any residual antibodies. Cells were incubated in fresh SF and PF media at 37°C and 5% CO2. For Western blots, cell media was collected two days after inhibition and concentrated using a protein concentrator (Thermo Fisher Scientific).
To inhibit TIMP–1 and TIMP–2 after hMSCs were encapsulated in the hydrogel scaffold, 45 µg mL−1 of TIMP–1 antibody and 30 µg mL−1 of TIMP–2 antibody was added to the SF and PF media and is put in the inner chamber of the petri dish to completely cover the hydrogel (1 mL) at room temperature. After 1.5 hrs this media was removed and normal growth media (without hFGF) was used to cover the entire volume of the petri dish (4 mL). Hydrogels were then incubated at 37°C and 5% CO2 overnight before MPT data were collected.
2.6 Western blot
Gel electrophoresis was used to detect TIMPs in cell media collected two days after incubation with TIMP–1 and –2 antibodies. Protein samples were subjected to reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and after protein separation by molecular weight, transferred into a nitrocellulose membrane (Life Technologies). For this work two different amounts of protein were used, 7 µg of protein is run of untreated cell media to determine which TIMPs are secreted by hMSCs and 2 µg of protein is run for TIMP inhibition experiments. Membranes were blocked in 5% nonfat milk/Tris-buffered saline (TBS) with 0.25% Tween-20 (TBS-T, Tween-20 from Fisher Scientific) for 1 hr. 1 L of TBS was made by dissolving 88 g sodium chloride (ThermoFisher Scientific) and 24 g of tris base (ThermoFisher Scientific) in 900 mL of deionized water. The pH was adjusted to 7.6 by adding hydrochloric acid (1 M, Fisher Scientific). The final volume of the buffer was adjusted to 1 L using deionized water.
After incubation in 5% nonfat milk/TBS-T, the membrane was then incubated at 4°C with primary TIMP–1 and –2 antibodies (Polyclonal Goat IgG, 1 µg mL−1, Research and Diagnostic Systems, Inc.) overnight. After washing the membranes with TBS-T, membranes were incubated for 45 mins in conjugated horseradish peroxidase (HRP) secondary antibody (1:2000 in 5% nonfat milk/TBS-T, Research and Diagnostic Systems, Inc.). A chemilumiscent substrate (Invitrogen) was applied to the membrane for 5 mins. Finally, bands were detected with a ChemiDoc MP imaging system (Bio-Rad Laboratories).
2.7 Multiple particle tracking microrheology
Spatio-temporal dynamic changes in hydrogel rheological properties were characterized using multiple particle tracking microrheology. In MPT, the Brownian motion of fluorescently labeled probe particles embedded in the hydrogel scaffold is captured using video microscopy45,47,48,52,56,57. 1 µm carboxylated polystyrene probe particles were added to the precursor solution prior to photopolymerization in the sample chamber. Once the hydrogel was polymerized probe particles were distributed throughout the hydrogel scaffold. An inverted microscope (Zeiss Observer Z1, Carl Zeiss AG) with a 63× water immersion objective (N.A. 1.3, 1× optovar, Carl Zeiss AG) was used to collect MPT data. Videos of probe particle motion were captured using a high speed camera (1024×1024 pixels, Miro M120, Vision Research Inc.) for 800 frames at a frame rate of 30 frame s−1 with an exposure time of 1000 µs47,49,51,56. The frame rate and exposure time are chosen to minimize static and dynamic particle tracking errors57. This microscope is fitted with an incubation chamber to maintain the environment during data acquisition at 37°C and 5% CO2, which is necessary for cell survival.
After data acquisition, videos of probe particle movement were tracked using classical tracking algorithms47,58. Particle positions were determined using the brightness-weighted centroid. The position of each particle was identified in each frame of the video. Then all positions for a single particle were linked into a trajectory, using a probability distribution function that accounts for Brownian motion47,58. These trajectories show particle movement throughout the video. From the particle trajectories within the video, the ensemble-averaged mean-squared displacement (MSD), 〈Δr2(τ)〉, of the probe particles was calculated46,51,58–61. MSDs were calculated as a function of a lag time, τ, which is the separation time between frames in the movie. For our measurements, the two dimensional MSD was calculated using 〈Δr2 (τ)〉 = 〈Δx2 (τ)〉 + 〈Δy2 (τ)〉. The MSD is directly related to the rheological properties of the material using the Generalized Stokes-Einstein relation (GSER), , where kBT is the thermal energy, a is the radius of the probe particles and J (t) is the creep compliance46,49,59,60,62–64. The state of the material can also be quantitatively determined by the logarithmic slope of mean-squared displacement, 46,51,52,59,64,65. This will be discussed further in the Results and Discussion.
For each time point of MPT data acquisition, the position of the cell was determined using brightfield microscopy. A brightfield image was collected prior to MPT data acquisition. Immediately after locating a cell (t = 0), MPT data were collected approximately every 5 – 6 mins in the same field of view. The total time of data collection around each cell in the hydrogel scaffold is 20 – 60 mins. The total acquisition time is limited by cell movement out of the field of view and in the z direction and photobleaching of probe particles. Brightfield images were used to determine the cell center for each time point of experiment, (xi, yi) using ImageJ. Cell speed, vcell, was calculated using . where t is time, 0 indicates the initial and f is the final time and positions.
2.8 Bulk rheology
Bulk rheology was used to measure the elastic modulus (G′) of the hydrogel. All the hydrogels have a thiol:ene ratio of 0.65. The unswollen hydrogel is measured first. Unswollen hydrogels were loaded on the bulk rheometer (TA Instrument, Ares G2) at 37°C. The modulus was measured with a 8 mm sandblasted parallel plate at 1% strain using a frequency sweep between 0.1 and 10 Hz. G′ was determined in the linear viscoelastic regime and the reported value is the average and standard deviation of measurements of three separate hydrogels.
To measure the modulus of a swollen gel, the hydrogel was made in a tube of PDMS (PDMS tube dimensions are inner diameter of 8 mm and outer diameter of 10 mm) in a petri dish. After hydrogel formation, PDMS tubes were removed to allow the gel to swell in all directions when media was added to the petri dish. After complete swelling at 37°C, ~ 24 hrs, the hydrogel was cut with an 8 mm disposable biopsy punches (Acuderm Inc.) to completely fit under the 8 mm parallel plate. Hydrogels were loaded into an immersion cup (TA Instrument), which allows the sample to be measured while fully immersed in media. This was done to prevent water evaporation from the hydrogel during the experiment at 37°C. The same frequency sweeps described above were used to measure the modulus of swollen hydrogel samples.
3 Results and discussion
Cell-laden hydrogel scaffolds are measured to determine the extent of degradation in the pericellular region as a function of the distance away from the cell. The goal of this work is to determine if previously measured degradation profiles are due to tissue inhibitor of metalloproteinases inhibiting matrix metalloproteinase activity and, thereby, inhibiting degradation around the cell. To determine the role of TIMPs in cell-mediated degradation we used multiple particle tracking microrheology to characterize the spatio-temporal rheological changes that occur around an encapsulated hMSC. We will define ‘untreated hMSCs’ as cells that have been encapsulated in the hydrogel with no further treatment and ‘TIMP inhibited hMSCs’ as hMSCs that have been incubated with TIMP–1 and –2 antibodies. The rheological changes in the pericellular region have been previously reported for untreated hMSCs and will be discussed briefly below. The pericellular region is characterized around TIMP inhibited hMSCs to determine how the degradation profile around the cell changes when MMPs are not inactivated due to MMP–TIMP binding. We find that the degradation profile around TIMP inhibited hMSCs reverses the previously measured degradation profile and has a reaction-diffusion type profile. We also measure increased migration speeds and degradation due to increased activity of MMPs around TIMP inhibited hMSCs.
Prior to hMSC encapsulation, the hydrogel scaffold is characterized using bulk rheology. The hydrogel scaffold used for all cell-mediated degradation experiments has a thiol:ene ratio of 0.65. The unswollen modulus of our hydrogel scaffold is G′ = 750.3 ± 28.2 Pa and the swollen modulus is , which is similar to the elasticity of soft tissue in the body, such as lung and breast tissue66,67. This low modulus enables cells to degrade the pericellular region rapidly and begin motility. Previous work by Kyburz et al. demonstrated that the average speed of hMSCs increased from 7.9±0.5 µm h−1 to 17.6±0.9 µm h−1 by decreasing the thiol:ene ratio from to . Also, the percentage of hMSCs migrating increased from 20% to 59% by decreasing the cross-link density3. After encapsulation cell-laden hydrogels are incubated in growth media overnight before MPT data acquisition, all data is collected 18–48 hrs after encapsulation. This is sufficient time for the hydrogel to completely swell and for encapsulated hMSCs to adjust to their microenvironment and initiate matrix degradation and motility. To further investigate microenvironmental changes due to cell-mediated degradation we characterize the pericellular region using multiple particle tracking microrheology.
In MPT, the logarithmic slope of the MSD, , quantitatively determines the state of the material. When α = 1 probe particles are freely diffusing and the material is a liquid. When α → 0 the probes are completely arrested in the gel network. Values between 0 and 1 indicate that material is a viscoelastic gel or sol46,52,60,65,68. The state of the material in the viscoelastic region is determined by comparing α to the critical relaxation exponent, n46,48,52,60,65,68–71. The value of n is determined by analyzing the ensemble-averaged mean-squared displacements taken throughout a degradation reaction with time-cure superposition (TCS). TCS is the superposition of viscoelastic functions at different extents of reaction46,48,60,68–70,72. n is a material property that is constant for each hydrogel scaffold and pinpoints the transition from a gel to sol. A gel is defined as a sample spanning network cluster and the gel-sol transition occurs when the last sample spanning network cluster breaks. If n > α, the material is a viscoelastic solid and if n < α the material is a viscoelastic fluid. The value of n also provides information about the network connectivity. 0.1 < n < 0.5 means that the gel is a densely cross-linked network and 0.5 < n < 1 indicates that gel is an open, loosely cross-linked network46,48,49,51,52,59,60,65,68–71. For this hydrogel scaffold, previous work degraded the PEG-norbornene scaffold uniformly with collagenase (a mixture of enzymes that degrades the peptide cross-linker) in the absence of hMSCs. The value of n is previously reported and confirmed through our measurements as n = 0.25 ± 0.0523,51. For this hydrogel scaffold, the value of n is less than 0.5 indicating a tightly cross-linked network. This value is used to quantitatively determine the state of the material in the measured spatial maps around encapsulated hMSCs.
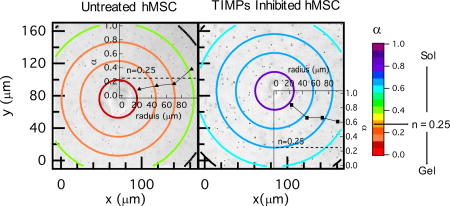
Changes in the material properties in the pericellular region due to cell-mediated degradation are measured using MPT. MPT measurements around encapsulated hMSCs are collected for a total time of 20 – 60 mins taken every 5 – 6 mins. Previous work characterized the degradation profile of untreated hMSCs in this hydrogel scaffold10,23. These experiments were repeated to characterize the degradation profile for comparison to profiles around TIMP inhibited hMSCs and are discussed here briefly. Figure 2a–c shows spatial and temporal changes around an untreated hMSC encapsulated in the PEG-norbornene hydrogel scaffold through time. The central ring has a radius of 23 µm (≈ 150 pixels) and the remaining rings increase in radius by 23 µm (≈ 150 pixels). Particles are measured within the central circle or the ring they are identified in at the beginning of data acquisition. No particle movement is used in more than one spatial measurement to calculate α. This radius is chosen to give a large enough area to measure a sufficient number of particles for statistically significant MPT measurements10. The logarithmic slope of the MSD, α, is calculated for the particles within each specified area.
Fig. 2.

Spatial degradation profiles around an encapsulated hMSC prior to motility. The left column are MPT data. MPT data are collected through time after locating the hMSC at (a) 0, (b) 12 and (c) 26 mins. The color of each ring represents the logarithmic slope of mean-squared displacement, , in the hydrogel. (d) Spatial profile of degradation at 12 mins with the α versus distance away from the cell center graph overlaid. For the overlaid graph, the x–axis is the radius of the bounding ring from the cell center and 0 is defined at the cell center. α is increasing with distance away from the cell center, showing that the cross-link density is decreasing with increasing distance from the cell. (e) α versus distance away from the cell center through time in the pericellular region, 0 is defined at the center of the cell. (f) Profile of degradation at 12 mins coupled with the normalized rate of MMP–TIMP unbinding calculated from a competitive inhibition Michaelis-Menten model23. The model indicates that TIMPs are inhibiting MMPs close to the cell and the maximum MMP–TIMP unbinding rate occurs 50 µm away from the cell, agreeing with MPT data. After the maximum unbinding rate MMP–TIMP unbinding decreases.
In Figure 2a–c the color of the bounding ring is the value of α. Warm colors indicate that the material is in the gel state. Conversely, cool colors indicate that the materials is degraded and probes are freely diffusing in the sol state. In these maps, the hMSC is outlined in black. The gel-sol transition occurs at n = 0.25 which is indicated by an orange color. Figure 2a–c shows that at the time the cell is identified and data acquisition is begun (t = 0), the area around the cell is in the gel phase and α → 0 across the measured field of view, Figure 2a. As time passes the hMSC secretes MMPs to degrade the scaffold and become motile, Figure 2b–c. For untreated hMSCs, the area directly around to the cell has the highest cross-link density and the cross-link density decreases as a function of increasing distance from the cell. This is the opposite of what was expected; since the source of the MMPs is the encapsulated hMSC, there was a general expectation that the greatest matrix degradation would occur directly around the cell. The measured degradation profile is counter-intuitive to the generally held expectation, but is consistent with previous MPT measurements of the pericellular region10,23.
To visualize the trend in scaffold degradation we overlay a plot of α for each ring versus the radius from the cell center for an encapsulated hMSC at t = 12 mins, Figure 2d (this is the same spatial map as Figure 2b). This figure shows that as the color of the ring changes from warm (gel state) to cool (liquid state), α is increasing, which indicates a decrease in scaffold connectivity. Figure 2e shows temporal changes in α as a function of the distance from the cell. At t = 0 the value of α → 0 and is uniform across the field of view and there is no scaffold degradation. As time increases, the hMSC is degrading the scaffold at the edge of the field of view and α is increasing as a function of increasing distance from the cell. The magnitude of these α values continue to increase through time and the spatial degradation trend remains the same. This indicates that the cell is limiting degradation of the network directly around it, most likely to spread and attach prior to motility. In our previous work we determined that cytoskeletal tension plays a minimal role in this degradation profile23.
From untreated hMSC degradation profiles we hypothesized that the measured degradation profile around untreated cells is due to hMSCs secreting both MMPs and TIMPs which create MMP–TIMP complexes that render MMPs inactive. A Michaelis-Menten competitive inhibition model was previously developed to describe MMP–TIMP unbinding and is shown in Figure 2f23. This graph shows that maximum unbinding occurs where maximum scaffold degradation is measured with MPT. By simultaneously secreting TIMPs and MMPs the cells are able to maintain scaffold stiffness directly around the cell enabling attachment to the network and spreading. After the hMSCs have spread, they degrade the scaffold and move rapidly through the network10,23. To determine the role of TIMPs in creating this degradation profile, we must first determine if they are being secreted by hMSCs and then inhibit them.
To determine the presence of TIMPs, the media of untreated hMSCs after 4 days of incubation is analyzed using Western blotting, Figure 3a. hMSCs derived from bone marrow secrete only TIMP–1 and –218,27. Western blots detect the presence of TIMP–1 and –2 while TIMP–3 and –4 are not detected in the cell media, Figure 3a. The presence of TIMPs is shown by the dark bands that form at 29 kDa and 21 kDa, which is the molecular weight of TIMP–1 and –2, respectively. To determine the role of TIMPs in the cell-mediated degradation profiles measured, TIMPs are inhibited. TIMP antibodies are used to inhibit TIMP–1 and –2 in the media. Figure 3b shows Western blots for untreated cells showing TIMP–1 and –2 in the cell media after 2 days (left column). The right column of Figure 3b shows the disappearance of these bands in cell media 2 days after treatment with inhibitors. Therefore, using these TIMP antibodies we can effectively inhibit TIMPs over our data acquisition window.
Fig. 3.

Analysis of cell media (CM) samples using Western blotting. a) Day 2 CM samples are analyzed with Western blotting for TIMP–1, –2, –3 and –4 expression. b) Day 2 CM samples treated with TIMP-neutralizing antibodies. The left column (CM) shows the presence of TIMP–1 and –2 by the bands in front of their molecular weight. The right column shows the disappearance of these bands.
To determine the role of TIMPs in matrix degradation we neutralize TIMPs in cell laden hydrogel scaffolds. Hydrogels are incubated with TIMP antibodies for 1.5 hrs, which is the time required for antibodies to diffuse into the hydrogel. Diffusivity of TIMP antibodies from SF and PF media into the hydrogel is calculated using, , where a is the hydrodynamic radius of TIMP antibodies, approximately 5.3 nm, and η is the viscosity of the solution73,74. The approximate diffusivity at T = 298 K is D ~ 5.52 × 10−11 m2 s−1. The time of diffusion, tD, into the hydrogel is calculated using equation, where L is the hydrogel thickness10,75,76. The time of diffusion for this hydrogel scaffold is ~1.2 hrs, therefore, incubation for 1.5 hrs ensures that the TIMP antibodies completely diffuse through the hydrogel scaffold and effectively treat hMSCs to inhibit TIMPs.
The logarithmic slope of the mean-squared displacement, α, is plotted over time for different cells for both cell treatments, untreated and TIMP inhibited hMSCs, Figure 4a and b. Figure 4a shows data around five different untreated hMSCs encapsulated in one hydrogel. Each line represents changes in the α value throughout time around a single encapsulated hMSC. The different colors on the graph represent data around different hMSCs. Figure 4b shows data for TIMP inhibited hMSCs. Here, data are collected in two different hydrogels and are plotted on the graph and represented by different symbols. The logarithmic slope of MSD shows that the state of the material is changing over time, from a gel to a sol. For untreated and TIMP inhibited hMSCs, two profiles of enzymatic degradation are measured and shown in Figure 4a and b. First, when data acquisition begins around an hMSC (t = 0) the hydrogel is in the gel phase, α → 0. Over time the hMSC is secreting MMPs which degrades the network and the value of α is increasing until it passes 0.25. When α = n = 0.25, the critical relaxation exponent for this hydrogel, the scaffold is transitioning from the last sample-spanning gel network to a sol. Then α > 0.25 is when the material is in the viscoelastic sol phase. The second profile of degradation is a relatively flat line when α = 0.2 for untreated cells and α = 0.75 for TIMP inhibited hMSCs and is not changing over time. For untreated hMSCs, the α value is within error of n and is at the gel-sol transition.
Fig. 4.

Changes in the logarithmic slope of mean-squared displacement, , over time for cell-laden hydrogels for (a) untreated hMSCs and (b) TIMP inhibited hMSCs. Dashed lines in both graphs (a, b) indicate the critical relaxation exponent, n, that quantitatively determines the transition from a gel to a sol. Each line represents changes in the α value throughout time around a single encapsulated hMSC. The colors on the graph represent data around different hMSCs. Two separate hydrogels are shown in (b) indicated by the –△– and –○– symbols. The average migration speed of hMSCs for (c) untreated hMSCs and (d) TIMP inhibited hMSCs. The cell speed increased significantly after TIMP inhibition, *p < 0.05.
For the motile TIMP inhibited hMSCs, the hydrogel has already substantially degraded the pericellular region when data acquisition is begun around the hMSC. In MPT, we measure probe particle diffusion, which is slightly restricted free diffusion, in the pericellular region. For both experiments, the trends in cell-mediated scaffold degradation in the pericellular region is similar. Although the trends are similar, the timing of degradation differs between the experiments. Untreated hMSCs degrade the scaffold slowly over approximately one hour, Figure 4a. After TIMP inhibition, MMP activity is increased and degradation occurs over a shorter time of approximately 20 mins, Figure 4b. The final α value after TIMP inhibited hMSC-mediated degradation is also higher than the value measured for untreated hMSCs. For TIMP inhibited hMSCs, MPT measures greater probe particle diffusion and, therefore, extent of scaffold degradation. This supports the assumption that MMP activity is greater when TIMPs are inhibited enabling faster degradation kinetics and overall greater scaffold degradation. The extent of degradation directly impacts the speed of hMSC motility.
To compare cell speeds between the untreated and TIMP inhibited hMSCs we have established three different populations. These three different populations are: the (1) slow motility (0 – 25 µ m h−1), (2) fast motility (25 – 100 µm h−1) and (3) super fast (> 100 µm h−1) groups. Two speeds of cell motility are measured for untreated hMSCs encapsulated in the hydrogel, Figure 4c. We have named these groups ‘untreated slow’ and ‘untreated fast’. The untreated slow group of cells are moving with an average speed of 9.1 ± 6.2 µm h−1. The untreated fast group is moving at an average speed of 27 ± 3 µm h−1, Figure 4c. To compare with work by Kyburz et al., which measured hMSCs speed in the same hydrogel over several days, we calculate the average of the untreated slow and untreated fast motility groups. The average hMSC speed in our scaffold is 11.7 ± 8.8 µm h−1. Kyburz et al. measure an average speed of 17.6 ± 0.09 µm h−1. These values are within error of each other, which indicates that the previous work has averaged the speed between slow and fast moving hMSCs3. The untreated slow group is the hMSCs that are degrading the scaffold past the gel-sol transition and the untreated fast group are the hMSCs that are in a viscoelastic sol and are moving through the scaffold. This shows that hMSCs have two steps during scaffold degradation: (1) the hMSC is degrading the scaffold far from it and limiting degradation of the network around the cell to enable spreading and attachment and is not highly motile and (2) the hMSC degrades the scaffold and migrates quickly through the pericellular region10,23. TIMP inhibited hMSCs differ in speed from the untreated hMSCs. Three groups of cell migration speeds are measured, ‘slow’, ‘fast’ and ‘super fast’, Figure 4d. All cell migration speeds are faster than those measured for untreated hMSCs. The average cell speed for slow migration is 16.3 ± 7.3 µm h−1 and for fast migration is 49.5 ± 18.6 µm h−1. For TIMP inhibited hMSCs, we also measure cells moving at very high speeds 630 ± 308 µm h−1, Figure 4d. By inhibiting TIMP activity and the ability for TIMPs to bind to MMPs making them inactive, the degradation of the scaffold is much more rapid resulting in more rapid cell motility. Therefore, we have greatly reduced the first step measured in hMSC motility, the low motility cell spreading and attachment, and have enhanced the motility of these cells through the scaffold.
Changes in hydrogel properties after TIMP inhibition are measured on hydrogel scaffolds 18 – 48 hrs after encapsulation. This incubation time allows the hMSCs to adjust to their surrounding after encapsulation and begin to migrate. MPT data are collected around hMSCs during motility using the same techniques developed to measure the pericellular region around untreated cells. Figure 5 shows the spatial changes in the rheological properties over time for three different cells. Figure 5a–c shows the spatial rheological changes around a cell in the slow motility group, Figure 5d–f is in the fast motility group and Figure 5g–i is the super fast motility group. A copy of Figure 5 with all quantitative α values included over the bounding ring is provided in the ESI† as Figure 10S. For all TIMP inhibited hMSCs, the spatial trend in the degradation profile is consistent. MPT data shows that by inhibiting both TIMP–1 and –2 the spatial hMSC-mediated degradation profile results in a reaction-diffusion type gradient. In a reaction-diffusion type mechanism for our system, MMPs are reacting with cross-links in the hydrogel scaffold while they are diffusing through the matrix. In the pericellular region, we measure the greatest degradation closest to the cell and the cross-link density increases with increasing distance from the cell center. Therefore, the MMPs are reacting with the cross-linkers directly around the cells, leaving fewer MMPs to diffuse through the matrix and react with cross-linkers far from the cell center.
Fig. 5.

Spatial degradation profiles around three different TIMP inhibited hMSC migration speed populations. The left column is MPT data for an hMSC in the slow motility group, the middle column shows the data around an hMSC in the fast motility group and the right column is MPT data for an hMSC in the super fast motility group. MPT data are collected through time for each hMSC after identification at (a) 0, (b) 32 and (c) 57, (d) 0, (e) 4 and (f) 8, (g) 0, (h) 8 and (i) 16 mins. The color of the each rings represents, , which determines the state of the material in the hydrogel. This plot with all the quantitative α values written on each bounding ring is also provided as Figure 10S in the ESI.†.
In the slow motility group, the first measurement is in the gel phase, Figure 5a. The spatial gradient in scaffold properties is illustrated for all hMSCs in the degradation profiles and is shown in Figure 5b–c. TIMP inhibition results in increased MMP activity that aggressively degrades the matrix. After this simple treatment, hMSCs cannot limit degradation of the network directly around the cell to attach and spread due to the enhanced cell-mediated degradation. Due to this, hMSCs migrate rapidly in the scaffold. There are two possible reasons for this rapid migration: (1) hMSCs are migrating towards stiffer regions, a process known as durotaxis77–79 or (2), due to increased material degradation there is less of a physical barrier directly around cell enabling increased motility.
Figure 5d–f is a spatial map for a TIMP inhibited hMSC that is in the fast motility group and has degraded the network past the gel-sol transition. This profile also shows a reaction-diffusion type degradation profile and is the reverse of degradation profiles measured around untreated hMSCs. Similar to the pericellular region in Figure 5a–c, directly around the cell has the lowest crosslink density and cross-link density is increasing as a function of distance from the cell. This profile is also measured in Figure 5g–i, which is in the super fast motility group. Additionally, due to the rapid motility we measure greater degradation and a uniform viscoelastic sol in the pericellular region.
To further illustrate the change in the degradation profile, we overlay the graph showing α as a function of distance from the cell center on a spatial degradation profile in Figure 6a (the complete degradation profile for this cell is Figure 8S in the ESI†). Here, the change in α is the opposite of what is measured for untreated hMSCs. α in the region directly around the cell has the highest value indicating the greatest probe particle movement and scaffold degradation and as the distance from the hMSC is increased the value of α decreases, indicative of restricted probe particle diffusion and increased network connectivity. This trend is shown for different time points during data acquisition around this TIMP inhibited hMSC, Figure 6b. Here, all measurements of α are in the viscoelastic sol with no measurements in any region around the encapsulated hMSC in the gel phase. This trend is prevalent in all degradation profile (for all cell speeds) and contributes to increased motility of these hMSCs.
Fig. 6.

Spatial degradation profiles around an encapsulated hMSC treated with TIMP–1 and –2 antibodies. a) Profile of degradation at 16 minutes with the logarithmic slope of mean-squared displacement, , versus distance away from the cell center graph overlaid. α is decreasing as the distance from center of the cell is increasing. b) α versus distance from the cell center for different times during MPT data acquisition in the pericellular region for a TIMP inhibited hMSC. This plot is showing the same decreasing trend in α for all different time point of data acquisition. The complete degradation profile around this hMSC is added as Figure 8S in the ESI†.
4 Conclusions
In this work, we use multiple particle tracking microrheology to measure spatio-temporal changes in the pericellular region around encapsulated hMSCs in well-defined MMP degradable poly(ethylene glycol)-peptide hydrogel scaffolds. We also develop a novel sample chamber to enable MPT characterization, limit directed motion of probe particles and minimize the volume of high value antibodies used for cell treatments. Using MPT, we determine the rheological properties in the pericellular region around untreated and TIMP inhibited hMSCs. For untreated hMSCs, MPT measurements characterize a degradation profile where directly around the cell the hydrogel has the greatest cross-link density and remains in a gel state and the cross-link density decreases as the distance from the cell center is increased. From previous work, we determine that cytoskeletal tension plays a minimal role in creating this degradation profile, therefore, MMPs are being inhibited close to the cell. MMPs are inhibited by TIMPs, which bind to MMPs making them inactive. This MMP–TIMP binding occurs directly after cell secretion and unbinds after diffusing away from the hMSC. This enables hMSCs to spread and attach to the network before motility.
To determine the role of TIMPs we inhibit them using antibodies and verify that they are no longer present in the cell media using Western blotting. After TIMP–1 and –2 inhibition, the degradation profile is reversed and is a reaction-diffusion type profile. Around TIMP inhibited hMSCs, the greatest degradation is directly around the cell and the cross-link density increases with increasing distance from the cell center. After treatment, MMP activity is no longer inhibited and the scaffold is degraded immediately after secretion. The cell speeds also change between these two experiments. Untreated hMSCs move slower on average than TIMP inhibited hMSCs. The increase in cell speed is due to durotaxis, where the TIMP inhibited hMSCs are migrating along the modulus gradient to stiffer materials, or decreased physical barrier to migration. By simply inhibiting TIMPs, we are able to enhance hMSC motility which can be useful in applications that require the delivery of cells, such as during wound healing. Additional degradation of the scaffold can be advantageous for implantable scaffolds but may also be a disadvantage if the scaffold is needed to give structure to a wound.
Overall, this work gives new insight into how simple treatments of hMSCs can modify degradation strategies and change the time scale of scaffold degradation. This work expands the use of MPT in quantification of dynamic biomaterial degradation and cell-material interactions. The development of a new sample chamber will also broaden the use of this technique to explore more expensive cell treatments. With these techniques, we have gained new knowledge about the complex interplay with the scaffold and cell-mediated remodeling of the pericellular region. This knowledge will lead to the design of new materials that recapitulate aspects of the native ECM and use highly designed pericellular regions to direct and manipulate basic cellular processes.
Supplementary Material
Acknowledgments
We thank Dr. Susan Perry from the Department of Bioengineering at Lehigh University for her useful discussion on Western Blot experiments. We also thank Michelle Mazzeo for bulk rheology data of swollen hydrogels. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R15GM119065. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Conflict of interest
There are no conflicts to declare.
References
- 1.West JL, Hubbell JA. Macromolecules. 1999;32:241–244. [Google Scholar]
- 2.Lutolf MP, Lauer-Fields JL, Schoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. PNAS. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kyburz KA, Anseth KS. Acta Biomaterialia. 2013;9:6381–6392. doi: 10.1016/j.actbio.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira LS, Gerecht S, Fuller J, Shieh HF, Vunjak-Novakovic G, Langer R. Biomaterials. 2007;28:2706–2717. doi: 10.1016/j.biomaterials.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv. Mater. 2006;18:1345–1360. [Google Scholar]
- 6.Anderson SB, Lin C-C, Kuntzler DV, Anseth KS. Biomaterials. 2011;32:3564–3574. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guvendiren M, Burdick JA. Curr. Opin. in Biotech. 2013;24:841–846. doi: 10.1016/j.copbio.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz MP, Fairbanks BD, Rogers RE, Rangarajan R, Zaman MH, Anseth KS. Integr Biol. 2010;2:32–40. doi: 10.1039/b912438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benton JA, Fairbanks BD, Anseth KS. Biomaterials. 2009;30:6593–6603. doi: 10.1016/j.biomaterials.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz KM, Kyburz KA, Anseth KS. PNAS. 2015;112:E3757–E3764. doi: 10.1073/pnas.1511304112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metzger S, Blache U, Lienemann PS, Karlsson M, Weber FE, Weber W, Ehrbar M. Macromolecular Bioscience. 2016;16:1703–1713. doi: 10.1002/mabi.201600223. [DOI] [PubMed] [Google Scholar]
- 12.Engler AJ, Sen S, Lee Sweeney H, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 13.Wolf K, te Lindert M, Krause M, Alexander S, te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. Cell Biology. 2013;7:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caplan AI. Journal of Pathology. 2009;217:318–324. doi: 10.1002/path.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singer AJ, Clark RA. The New England Journal of Medicine. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 16.Bryant SJ, Anseth KS. Biomedical Materials Research. 2003;64A:70–79. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 17.Mackenzie TC, Flake AW. Blood Cells, Molecules and Diseases. 2001;27:601–604. doi: 10.1006/bcmd.2001.0424. [DOI] [PubMed] [Google Scholar]
- 18.Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P. Blood. 2007;109:4055–4063. doi: 10.1182/blood-2006-10-051060. [DOI] [PubMed] [Google Scholar]
- 19.Kawada H, Fujita J, Kinjo K, Matsuzaki Y, Tsuma M, Miyatake H, Muguruma Y, Tsuboi K, Itabashi Y, Ikeda Y, Ogawa S, Okano H, Hotta T, Ando K, Fukuda K. blood. 2004;104:3581–3587. doi: 10.1182/blood-2004-04-1488. [DOI] [PubMed] [Google Scholar]
- 20.Tibbitt MW, Anseth KS. Biotechnol. Bioeng. 2009;103:655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brew K, Nagase H. Biochimica et Biophysica Acta. 2010;1:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vu TH, Werb Z. Genes and development. 2000;14:2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- 23.Daviran M, Caram HS, Schultz KM. ACS Biomatterials. 2018;4:468–472. doi: 10.1021/acsbiomaterials.7b01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y, Su J, Roberts AI, Shou P, Rabson AB, Ren G. Cell Press. 2012;33:136–143. doi: 10.1016/j.it.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mountziaris PM, Mikos AG. Tissue Eng. 2008;14:179–186. doi: 10.1089/ten.teb.2008.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lozito TP, Tuan RS. Cell Physiology. 2011;227:534–549. doi: 10.1002/jcp.22744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lozito TP, Jackson WM, Nesti LJ, Tuan RS. Matrix biology. 2014;34:132–143. doi: 10.1016/j.matbio.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Brew K, Dinakarpandian D, Nagase H. Biochemica et Biophysica Acta. 2000;1477:267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- 29.Mannello F. Stem Cells. 2004;24:1904–1907. doi: 10.1634/stemcells.2005-0608. [DOI] [PubMed] [Google Scholar]
- 30.Bassi EJ, Candido de Almeida D, Moraes-Vieira PMM, Camara NOS. Stem Cell Rev and Rep. 2012;8:329–342. doi: 10.1007/s12015-011-9311-1. [DOI] [PubMed] [Google Scholar]
- 31.Olson MW, Gervasi DC, Mobashery S, Fridman R. Biological Chemistry. 1997;272:29975–29983. doi: 10.1074/jbc.272.47.29975. [DOI] [PubMed] [Google Scholar]
- 32.Aimetti AA, Machen AJ, Anseth KS. Biomaterials. 2009;30:6048–6054. doi: 10.1016/j.biomaterials.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS. Biomaterials. 2010;22:3484–3494. doi: 10.1002/adma.200904179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peyton SR, Raub CB, Keschrumrus VP, Putnam AJ. Biomaterials. 2006;27:4881–4893. doi: 10.1016/j.biomaterials.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 35.Grim JC, Marozas IA, Anseth KS. Controlled Release. 2015;219:95–106. doi: 10.1016/j.jconrel.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patterson J, Hubbell J. Biomaterials. 2010;31:7836–7845. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 37.Raeber GP, Lutolf MP, Hubbell JA. Acta Biomater. 2007;3:615–629. doi: 10.1016/j.actbio.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Longhurst CM, Jennings LK. Cellular and molecular life science. 1998;54:514–526. doi: 10.1007/s000180050180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaman MH, Trapani LM, Sieminski AL, MacKellar D, Gong H, Kamm RD, Wells A, Lauffenburger DA, Matsudaira P. PNAS. 2005;103 doi: 10.1073/pnas.0604460103. year. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SH, Moon JJ, Miller JS, West JL. Biomaterials. 2007;28:3163–3170. doi: 10.1016/j.biomaterials.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 41.Leight JL, Tokuda EY, Jones CE, Lin AJ, Anseth KS. PNAS. 2015;112:5366–5371. doi: 10.1073/pnas.1505662112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maskarine SA, Franck C, Tirrell DA, Ravichandran G. PNAS. 2009;106:22108–22113. doi: 10.1073/pnas.0904565106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Legant WR, Miller JS, Blakely BL, Cohen DM, Genin GM, Chen CS. Nat. Methods. 2010;7:969–973. doi: 10.1038/nmeth.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mason TG, Ganesan K, van Zanten JH, Wirtz D, Kuo SC. Phys. Rev. Lett. 1997;79:3282–3285. [Google Scholar]
- 45.Waigh TA. Rep. Prog. Phys. 2005;68:685–742. [Google Scholar]
- 46.Larsen TH, Furst EM. Phys. Rev. Lett. 2008;100:146001–4. doi: 10.1103/PhysRevLett.100.146001. [DOI] [PubMed] [Google Scholar]
- 47.Crocker JC, Grier DG. Journal of colloid and interface science. 1996;179:298–310. [Google Scholar]
- 48.Furst EM, Squires TM. Microrheology. 1. Oxford University Press; 2017. [Google Scholar]
- 49.Schultz KM, Furst EM. Soft Matter. 2012;8:6198–6205. [Google Scholar]
- 50.Winter HH. Polym. Eng. Sci. 1987;27:1698–1702. [Google Scholar]
- 51.Schultz KM, Anseth KS. Soft Matter. 2013;9:1570–1579. [Google Scholar]
- 52.Wehrman MD, Lindberg S, Schultz KM. Soft Matter. 2016;12:6463–6472. doi: 10.1039/c6sm00978f. [DOI] [PubMed] [Google Scholar]
- 53.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller JS, Shen CJ, Legant WR, Baranski JD, Blakely BL, Chen CS. Biomaterials. 2010;31:3736–3743. doi: 10.1016/j.biomaterials.2010.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jackson WM, Nesti LJ, Tuan RS. Stem cell translational medicine. 2012;1:44–50. doi: 10.5966/sctm.2011-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mason TG, Weitz DA. Physcical review letter. 1995;74:1250–1253. doi: 10.1103/PhysRevLett.74.1250. [DOI] [PubMed] [Google Scholar]
- 57.Savin T, Doyle PS. Biophys. J. 2005;88:623–638. doi: 10.1529/biophysj.104.042457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crocker JC, Weeks ER. 2018 http://www.physics.emory.edu/faculty/weeks//idl/
- 59.Schultz KM, Baldwin AD, Kiick KL, Furst EM. Macro letters. 2012;1:706–708. doi: 10.1021/mz300106y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larsen T, Schultz K, Furst EM. Rheology journal. 2008;20:165–173. [Google Scholar]
- 61.Squires TM, Mason TG. Annu. Rev. Fluid Mech. 2010;42:413–438. [Google Scholar]
- 62.Ferry JD. Viscoelastic Properties of Polymers. Wiley; New York: 1980. [Google Scholar]
- 63.Palmer A, Xu J, Wirtz D. Rheol Acta. 1998;33:97–106. [Google Scholar]
- 64.Schultz KM, Bayles AV, Baldwin AD, Kiick KL, Furst EM. Biomacromolecules. 2011;12:4178–4182. doi: 10.1021/bm201214r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Escobar F, Anseth KS, Schultz KM. Macromolecules. 2017;50:7351–7360. [Google Scholar]
- 66.Buxboim A, Ivanovska IL, Discher DE. Cell Science. 2010;123:297–308. doi: 10.1242/jcs.041186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cox TR, Erler JT. Disease Models and Mechanisms. 2009;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Corrigan AM, Donald AM. Langmuir. 2009;25:8599–8605. doi: 10.1021/la804208q. [DOI] [PubMed] [Google Scholar]
- 69.Muthukumar M, Winter HH. Macromolecules. 1986;19:1284–1285. [Google Scholar]
- 70.Scanlan JC, Winter HH. Macromolecules. 1991;24:47–54. [Google Scholar]
- 71.Chambon F, Winter HH. J. Rheol. 1987;31:683–697. [Google Scholar]
- 72.Adolf D, Martin JE. Macromolecules. 1990;23:3700–3704. [Google Scholar]
- 73.Armstrong JK, Wenby RB, Meiselman HJ, Fisher TC. Biophysical Journal. 2004;87:4259–4270. doi: 10.1529/biophysj.104.047746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Research and Diagnostic Systems, Inc, Personal communication. 2017 [Google Scholar]
- 75.Naomi O, Noriko Y, Goto M, Kimiko W, Youhei Y, Kazutaka M. Applied and Environmental Microbiology. 2012 doi: 10.1128/AEM.00808-12. [DOI] [Google Scholar]
- 76.Weber LM, Lopez CG, Anseth KS. J. Biomed. Mater. Res. A. 2008;90A:720–729. doi: 10.1002/jbm.a.32134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vincent LG, Choi YS, Alonso-Latorre B, del Álamo JC, Engler AJ. Biotechnol. 2013;8:472–484. doi: 10.1002/biot.201200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tse JR, Engler AJ. PloS ONE. 2011;6:e15978. doi: 10.1371/journal.pone.0015978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bear JE, Haugh JM. Curr. Opin. in Cell Biology. 2014;30:78–82. doi: 10.1016/j.ceb.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.