

Abstract
Androgen excess is a hallmark of polycystic ovary syndrome (PCOS), a prevalent yet poorly understood endocrine disorder. Evidence from women and preclinical animal models suggests that elevated perinatal androgens can elicit PCOS onset in adulthood, implying androgen actions in both PCOS ontogeny and adult pathophysiology. Prenatally androgenized (PNA) mice exhibit a robust increase of progesterone-sensitive GABAergic inputs to gonadotropin-releasing hormone (GnRH) neurons implicated in the pathogenesis of PCOS. It is unclear when altered GABAergic wiring develops in the brain, and whether these central abnormalities are dependent upon adult androgen excess. Using GnRH-GFP–transgenic mice, we determined that increased GABA input to GnRH neurons occurs prior to androgen excess and the manifestation of reproductive impairments in PNA mice. These data suggest that brain circuit abnormalities precede the postpubertal development of PCOS traits. Despite the apparent developmental programming of circuit abnormalities, long-term blockade of androgen receptor signaling from early adulthood rescued normal GABAergic wiring onto GnRH neurons, improved ovarian morphology, and restored reproductive cycles in PNA mice. Therefore, androgen excess maintains changes in female brain wiring linked to PCOS features and the blockade of androgen receptor signaling reverses both the central and peripheral PNA-induced PCOS phenotype.
Keywords: Endocrinology, Neuroscience
Keywords: Fertility, Neuroendocrine regulation, Sex hormones
Hypothalamic circuit abnormalities in a preclinical model of PCOS are programmed by early androgen exposure yet remain plastic and androgen-dependent in adulthood.
Introduction
Polycystic ovary syndrome (PCOS) is a major endocrine disorder affecting around 1 in 10 women of reproductive age globally (1, 2). The disease is characterized by clinical and/or biochemical evidence of androgen excess (hyperandrogenism), menstrual dysfunction (oligo-anovulation), and polycystic ovarian follicle morphology (3). Most PCOS patients also have elevated luteinizing hormone (LH) secretion (4), which is likely in response to enhanced pulsatile secretion of gonadotropin-releasing hormone (GnRH) from hypophysiotropic GnRH neurons in the rostral forebrain (5, 6). Compared with normal women, PCOS patients require higher concentrations of estradiol and progesterone to suppress pulsatile LH release (5, 7, 8), demonstrating an impairment in the ability of steroid hormones to restrain the activity of the GnRH/LH pulse generator. Elevated LH release acts downstream to overstimulate the theca cells of the ovaries and favors greater androgen production when compared with normal subjects (9–11).
Evidence suggests that inappropriately high levels of androgens indirectly establish an impaired negative feedback in PCOS patients. Acute testosterone administration does not increase pulsatile LH levels in healthy women (12). Likewise, acute blockade of the androgen receptor (AR) with an antagonist does not restore normal LH secretion in women with PCOS (13). However, long-term treatment with an AR antagonist is able to reestablish the ability of estradiol and progesterone to suppress LH pulse frequency (13). This implies that androgen excess does not directly drive LH hypersecretion, but instead interferes with the ability of estradiol and progesterone to relay negative feedback signals to GnRH neurons and regulate LH release. However, little is known about the spatial and temporal mechanisms mediating this vicious cycle of androgen excess in the development and pathophysiology of PCOS.
Although the etiology of PCOS is undefined and likely to be multivariate (14, 15), exposure to elevated androgens in early development is known to trigger reproductive and metabolic traits of PCOS during adult life in both humans (16, 17) and in a variety of animal models (18–20). Notably, daughters of PCOS mothers, that have higher circulating androgen levels during pregnancy than non-PCOS women, are at higher risk of developing PCOS features and associated comorbidities, such as diabetes mellitus type 2, from early to late puberty (21, 22). Prenatally androgenized (PNA) mouse models can also recapitulate the clinical features of PCOS, including androgen excess, impaired steroid hormone feedback, and hyperactive GnRH/LH activity (23–25). PNA mice enable transgenic tools to be used to investigate the impact of androgen excess in the brain. Evidence to date has identified that PCOS traits in PNA mice are coincident with modifications in the GnRH neuronal network. GnRH neurons have significantly enhanced GABAergic input (26) and signaling (23) and that likely originates from neurons in the arcuate nucleus (26, 27). In support of these preclinical findings, women with PCOS have elevated cerebrospinal fluid levels of GABA, (28) and GABA agonists used in the treatment of epilepsy can result in the onset of PCOS features (29). While GABA typically acts as an inhibitory neurotransmitter, GnRH neurons can respond to GABA with depolarization due to a higher intracellular chloride concentration (30, 31). These observations suggest that increased GABA signaling in PCOS may be responsible for driving the hyperactive GnRH/LH system and associated downstream consequences at the ovary.
These findings have led to important questions about the development and maintenance of identified PCOS-associated brain circuit abnormalities. The present study aimed to determine, first, when GnRH neuronal network perturbations in PNA mice occur in relation to the development of androgen excess. Secondly, this study investigated the impact of long-term AR blockade on established GnRH neuronal network circuit changes and the associated PCOS phenotype in adulthood.
Results
Androgen excess occurs during early adulthood in PNA mice.
To determine the developmental timeline of androgen excess in the PNA PCOS-like mouse model, circulating levels of testosterone were measured throughout peripubertal development and into adulthood, from postnatal day (PND) 30 to PND 60. Testosterone levels were similar between groups at PND 30 (PNA: 0.11 ± 0.03 vs. control: 0.10 ± 0.02 ng/ml) and PND 40 (PNA: 0.14 ± 0.03 vs. control: 0.15 ± 0.03 ng/ml), corresponding to early and mid-pubertal stages (Figure 1). In contrast, by early adulthood at PND 50, average circulating testosterone levels of PNA mice were significantly elevated compared with controls (0.24 ± 0.06 vs. 0.06 ± 0.01 ng/ml; P < 0.01). Mean testosterone levels remained higher in adult PNA mice at PND 60 when compared with control mice (Figure 1; 0.24 ± 0.06 vs. 0.08 ± 0.01 ng/ml; P < 0.05; F3,51 [interaction] = 6.2; F1,17 [phenotype] = 7.3). Analysis of testosterone fluctuations over the pubertal transition into adulthood in control animals demonstrated that peak testosterone levels occur at PND 40, with levels falling significantly into adulthood (PND 50–60) (P < 0.05). In contrast, in PNA mice, peak testosterone levels were significantly higher at PND 50 compared with PND 30 (P < 0.05) (Figure 1). While control mice all exhibited relatively low circulating testosterone in adulthood, testosterone levels in PNA mice were highly variable. Despite variable testosterone levels at PND 50 and 60, vaginal cytology confirmed that all adult PNA mice from PND 60–80 exhibited marked disruption of estrous cycles and a complete absence of proestrous smears, suggesting the absence of ovulation (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.99405DS1). These data demonstrate that a short prenatal androgen exposure in female mice induces the development of high circulating testosterone levels that begin after pubertal onset and continue into adulthood, associated with reproductive dysfunction.
Figure 1. Androgen excess in PNA mice develops by PND 50.
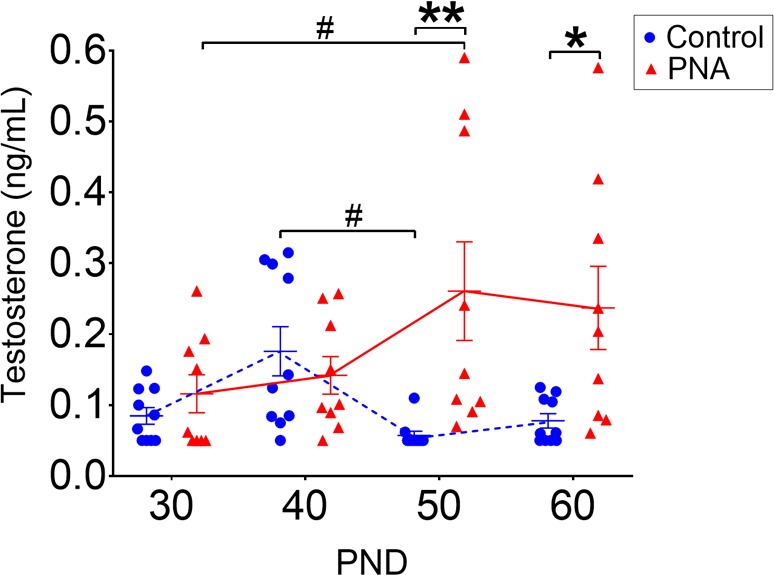
Circulating testosterone levels from postnatal day (PND) 30 to PND 60 in control (N = 10) and prenatally androgenized (PNA) (N = 9) mice. Data are shown as dot plots for each time point and bars represent mean ± SEM. Dashed blue lines indicate testosterone levels of control mice and red full lines indicate PNA mice. Differences between the groups are reported as *P < 0.05; **P < 0.01 and differences within each groups are reported as #P < 0.05. Data analysis was performed with repeated-measures 2-way ANOVA with Sidak’s post hoc test.
Enhanced GABAergic contact to GnRH neurons is evident prior to pubertal onset and androgen excess.
To address whether GnRH neuronal network changes observed in PCOS-like mice were programmed through early androgen exposure or driven by adult androgen excess, the brains of control and PNA GnRH-GFP mice were evaluated at PND 25, prior to pubertal onset. All PNA (N = 13) and control (N = 8) mice exhibited a complete absence of vaginal opening, a marker of pubertal onset, and LH levels were undetectable (<0.04 ng/ml), confirming PND 25 as a prepubertal time point in both groups. In this cohort of mice, circulating testosterone levels at PND 25 were not different between PNA and control groups (PNA: 0.20 ± 0.01 vs. control: 0.22 ± 0.03 ng/ml; Student’s t test; Supplemental Figure 2).
Close appositions of vesicular GABA transporter (VGAT) immunoreactive (-ir) puncta to GFP-ir GnRH neurons in the rostral preoptic area (rPOA) of the hypothalamus, indicative of putative GABA synaptic input (32, 33), were quantified with confocal microscopy (Figure 2A). The dendrites of bipolar GnRH neurons were classified as either primary, the thicker dendrite (larger sectional area) at the point of origin from the soma, or secondary, the thinner dendrite, according to reported morphological criteria (34) (Figure 2B). The locations of putative GABA synapses were classified as onto spines, crucial membrane structures which harbor excitatory inputs (35, 36), or onto the somatic or dendritic shaft membrane without spines, here referenced as non-spiny membrane.
Figure 2. Increased GABAergic contact to GnRH neurons is present in prepubertal PNA mice.

(A) Confocal images of control (N = 4; 45 neurons total) and prenatally androgenized (PNA) (N = 5; 51 neurons total) mice showing GnRH-GFP neurons (green) and vesicular GABA transporter–immunoreactive (VGAT-ir) puncta (red) in the rostral preoptic area. Scale bars: 10 μm. Inset images depict a selected confocal image stack of 1.15 μm thickness illustrating VGAT appositions at the level of GnRH neuron soma and dendrite. Red puncta (VGAT-ir) in close apposition to green GnRH neurons can appear as yellow or with a yellow halo as a result of overlap in confocal projections. White arrowheads point to putative GABA inputs onto the non-spiny GnRH neuron membrane and blue arrowheads show GABA inputs onto spines. Scale bars: 5 μm. (B) Neuronal compartments of bipolar GnRH neurons. Morphological criteria classified the primary dendrite as the thicker dendrite (with largest sectional area leaving the soma) and secondary dendrite as the thinner dendrite. Histograms depict total VGAT-ir apposition density on GnRH neurons (C) and per neuronal compartment (D). Total VGAT-ir apposition density onto the non-spiny GnRH neuron membrane (E), and per neuronal compartment (F). Total VGAT-ir apposition density onto GnRH neuron spines (G), and per neuronal compartment (H). Histogram values are represented as mean ± SEM with dot plot of individual values. *P < 0.05; Mann-Whitney U test. GnRH, gonadotropin-releasing hormone.
We discovered that at PND 25, the density of VGAT appositions to GnRH neurons was already significantly increased in PNA mice (N = 5; 51 total neurons) when compared with controls (N = 4; 45 total neurons) (P < 0.05; Mann-Whitney U test; Figure 2C). In addition, the density of VGAT appositions to GnRH neurons was significantly elevated in all of the neuronal compartments (Figure 2D). The density of VGAT close appositions to the non-spiny membrane of the soma and dendrites was also significantly higher in prepubertal PNA mice than in controls (P < 0.05; Figure 2E), and specifically at the level of the soma (P < 0.05) and primary dendrite (P < 0.05) (Figure 2F). The total density of VGAT appositions to GnRH neuron spines was not different between groups (P = 0.19; Figure 2G); however, a greater number of GABAergic inputs on GnRH neuron somatic spines was detected in prepubertal PNA mice when compared with controls (P < 0.05; Figure 2H). GnRH neuron spine density, a general index of synaptic input, was similar between groups at PND 25 (PNA: 0.24 ± 0.01 vs. control: 0.23 ± 0.03 spines/μm). Our data reveal that prenatal androgen exposure is sufficient to program circuitry changes in the female GnRH neuronal network that occur prior to the postpubertal development of circulating androgen excess and reproductive deficits in PNA mice.
Long-term AR blockade reverses GnRH neuron circuit abnormalities in adult PNA mice.
Long-term treatment with the AR antagonist flutamide (Flut) restores ovarian steroid-mediated feedback sensitivity in adult PCOS patients (13). Likewise, Flut treatment decreases GABA-mediated postsynaptic currents to control levels in PNA mice (23). Therefore, despite the indication that GABA-to-GnRH neuron wiring abnormalities are programmed early, suggestive of developmentally hard-wired circuitry, we tested the hypothesis that long-term blockade of AR signaling from early adulthood could reverse the established increase in GABA-to-GnRH neuron inputs in PNA mice. To test this hypothesis, control and PNA mice were treated with Flut (25 mg/kg/day) or an oil vehicle from PND 40 to PND 60. This effective dose was established in a pilot study in males and females. In males, 25 mg/kg/day of Flut for 15 days significantly decreased seminal vesicle weight (P < 0.05) and adrenal gland weight (P < 0.05) compared with oil-treated controls, confirming the antiandrogenic effect of this dose (Supplemental Figure 3A). In healthy females, the same treatment protocol had no effect on ovarian or adrenal gland weight (Supplemental Figure 3B).
Applying the same technical approach used in prepubertal mice, GABA inputs to GnRH neurons were assessed in control+oil (N = 6; 61 total neurons), control+Flut (N = 7; 72 total neurons), PNA+oil (N = 5; 53 total neurons), and PNA+Flut (N = 7; 72 total neurons) groups (Figure 3, A–G). As expected and reported previously (26), PNA mice receiving an oil vehicle (PNA+oil) exhibited significantly increased VGAT appositions to GnRH neurons at the soma (P < 0.0001), primary dendrite (P < 0.0001), and secondary dendrite (P < 0.001) compared with control mice (Figure 3H; F1,21 = 33.8). Remarkably, AR blockade decreased the density of GABA inputs to GnRH neurons in PNA mice to levels found in control+oil and control+Flut groups (Figure 3H; F1,21 [treatment] = 37.8). Similar patterns were found when VGAT contacts were identified onto the non-spiny membrane (Figure 3I) or onto spines (Figure 3J), although VGAT apposition density on the non-spiny membrane of secondary dendrites was not different among the groups (F1,21 [treatment] = 0.7).
Figure 3. Blockade of androgen receptor (AR) signaling reverses GABA-to-GnRH circuit abnormalities in adult PNA mice.

(A) Confocal images of adult diestrus control and prenatally androgenized (PNA) mice showing GnRH-GFP neurons (green) and vesicular GABA transporter–immunoreactive (VGAT-ir) contacts representing GABA inputs (red puncta) in the rostral preoptic area. Red puncta (VGAT-ir) in close apposition to green GnRH neurons can appear as yellow or with a yellow halo as a result of overlap in confocal projections. White arrowheads indicate putative GABAergic inputs to GnRH neurons. Scale bars: 10 μm. (B and E) Projected 3D reconstruction of a GnRH-GFP neuron from an adult PNA mouse illustrating VGAT-ir puncta contacts, with white arrowheads indicating VGAT contact onto non-spiny GnRH neuron membrane, and blue arrowheads indicating inputs onto somatic and dendritic spines. Rotated and zoomed inset images of VGAT contact with a somatic spine (C), front and back views of dendritic spines in the distal dendrite covered with GABA inputs (D and F), and VGAT contacts onto the proximal dendrite of a GnRH neuron (G). (H) Total VGAT-ir density on GnRH neurons of control+oil (N = 6; 61 neurons), control+Flut (N = 7; 72 neurons), PNA+oil (N = 5; 53 neurons), and PNA+Flut (N = 7; 72 neurons) groups. VGAT appositions are shown for each neuronal compartment. (I) Total VGAT apposition density onto non-spiny GnRH neuron membrane. (J) Total VGAT-ir apposition density onto GnRH neuron spines. (K) Number of GnRH neuron spines. Histogram values are represented by mean ± SEM with dot plots of individual values. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.01; 2-way ANOVA with Tukey’s post hoc test. Flut, flutamide; GnRH, gonadotropin-releasing hormone.
Spine density was also remodeled by long-term AR blockade in PNA mice. GnRH neuron spine density in PNA+Flut was significantly decreased compared with the PNA+oil group (soma, P < 0.05; primary dendrite, P < 0.01; secondary dendrite, P < 0.01; F1,21 [interaction] = 19.2; F1,21 [treatment] = 23.2) (Figure 3K). In contrast to earlier reports (25, 26), spine density was not statistically different between control+oil and PNA+oil groups in this cohort, although PNA+oil spine density was significantly higher than control+Flut group at the soma (P < 0.05; F1,21 = 9.3) and primary dendrite (P < 0.01; F1,21 = 16.1) (Figure 3K).
Comparisons between prepubertal and adult mice revealed that PNA treatment was the major determinant for GABA-to-GnRH neuron circuit abnormalities (phenotype: PNA vs. control; F1,16 = 29.96; P < 0.0001), independent of age (age: PNA vs. control; F1,16 = 0.38; P = 0.55) (Supplemental Figure 4). Blocking AR signaling reduced the total VGAT apposition density on GnRH neurons by 53%, to a level comparable to controls. These data suggest that although enhanced GABAergic innervation to GnRH neurons in PCOS-like mice is programmed early, the system remains plastic in adults and can be completely reversed by AR signaling blockade.
Long-term AR blockade restores estrous cyclicity in PNA mice.
To determine whether the reversal in GABAergic brain wiring to GnRH neurons was associated with improved reproductive measures in PNA mice, estrous cycles were followed over the 20-day treatment period. All PNA+oil mice were acyclic with a complete absence of proestrus stage vaginal smears. In contrast, the PNA+Flut group displayed recurrent proestrus stage vaginal cytology (Figure 4A), at a frequency not different from controls (Figure 4B), suggesting at least partial restoration of ovulation and estrous cyclicity.
Figure 4. Blockade of androgen receptor (AR) signaling restores reproductive cycles in prenatally androgenized (PNA) mice.
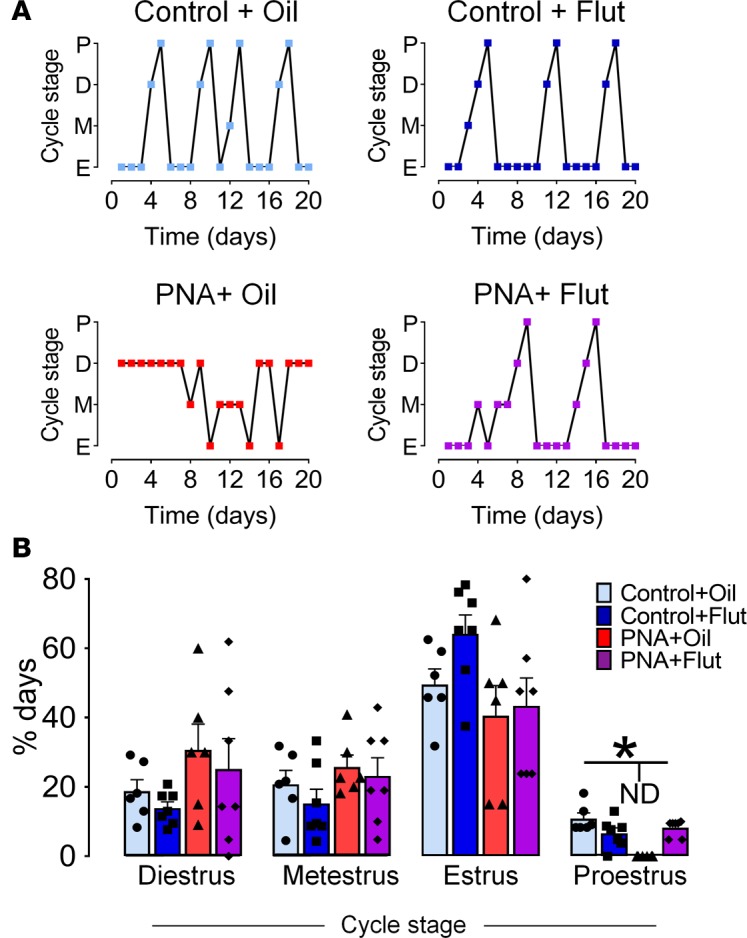
AR signaling blockade was achieved with the administration of flutamide (Flut) s.c. 25 mg/kg/day during 20 days. (A) Representative estrous cycle plots over 20 days from control+oil (N = 6), control+Flut (N = 7), PNA+oil (N = 6), and PNA+Flut (N = 7) groups. P, proestrus; D, diestrus; M, metestrus; E, estrus. (B) Histogram showing mean ± SEM percentage of days spent in each stage of the estrous cycle. ND, not detected.*P < 0.05; F1,23 = 20.5; 2-way ANOVA with Tukey’s post hoc test.
Long-term AR blockade improves the recruitment of preovulatory follicles and their morphological features in the ovaries of PNA mice.
PNA mice exhibit altered ovarian morphology, including small follicle arrest, decreased corpora lutea (CL), and theca cell hyperplasia, similar to PCOS women (25, 37). We investigated whether restoration of normal GnRH neuronal circuitry following AR blockade was associated with an improvement of ovarian morphology (Figure 5A). Investigating the first steps of folliculogenesis, we discovered that PNA mice, irrespective of Flut treatment, had a greater number of primordial follicles than controls (Figure 5B) (effect of the phenotype: P < 0.001; F1,21 = 23.84; effect of treatment: P = 0.89; F1,21 = 0.02). Confirming follicular arrest, PNA groups showed significantly fewer primary follicles when compared with the control+oil group (Figure 5B; P < 0.05). Flut treatment in PNA mice increased the number of primary follicles to levels similar to the control+Flut group (control+oil: 39.00 ± 3.56; control+Flut: 35.71 ± 3.30; PNA+oil: 16.60 ± 0.75; PNA+Flut: 26.17 ± 3.55 primary follicles per ovary), although numbers were not completely restored to control levels. The numbers of secondary and early antral follicles were not different across the 4 groups (Figure 5B). In addition, the total number of atretic follicles, regardless the stage of atresia, was similar among the 4 groups (Supplemental Figure 5B). These results reaffirm that PNA mice exhibit altered folliculogenesis and suggest that pharmacological blockade of androgen-mediated actions has minimal impact on the arrest of small follicles.
Figure 5. Androgen receptor (AR) blockade improves the recruitment of preovulatory follicles and their features in PNA mice.

Ovarian morphology of adult control and prenatally androgenized (PNA) mice undergoing s.c. injection with oil-vehicle or flutamide (Flut) 25 mg/kg/day from postnatal day (PND) 40 to PND 60. (A) First column: 5-μm-thick ovarian sections from control and PNA mice in diestrus. Corpora lutea are indicated by black asterisks. Scale bars: 500 μm. Second column: representative images of a preovulatory follicle wall from each group; yellow dashed lines delineate the theca cell layer (TLC) from granulosa cell layer (GCL). Scale bars: 50 μm. (B) Total number of primordial, primary, secondary, and antral follicles. (C) Total number of preovulatory follicles and corpora lutea. (D) Percentage of the follicle wall area made up of GCL and TLC from the largest preovulatory follicle. Different letters indicate significant statistical differences with P < 0.05; 2-way ANOVA followed by Tukey’s post hoc test. Histograms show mean ± SEM data from control+oil (N = 6), control+Flut (N = 7), PNA+oil (N = 5), and PNA+Flut (N = 6) groups.
Competent preovulatory follicles are able to ovulate in response to coordinated gonadotropin actions and an LH surge, which occurs on the evening of the proestrus stage in mice. In PCOS, LH hypersecretion and hyperandrogenic ovaries do not favor the appropriate recruitment of preovulatory follicles, leading to infertility (38, 39). As expected, we observed significantly fewer preovulatory follicles in PNA+oil mice compared with control+oil mice (PNA+oil: 7.40 ± 2.89 vs. control+oil: 26.86 ± 3.15 follicles per ovary; P < 0.001). Flut treatment resulted in divergent effects in PNA and control animals. Flut increased the number of preovulatory follicles in PNA mice (P < 0.01) to levels not different from control+oil animals, while the number of preovulatory follicles was reduced in control+Flut animals compared with controls treated with oil (F1,20 = 40.3; P < 0.01) (Figure 5C). Thus, AR signaling blockade can restore the number of preovulatory follicles in a hyperandrogenic state; however, direct androgen actions in the ovaries are also critical for the normal final steps of folliculogenesis (28).
In line with an absence of the proestrous cycle stage detected by vaginal cytology, most PNA mice showed dramatically fewer, and typical completely absent, CL compared with control mice (PNA+oil: 0.60 ± 0.24 vs. control+oil: 9.57 ± 1.09 CL per ovary; P < 0.0001). Although not identified as significantly different, PNA+Flut mice displayed an approximately 4-fold increase in the mean number of CL when compared with the PNA+oil group (PNA+oil: 0.6 ± 0.24 vs. PNA+Flut: 3.79 ± 0.8 CL per ovary; Figure 5C). The proportion of the ovary composed of CL was significantly lower in PNA+oil compared with control+oil (P < 0.001) and control+Flut groups (P < 0.05) (Supplemental Figure 5A). Flut treatment restored mean CL area to levels not different from control groups; however, the distribution of group data suggests that this period of Flut has fully restored reproductive capacity in some animals, while others are not fully restored. Together, these data suggest that long-term AR signaling blockade can increase the recruitment of preovulatory follicles, and ultimately support the return of ovulation, estrous cyclicity, and the capacity for fertility.
The composition of the preovulatory follicle wall thickness is indicative of gonadotropin-mediated actions in the ovaries (9, 25). Hence, the thickness of the granulosa cell layer (as a mean of determining follicle-stimulating hormone actions) and the theca cell layer (as a mean of determining LH actions) was measured. The granulosa cell layer was significantly thinner in PNA+oil mice compared with control groups (P < 0.05). Flut treatment restored the granulosa cell layer thickness to levels not different from controls (Figure 5D). The theca cell layer was significantly thicker in PNA+oil mice compared with controls (PNA+oil: 18.60% ± 1.51% vs. control+oil: 12.21% ± 0.57% vs. control+Flut: 12.51% ± 0.82% of the preovulatory follicle wall area; P < 0.05). This resembles the condition of ovarian hyperthecosis, a well-known feature of PCOS preovulatory follicles (10, 25, 40). Notably, AR signaling blockade significantly reduced the thickness of the theca cell layer in PNA mice to control levels (11.06% ± 0.97%; Figure 5D), suggesting the normalization of gonadotropin-mediated actions and by long-term AR blockade.
Discussion
Although defined by ovarian dysfunction, there is both long-standing and more recent evidence that the brain plays a critical role in the development and pathophysiology of PCOS (7, 8, 27). The present study elucidated the developmental timing and adult plasticity of disordered GABAergic innervation to GnRH neurons in a well-characterized PNA model of PCOS (23, 26, 41). This work demonstrated that enhanced GABAergic wiring onto GnRH neurons is established early, prior to the development of postpubertal circulating androgen excess, suggesting that early programming defects in the neural regulation of the hypothalamic-pituitary-gonadal axis precede the manifestation of neuroendocrine dysfunction in PCOS. This work also found that, despite early programming, disordered GABA-to-GnRH wiring and can be restored to control levels with long-term AR blockade, coincident with the resumption of estrous cyclicity and improved ovarian morphology.
Clinical and preclinical studies support the notion that high in utero androgen levels lead to the installation of a biological program in female offspring to develop PCOS (18, 21, 42), supporting the use of PNA to model PCOS here. This is most dramatically evident in women with congenital adrenal hyperplasia (CAH). Adrenal hyperplasia in utero leads to abnormally high prenatal androgen exposure and CAH women manifest PCOS symptoms in adulthood, including ovarian dysfunction, hyperandrogenism, and elevated LH levels (16, 17). In addition, exogenous administration of testosterone or dihydrotestosterone (DHT) to pregnant animal models can drive the development of PCOS-like ovarian and endocrine traits; this is well defined in sheep (43), monkeys (44), rats (45, 46), and mice (25, 26). In women, although the route of fetal exposure is not clearly defined, elevated maternal testosterone levels are linked to the development of PCOS in their daughters (21, 47). Serum testosterone levels increase by approximately 70% in pregnancy (48), and maternal testosterone is even greater in women with PCOS (49, 50). It is well known that the architecture of the fetal mammalian brain is exquisitely sensitive to androgen exposure and can program adult physiology (51). Low levels of ARs are expressed in the female hypothalamus during embryonic development (52, 53), and prenatal androgen exposure is well known to program life-long changes in synaptic patterning and adult sex behavior in rodents (54). Therefore, PNA-induced PCOS ontogeny may very well begin in the brain.
PNA mice model PCOS-like reproductive dysfunction, including disruption of estrous cycles, sub- and infertility, hyperandrogenism, elevated LH pulse frequency, impaired ovarian steroid-mediated negative feedback, and altered ovarian morphology (23, 25, 26, 41). Transgenic approaches in this model have identified greater GABAergic transmission (23) and innervation (26) to GnRH neurons that may underpin the desensitization of the GnRH pulse generator to steroid hormone feedback described in PCOS women (39). GnRH neurons do not express the hormone receptors required to respond directly to steroid hormone feedback, making them dependent on a steroid hormone–sensitive afferent neuronal network (55). GABA neurons in the arcuate nucleus project extensively to GnRH neurons and exhibit, in the PCOS-like condition, enhanced connections with GnRH neurons (26). Arcuate nucleus GABA neurons also express progesterone receptors and this expression is dramatically reduced in PNA mice (26). These data suggest that steroid hormone desensitization in arcuate nucleus GABA neurons is linked with plastic changes in GnRH afferents that may underlie the neuroendocrine impairments in PCOS. As GnRH neurons can respond to GABA with excitation (30, 31), increased GABA innervation may play a role in GnRH/LH hyperactivity in the PCOS condition. We found here that increased GABAergic contact to GnRH neurons is evident by PND 25 in PNA mice, well after prenatal exposure to elevated androgens, but prior to the development of postpubertal androgen excess (Figure 6). Although circulating levels of testosterone are not different in PNA animals at PND 25, it is currently unknown if local changes in androgen production in the brain over this period could contribute to the establishment or maintenance of these wiring changes.
Figure 6. Summary schematic of the ontogeny and plasticity of GABA-to-GnRH neuron circuit abnormalities in a preclinical model of PCOS.

Typically, female androgen levels are relatively low throughout pubertal development and into adulthood, with the exception of a small peripubertal rise (blue line). Elevated perinatal androgens can drive a hyperandrogenic polycystic ovary syndrome (PCOS) condition in adulthood (red line). GABA-to-GnRH neuron circuit abnormalities are present before the onset of puberty in prenatally androgenized PCOS-like mice, suggesting early programming of this aberrant wiring. Adult PCOS-like mice remain with enhanced GABAergic inputs onto GnRH neurons associated with an impairment of steroid hormone–mediated negative feedback, disruption of reproductive cycles, and ovarian dysfunction. Remarkably, GABAergic wiring to GnRH neurons is still plastic through androgen receptor signaling blockade, which also positively impacts reproductive cycles, and improves the recruitment and features of preovulatory follicles. AR, androgen receptor; GnRH, gonadotropin-releasing hormone.
In PND 25 PNA animals, GABA contact to GnRH neurons was significantly increased to levels similar to those identified in adult PNA mice. In contrast to what has been previously reported in PNA adults (25), GnRH neuron spine density was not increased by PND 25. In adults, the proportion of GABAergic inputs onto GnRH neuron spines is increased in addition to increased spine density (26). Here, VGAT appositions to spines in PND 25 animals were elevated only at somatic spines, while the majority of increased presynaptic GABA contact to GnRH neurons was to the non-spiny portion of the somatic cell membrane and dendritic shaft. In most immature neurons, GABA actions via GABAAR are excitatory through a differential chloride (Cl–) homeostasis, by which cells maintain higher intracellular Cl– concentration and a more depolarizing reversal potential for Cl– than the resting membrane potential (56). Throughout the brain, excitatory actions of GABA during early development are critical to induce de novo synapse formation and increase the number of dendritic spines through the regulation of intracellular calcium signaling (36, 57). The enhanced GABAergic input identified at PND 25 may precede an increase in spine density that develops as a consequence of elevated responses to GABAergic signaling that develop later in adulthood. In addition, while these data show that VGAT contact is elevated at GnRH neurons, it remains to be determined if the majority of this elevated input comes from GABA neurons originating in the arcuate nucleus as shown previously in adults (24).
Although LH is low throughout prepubertal life, GnRH neurons are highly active throughout this period (58) and undergo dramatic morphological and synaptic plasticity prior to pubertal onset (33). Two very recent reports out of the Moenter laboratory have reported that both GnRH neuron firing activity (58) and GABAergic transmission to GnRH neurons (59) are present at 1 week of age and peak around 3 weeks of age, corresponding closely to the time point investigated here. Berg et al. have shown that GABAergic inhibitory post-synaptic currents (IPSCs) in PNA females are higher than controls at 3 weeks of age, lending support to our anatomical findings. However, they also report that GnRH firing frequency and response to GABA application was unexpectedly blunted in PNA mice when compared with control females (58, 59). It is tempting to speculate that increased GABAergic synaptic contact and synaptic transmission would be coincident with increased GnRH neuron activity even in the juvenile period. However, the relationship between GnRH neurons and their developing neuronal network might be highly dynamic throughout the prepubertal period, and it is unclear whether GnRH neuron activity or afferent GnRH neuronal network stability comes first. The lack of increased GnRH neuron firing activity at 3 weeks of age despite increased synaptic transmission (59) is suggestive of compensatory postsynaptic mechanisms that remain to be clearly determined. These compensatory mechanisms are lost over pubertal development, as responses to GABA are not different in adults and postpubertal PNA mice display both increased GABAergic postsynaptic currents in GnRH neurons and elevated GnRH firing frequency (23). Together, these data suggest that aberrant synaptic organization develops early in PNA mice, established by 3 weeks of age, but remains under homeostatic control mechanisms that are ultimately lost over pubertal development. Understanding the mechanisms underpinning this loss remain will be important in understanding what ultimately enables a hyperactive GnRH/LH system in the PNA PCOS-like condition.
Serial measurements of blood testosterone levels in PNA mice indicated that circulating androgen excess does not develop until PND 50. Although pubertal onset is reported as both precocious and delayed in this model, all mice display a first estrous by PND 40 or earlier (25). Androgen excess is therefore likely to be downstream from altered GnRH/LH secretion that drives the establishment of hyperandrogenic ovaries in PCOS (3, 10, 40). In girls, prepubertal adrenarche leading to precocious exposure to increased androgen levels correlates with a higher risk of developing PCOS and hyperandrogenic ovaries (60, 61). In rodents, the adrenal glands do not possess the molecular machinery necessary to synthesize androgens (62), indicating that PNA-induced androgen excess develops largely from an ovarian source. The brain also produces androgens, expressing significant levels of P450 aromatase to convert local or circulating estrogens into testosterone in situ. Fluctuations in brain androgen levels are evident in the perinatal period in female rodents, likely impacting synaptogenesis, but then remain relatively low and unchanging in adulthood (63). It is currently unknown whether PNA changes the production of androgens in the brain at any stage. Regardless of the source, precocious exposure to androgens leads to aberrant GnRH neuron wiring that precedes the development of postpubertal hyperandrogenic ovaries, supporting a developmental programming of disease progression. In support of a top-down manifestation of the peripheral disorder, large genetic analysis studies have identified polymorphisms in the gonadotropin receptors associated with neuroendocrine dysfunction in PCOS (64, 65).
Despite the evidence supporting early organization of wiring defects, long-term androgen signaling blockade from pubertal onset completely reversed the elevated GABA-to-GnRH neuron wiring and dramatically ameliorated PCOS reproductive dysfunction (Figure 6). Previous studies have indicated that AR blockade of androgen signaling can alleviate PCOS-associated dysfunction. One week of pharmacological AR blockade with Flut can prevent the increased GABAergic postsynaptic currents measured in adult GnRH neurons from PNA mice (23). Additionally, long-term Flut treatment in women with PCOS can improve progesterone sensitivity to restore the ability of estrogen and progesterone to reduce LH pulse frequency (13). Impaired progesterone-mediated negative feedback is evident in PNA mice and progesterone receptor expression is reduced in GABA neurons that project to GnRH neurons (26). It is unclear whether AR blockade directly improves the progesterone sensitivity of GABA neurons; however, the present findings suggest that remodeling of GABA inputs to GnRH neurons is associated with a restoration of gonadal steroid hormone negative feedback. In addition, AR blockade was able to restore estrous cyclicity in all PNA mice. Although Flut will block androgen actions in multiple tissues, including the ovary, recent evidence shows that AR signaling in the brain is required for hyperandrogenism to drive the full presentation of PCOS-like features in a postnatal model of the disease (66). This suggests that Flut blockade of AR signaling in the brain is essential to the reversal of the PCOS-like phenotype demonstrated here. It will be important to assess in future studies the impact of androgen excess and AR blockade on a cellular level in the brain.
Prenatal exposure to androgens ultimately leads to ovarian dysfunction; this is evident in women with CAH (16) and in different preclinical models of PCOS (18, 25, 67). While AR blockade did not change the follicular arrest of small follicles (primordial and primary) typical of PCOS ovaries (37), a marked effect was observed in recovering the number of preovulatory follicles. AR signaling blockade correlated with a recovery of normal granulosa and theca cell layer thickness. Theca cell hyperplasia is directly related to increased theca cell androgen production and is typical of PCOS ovaries (10, 40). Long-term AR blockade reduced the thickness of the theca cell layer to control levels, probably reflecting a reduced LH pulse frequency drive and resulting in decreased androgen production. Granulosa cells are the main target of androgen actions in preovulatory follicles (68); however, granulosa cell–specific AR-knockout does not protect mice from developing the PCOS phenotype with chronic DHT administration (66). This indicates that the restoration of normal follicle morphology after AR blockade is more likely to be indirect, through the normalization of gonadotropin action, which may reflect normalized regulation of GnRH neuron activity by GABAergic neurons.
The link between GABA actions, androgen excess, and ovarian dysfunction are not only evident in preclinical models of PCOS. Women with PCOS have recently been reported to have elevated cerebrospinal fluid levels of GABA associated with androgen excess (28). There is also evidence that PCOS patients have increased levels of allopregnanolone, a GABAAR positive modulator, and display impaired sensitivity to its sedation effects (69). Likewise, epileptic patients undergoing treatment with valproate drugs to boost GABAergic tone in the brain for the control of seizures, usually present PCOS features such as hyperandrogenism and menstrual irregularities (29, 70). Therefore, altered GABAergic signaling may be central to the development of PCOS reproductive abnormalities in women. Our findings showed that altered GABA wiring to GnRH neurons develops prior to the onset of PCOS-like reproductive dysfunction and supports the idea that these abnormalities may be a primary mediator of disease development downstream from prenatal androgen exposure. The ability of AR blockade to restore normal GABA-to-GnRH innervation and rescue downstream reproductive function indicate that circuit abnormalities remain plastic in adulthood and are dependent on ongoing androgen signaling. Together, our observations suggest that early blockade of androgen signaling in PCOS might be able to restore a normal GnRH neuronal network and highlight AR antagonism as a therapeutic target.
Methods
Animals.
Female C57BL/6J and GnRH-GFP mice (71) were housed with ad libitum access to water and food, and under a 12-hour light/dark cycle. To generate PNA mice, we used established protocols as previously described (23, 25). Briefly, time-mated pregnant dams were injected with 100 μl of DHT (250 μg) in oil, or oil alone to serve as a control, on gestational days 16, 17, and 18.
Experiment 1: Temporal assessment of plasma testosterone and estrous cyclicity.
Control (N = 10) and PNA (N = 9) mice were followed from PND 30 to PND 80. At PND 30, 40, 50, and 60, mice were handled and tail-tip blood samples were collected from 9–10 AM. Blood samples were spun at 4 g for 10 minutes at room temperature and plasma was collected and kept at –20°C until the day of hormone measurement. From PND 60 to PND 80, estrous cyclicity was followed by daily vaginal smears and examination of the vaginal cytology (72).
Experiment 2: Prepubertal assessment of GABAergic input to GnRH neurons.
On PND 25, control (N = 4) and PNA (N = 5) GnRH-GFP female offspring were euthanized by an overdose of sodium pentobarbital (30 mg/ml). Blood was collected from the vena cava just prior to transcardial perfusion with 4% buffered paraformaldehyde (0.1 M phosphate buffer-PFA 4%; pH 7.6). Brains were dissected, postfixed for 1 hour at room temperature, and saturated in 30% Tris-buffered sucrose at 4°C until processed for immunohistochemistry.
Experiment 3: Establishing the antiandrogenic effect of Flut dose.
Male (N = 4) and female (N = 4) C57BL6 mice were injected daily with 100 μl of Flut at a dose of 25 mg/kg/day for 15 days. Control vehicle animals received 100 μl of sesame oil during the same period. Male mice were used as biological control to assess the effectiveness of Flut treatment according to previously published studies (73, 74). Seminal vesicles and adrenal gland weights were measured from treated intact male mice, and ovaries and adrenal gland weights were measured from treated female mice.
Experiment 4: Assessing androgen dependence of central and peripheral PCOS features.
Control and PNA GnRH-GFP mice received daily 50 μl injections of either Flut (Sigma-Aldrich; s.c. 25 mg/kg/day) or a vehicle control (sesame oil) for 20–22 days from PND 40. Groups were identified as control+oil (N = 6), control+Flut (N = 7), PNA+oil (N = 5), and PNA+Flut (N = 7). All mice were monitored daily for estrous cyclicity through vaginal smears. Following at least 20 days of treatment, animals in diestrus were euthanized and perfused as above. Brains were dissected and treated as above until processing for immunohistochemistry. Ovaries were postfixed for 24 hours and then transferred to 70% ethanol at room temperature until paraffin embedding for histology.
Immunohistochemistry.
Fixed brain tissue was cut into 3 sets of 30-μm-thick coronal sections using a freezing microtome. Coronal brain sections including the rPOA and organum vasculosum of lamina terminalis (OVLT) of the hypothalamus were selected according to the mouse brain atlas (75). Free-floating immunohistochemistry was performed as previously described (26, 33) on one set of sections (including every third brain section). Primary antibodies were used at the respective concentrations: polyclonal chicken anti-GFP (1:2,500; Aves Labs Inc., catalog GFP-1020) and polyclonal rabbit anti-VGAT (1:750, Synaptic Systems, catalog 131003). Brain sections were incubated with a cocktail of primary antibodies and 2% normal goat serum for 48 hours at 4°C. Alexa Fluor 488 goat anti-chicken (1:200, catalog A-11039) and Streptavidin 568 (1:200, catalog S11226) (both Molecular Probes, Invitrogen) were used as secondary antibodies. Primary antibody omission served as a negative control.
Confocal image analysis.
Following double immunofluorescent labeling of VGAT and GFP, the density of VGAT appositions onto GFP-ir GnRH neurons was quantified in 2 representative sections from the rPOA/OVLT region. Confocal images were collected on a Nikon A1R multi-photon confocal microscope with 488- and 543-nm diode lasers. Brain sections were imaged using a Plan NeoFluor ×40 oil objective (1.30 NA), taking Z-stacks of 0.5 μm thickness (pinhole at 1 AU) and analyzed with software NIS-Elements AR 4.00.00 (Nikon Instruments Inc.). Ten to 12 GnRH neurons located in the rPOA from each animal were randomly selected and scanned for analysis. VGAT apposition density was recorded for the GnRH neuron soma and in 15-μm intervals of the primary and secondary dendrite out to 75 μm as previously reported (26).
Ovarian histology analysis.
All ovaries were collected from control and PNA mice in diestrus and one ovary was randomly chosen for morphological analysis as previously reported (25). Groups were identified as control+oil (N = 6), control+Flut (N = 7), PNA+oil (N = 5), and PNA+Flut (N = 6). Briefly, ovarian sections of 5-μm thickness were collected in 50-μm intervals and stained with hematoxylin and eosin. Sections were visualized with a light microscope (Olympus BX51) and ovarian follicle types and numbers were assessed in every collected section from the whole ovary using ×10 and ×20 objectives (76). Follicle wall thickness was determined in the largest preovulatory follicle from the ovarian section with the largest radius. The total follicle wall area and the area made up of granulosa or theca cell layers were defined using ImageJ software (NIH) and the percentage of the layer thickness in relation to the total follicle area was calculated. Using ImageJ, the percentage area containing CL was calculated from the ovarian section with the largest radius.
Hormone measurements.
Plasma testosterone levels were determined using a commercially available mouse/rat ELISA kit (LDN). The assay sensitivity is 0.066 ng/ml at the 2 standard deviation confidence limit. Intra-assay coefficient of variation for testosterone was 10.3% and inter-assay coefficient of variation was 11.0%. Fifty microliters of whole blood were collected from the tail via a hematocrit tube to obtain 10 μl of plasma for the testosterone assay, run in duplicate samples. LH levels were determined using well-established ELISA methods (26, 77, 78). A mouse LH-RP reference was provided by Albert F. Parlow (National Hormone and Pituitary Program, Torrance, California, USA). The assay sensitivity of the LH ELISA was 0.04 ng/ml and intra-assay coefficient of variation was 4.3%.
Statistics.
Statistical analysis was performed with PRISM software 7.0 (GraphPad Software). Testosterone levels throughout pubertal development of control and PNA mice were compared using repeated-measures 2-way ANOVA with Sidak’s multiple comparison post hoc test. Evaluation of prepubertal PND 25 testosterone levels was performed with a 2-tailed Student’s t test. Analysis of VGAT appositions onto GnRH neurons from prepubertal control and PNA females used the Mann-Whitney U test. Adult control and PNA mice were compared using 2-way ANOVA with Tukey’s post hoc test when studying GABA inputs on GnRH neurons and ovarian morphology features. A 2-way ANOVA with Bonferroni’s post hoc test was used to compare prepubertal and adult groups. All data are represented as mean ± SEM. Statistical significance was accepted when P < 0.05. Graphical representation to display significant different are shown as * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001.
Study approval.
All protocols were approved by the University of Otago Animal Ethics Committee (Dunedin, New Zealand).
Author contributions
MSBS contributed to experimental design, conducted and analyzed the majority of experiments, and prepared the majority of the manuscript. MP was fundamental to developing the PCOS model, conducted some experiments, analyzed data, and edited the manuscript. REC conceived and funded the project, supervised the experiments and analysis, provided scientific oversight, and cowrote the manuscript with MSBS.
Supplementary Material
Acknowledgments
This work was supported by the Royal Society Marsden Fund and the Health Research Council of New Zealand. We are grateful to the following laboratory members for their editorial comments on this manuscript: Elodie Desroziers, Eulalia Coutinho, Bradley Jamison, Sarah Holland, and Sabine Hessler.
Version 1. 04/05/2018
Electronic publication
Funding Statement
14/077
15/097
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(7):e99405. https://doi.org/10.1172/jci.insight.99405.
References
- 1.Lizneva D, Suturina L, Walker W, Brakta S, Gavrilova-Jordan L, Azziz R. Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil Steril. 2016;106(1):6–15. doi: 10.1016/j.fertnstert.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81(1):19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Dumesic DA, Oberfield SE, Stener-Victorin E, Marshall JC, Laven JS, Legro RS. Scientific statement on the diagnostic criteria, epidemiology, pathophysiology, and molecular genetics of polycystic ovary syndrome. Endocr Rev. 2015;36(5):487–525. doi: 10.1210/er.2015-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rebar R, Judd HL, Yen SS, Rakoff J, Vandenberg G, Naftolin F. Characterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest. 1976;57(5):1320–1329. doi: 10.1172/JCI108400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83(2):582–590. doi: 10.1210/jcem.83.2.4604. [DOI] [PubMed] [Google Scholar]
- 6.Moore AM, Campbell RE. Polycystic ovary syndrome: Understanding the role of the brain. Front Neuroendocrinol. 2017;46:1–14. doi: 10.1016/j.yfrne.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Chhabra S, McCartney CR, Yoo RY, Eagleson CA, Chang RJ, Marshall JC. Progesterone inhibition of the hypothalamic gonadotropin-releasing hormone pulse generator: evidence for varied effects in hyperandrogenemic adolescent girls. J Clin Endocrinol Metab. 2005;90(5):2810–2815. doi: 10.1210/jc.2004-2359. [DOI] [PubMed] [Google Scholar]
- 8.Daniels TL, Berga SL. Resistance of gonadotropin releasing hormone drive to sex steroid-induced suppression in hyperandrogenic anovulation. J Clin Endocrinol Metab. 1997;82(12):4179–4183. doi: 10.1210/jcem.82.12.4402. [DOI] [PubMed] [Google Scholar]
- 9.Gilling-Smith C, Willis DS, Beard RW, Franks S. Hypersecretion of androstenedione by isolated thecal cells from polycystic ovaries. J Clin Endocrinol Metab. 1994;79(4):1158–1165. doi: 10.1210/jcem.79.4.7962289. [DOI] [PubMed] [Google Scholar]
- 10.Eisner JR, Barnett MA, Dumesic DA, Abbott DH. Ovarian hyperandrogenism in adult female rhesus monkeys exposed to prenatal androgen excess. Fertil Steril. 2002;77(1):167–172. doi: 10.1016/S0015-0282(01)02947-8. [DOI] [PubMed] [Google Scholar]
- 11.Jonard S, Dewailly D. The follicular excess in polycystic ovaries, due to intra-ovarian hyperandrogenism, may be the main culprit for the follicular arrest. Hum Reprod Update. 2004;10(2):107–117. doi: 10.1093/humupd/dmh010. [DOI] [PubMed] [Google Scholar]
- 12.Dunaif A. Do androgens directly regulate gonadotropin secretion in the polycystic ovary syndrome? J Clin Endocrinol Metab. 1986;63(1):215–221. doi: 10.1210/jcem-63-1-215. [DOI] [PubMed] [Google Scholar]
- 13.Eagleson CA, et al. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85(11):4047–4052. doi: 10.1210/jcem.85.11.6992. [DOI] [PubMed] [Google Scholar]
- 14.Azziz R, et al. Polycystic ovary syndrome. Nat Rev Dis Primers. 2016;2:16057. doi: 10.1038/nrdp.2016.57. [DOI] [PubMed] [Google Scholar]
- 15.Barber TM, Dimitriadis GK, Andreou A, Franks S. Polycystic ovary syndrome: insight into pathogenesis and a common association with insulin resistance. Clin Med (Lond) 2015;15 Suppl 6:s72–s76. doi: 10.7861/clinmedicine.15-6-s72. [DOI] [PubMed] [Google Scholar]
- 16.Hague WM, et al. The prevalence of polycystic ovaries in patients with congenital adrenal hyperplasia and their close relatives. Clin Endocrinol (Oxf) 1990;33(4):501–510. doi: 10.1111/j.1365-2265.1990.tb03887.x. [DOI] [PubMed] [Google Scholar]
- 17.Barnes RB, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79(5):1328–1333. doi: 10.1210/jcem.79.5.7962325. [DOI] [PubMed] [Google Scholar]
- 18.Dumesic DA, Abbott DH, Eisner JR, Goy RW. Prenatal exposure of female rhesus monkeys to testosterone propionate increases serum luteinizing hormone levels in adulthood. Fertil Steril. 1997;67(1):155–163. doi: 10.1016/S0015-0282(97)81873-0. [DOI] [PubMed] [Google Scholar]
- 19.Padmanabhan V, Veiga-Lopez A. Animal models of the polycystic ovary syndrome phenotype. Steroids. 2013;78(8):734–740. doi: 10.1016/j.steroids.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNeilly AS, Duncan WC. Rodent models of polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373(1–2):2–7. doi: 10.1016/j.mce.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Sir-Petermann T, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94(6):1923–1930. doi: 10.1210/jc.2008-2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maliqueo M, et al. Relationship of serum adipocyte-derived proteins with insulin sensitivity and reproductive features in pre-pubertal and pubertal daughters of polycystic ovary syndrome women. Eur J Obstet Gynecol Reprod Biol. 2012;161(1):56–61. doi: 10.1016/j.ejogrb.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 23.Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: implications for a common fertility disorder. Proc Natl Acad Sci USA. 2004;101(18):7129–7134. doi: 10.1073/pnas.0308058101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roland AV, Moenter SM. Prenatal androgenization of female mice programs an increase in firing activity of gonadotropin-releasing hormone (GnRH) neurons that is reversed by metformin treatment in adulthood. Endocrinology. 2011;152(2):618–628. doi: 10.1210/en.2010-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore AM, Prescott M, Campbell RE. Estradiol negative and positive feedback in a prenatal androgen-induced mouse model of polycystic ovarian syndrome. Endocrinology. 2013;154(2):796–806. doi: 10.1210/en.2012-1954. [DOI] [PubMed] [Google Scholar]
- 26.Moore AM, Prescott M, Marshall CJ, Yip SH, Campbell RE. Enhancement of a robust arcuate GABAergic input to gonadotropin-releasing hormone neurons in a model of polycystic ovarian syndrome. Proc Natl Acad Sci USA. 2015;112(2):596–601. doi: 10.1073/pnas.1415038112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore AM, Campbell RE. The neuroendocrine genesis of polycystic ovary syndrome: A role for arcuate nucleus GABA neurons. J Steroid Biochem Mol Biol. 2016;160:106–117. doi: 10.1016/j.jsbmb.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 28.Kawwass JF, Sanders KM, Loucks TL, Rohan LC, Berga SL. Increased cerebrospinal fluid levels of GABA, testosterone and estradiol in women with polycystic ovary syndrome. Hum Reprod. 2017;32(7):1450–1456. doi: 10.1093/humrep/dex086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bilo L, Meo R. Polycystic ovary syndrome in women using valproate: a review. Gynecol Endocrinol. 2008;24(10):562–570. doi: 10.1080/09513590802288259. [DOI] [PubMed] [Google Scholar]
- 30.DeFazio RA, Heger S, Ojeda SR, Moenter SM. Activation of A-type gamma-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16(12):2872–2891. doi: 10.1210/me.2002-0163. [DOI] [PubMed] [Google Scholar]
- 31.Herbison AE, Moenter SM. Depolarising and hyperpolarising actions of GABA(A) receptor activation on gonadotrophin-releasing hormone neurones: towards an emerging consensus. J Neuroendocrinol. 2011;23(7):557–569. doi: 10.1111/j.1365-2826.2011.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389(6653):870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- 33.Cottrell EC, Campbell RE, Han SK, Herbison AE. Postnatal remodeling of dendritic structure and spine density in gonadotropin-releasing hormone neurons. Endocrinology. 2006;147(8):3652–3661. doi: 10.1210/en.2006-0296. [DOI] [PubMed] [Google Scholar]
- 34.Herde MK, Herbison AE. Morphological characterization of the action potential initiation segment in GnRH neuron dendrites and axons of male mice. Endocrinology. 2015;156(11):4174–4186. doi: 10.1210/en.2015-1284. [DOI] [PubMed] [Google Scholar]
- 35.Harris KM, Weinberg RJ. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb Perspect Biol. 2012;4(5):a005587. doi: 10.1101/cshperspect.a005587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh WC, Lutzu S, Castillo PE, Kwon HB. De novo synaptogenesis induced by GABA in the developing mouse cortex. Science. 2016;353(6303):1037–1040. doi: 10.1126/science.aaf5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webber LJ, et al. Formation and early development of follicles in the polycystic ovary. Lancet. 2003;362(9389):1017–1021. doi: 10.1016/S0140-6736(03)14410-8. [DOI] [PubMed] [Google Scholar]
- 38.Dumesic DA, Richards JS. Ontogeny of the ovary in polycystic ovary syndrome. Fertil Steril. 2013;100(1):23–38. doi: 10.1016/j.fertnstert.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update. 2006;12(4):351–361. doi: 10.1093/humupd/dml017. [DOI] [PubMed] [Google Scholar]
- 40.Hughesdon PE. Morphology and morphogenesis of the Stein-Leventhal ovary and of so-called “hyperthecosis”. Obstet Gynecol Surv. 1982;37(2):59–77. doi: 10.1097/00006254-198202000-00001. [DOI] [PubMed] [Google Scholar]
- 41.Witham EA, Meadows JD, Shojaei S, Kauffman AS, Mellon PL. Prenatal exposure to low levels of androgen accelerates female puberty onset and reproductive senescence in mice. Endocrinology. 2012;153(9):4522–4532. doi: 10.1210/en.2012-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cresswell JL, Barker DJ, Osmond C, Egger P, Phillips DI, Fraser RB. Fetal growth, length of gestation, and polycystic ovaries in adult life. Lancet. 1997;350(9085):1131–1135. doi: 10.1016/S0140-6736(97)06062-5. [DOI] [PubMed] [Google Scholar]
- 43.Padmanabhan V, Veiga-Lopez A. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol. 2013;373(1–2):8–20. doi: 10.1016/j.mce.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update. 2005;11(4):357–374. doi: 10.1093/humupd/dmi013. [DOI] [PubMed] [Google Scholar]
- 45.Sun M, et al. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. Am J Physiol Endocrinol Metab. 2012;303(11):E1373–E1385. doi: 10.1152/ajpendo.00421.2012. [DOI] [PubMed] [Google Scholar]
- 46.Ramezani Tehrani F, Noroozzadeh M, Zahediasl S, Piryaei A, Hashemi S, Azizi F. The time of prenatal androgen exposure affects development of polycystic ovary syndrome-like phenotype in adulthood in female rats. Int J Endocrinol Metab. 2014;12(2):e16502. doi: 10.5812/ijem.16502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maliqueo M, et al. Adrenal function during childhood and puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94(9):3282–3288. doi: 10.1210/jc.2009-0427. [DOI] [PubMed] [Google Scholar]
- 48.O’Leary P, Boyne P, Flett P, Beilby J, James I. Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clin Chem. 1991;37(5):667–672. [PubMed] [Google Scholar]
- 49.Maliqueo M, et al. Placental STAT3 signaling is activated in women with polycystic ovary syndrome. Hum Reprod. 2015;30(3):692–700. doi: 10.1093/humrep/deu351. [DOI] [PubMed] [Google Scholar]
- 50.Caanen MR, et al. Mass spectrometry methods measured androgen and estrogen concentrations during pregnancy and in newborns of mothers with polycystic ovary syndrome. Eur J Endocrinol. 2016;174(1):25–32. doi: 10.1530/EJE-15-0699. [DOI] [PubMed] [Google Scholar]
- 51.Robinson J. Prenatal programming of the female reproductive neuroendocrine system by androgens. Reproduction. 2006;132(4):539–547. doi: 10.1530/rep.1.00064. [DOI] [PubMed] [Google Scholar]
- 52.Mogi K, Takanashi H, Nagasawa M, Kikusui T. Sex differences in spatiotemporal expression of AR, ERα, and ERβ mRNA in the perinatal mouse brain. Neurosci Lett. 2015;584:88–92. doi: 10.1016/j.neulet.2014.10.028. [DOI] [PubMed] [Google Scholar]
- 53.Brock O, De Mees C, Bakker J. Hypothalamic expression of oestrogen receptor α and androgen receptor is sex-, age- and region-dependent in mice. J Neuroendocrinol. 2015;27(4):264–276. doi: 10.1111/jne.12258. [DOI] [PubMed] [Google Scholar]
- 54.McCarthy MM. How it’s made: organisational effects of hormones on the developing brain. J Neuroendocrinol. 2010;22(7):736–742. doi: 10.1111/j.1365-2826.2010.02021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wintermantel TM, et al. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52(2):271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3(9):728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- 57.Kirmse K, Kummer M, Kovalchuk Y, Witte OW, Garaschuk O, Holthoff K. GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat Commun. 2015;6:7750. doi: 10.1038/ncomms8750. [DOI] [PubMed] [Google Scholar]
- 58.Dulka EA, Moenter SM. Prepubertal development of gonadotropin-releasing hormone neuron activity is altered by sex, age, and prenatal androgen exposure. Endocrinology. 2017;158(11):3943–3953. doi: 10.1210/en.2017-00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berg T, Silveira MA, Moenter SM. Prepubertal development of GABAergic Transmission to gonadotropin-releasing hormone (GnRH) neurons and postsynaptic response are altered by prenatal androgenization. J Neurosci. 2018;38(9):2283–2293. doi: 10.1523/JNEUROSCI.2304-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vuguin P, Linder B, Rosenfeld RG, Saenger P, DiMartino-Nardi J. The roles of insulin sensitivity, insulin-like growth factor I (IGF-I), and IGF-binding protein-1 and -3 in the hyperandrogenism of African-American and Caribbean Hispanic girls with premature adrenarche. J Clin Endocrinol Metab. 1999;84(6):2037–2042. doi: 10.1210/jcem.84.6.5722. [DOI] [PubMed] [Google Scholar]
- 61.Silfen ME, Manibo AM, Ferin M, McMahon DJ, Levine LS, Oberfield SE. Elevated free IGF-I levels in prepubertal Hispanic girls with premature adrenarche: relationship with hyperandrogenism and insulin sensitivity. J Clin Endocrinol Metab. 2002;87(1):398–403. doi: 10.1210/jcem.87.1.8143. [DOI] [PubMed] [Google Scholar]
- 62.van Weerden WM, Bierings HG, van Steenbrugge GJ, de Jong FH, Schröder FH. Adrenal glands of mouse and rat do not synthesize androgens. Life Sci. 1992;50(12):857–861. doi: 10.1016/0024-3205(92)90204-3. [DOI] [PubMed] [Google Scholar]
- 63.Konkle AT, McCarthy MM. Developmental time course of estradiol, testosterone, and dihydrotestosterone levels in discrete regions of male and female rat brain. Endocrinology. 2011;152(1):223–235. doi: 10.1210/en.2010-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Day FR, et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Commun. 2015;6:8464. doi: 10.1038/ncomms9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cui L, et al. Genotype-phenotype correlations of PCOS susceptibility SNPs identified by GWAS in a large cohort of Han Chinese women. Hum Reprod. 2013;28(2):538–544. doi: 10.1093/humrep/des424. [DOI] [PubMed] [Google Scholar]
- 66.Caldwell ASL, et al. Neuroendocrine androgen action is a key extraovarian mediator in the development of polycystic ovary syndrome. Proc Natl Acad Sci USA. 2017;114(16):E3334–E3343. doi: 10.1073/pnas.1616467114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith P, Steckler TL, Veiga-Lopez A, Padmanabhan V. Developmental programming: differential effects of prenatal testosterone and dihydrotestosterone on follicular recruitment, depletion of follicular reserve, and ovarian morphology in sheep. Biol Reprod. 2009;80(4):726–736. doi: 10.1095/biolreprod.108.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sen A, et al. Androgens regulate ovarian follicular development by increasing follicle stimulating hormone receptor and microRNA-125b expression. Proc Natl Acad Sci USA. 2014;111(8):3008–3013. doi: 10.1073/pnas.1318978111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hedström H, et al. Women with polycystic ovary syndrome have elevated serum concentrations of and altered GABA(A) receptor sensitivity to allopregnanolone. Clin Endocrinol (Oxf) 2015;83(5):643–650. doi: 10.1111/cen.12809. [DOI] [PubMed] [Google Scholar]
- 70.Hu X, Wang J, Dong W, Fang Q, Hu L, Liu C. A meta-analysis of polycystic ovary syndrome in women taking valproate for epilepsy. Epilepsy Res. 2011;97(1–2):73–82. doi: 10.1016/j.eplepsyres.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 71.Spergel DJ, Krüth U, Hanley DF, Sprengel R, Seeburg PH. GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci. 1999;19(6):2037–2050. doi: 10.1523/JNEUROSCI.19-06-02037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caligioni CS. Assessing reproductive status/stages in mice. Curr Protoc Neurosci. 2009;Appendix 4:Appendix 4I. doi: 10.1002/0471142301.nsa04is48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luo S, et al. Daily dosing with flutamide or Casodex exerts maximal antiandrogenic activity. Urology. 1997;50(6):913–919. doi: 10.1016/S0090-4295(97)00393-2. [DOI] [PubMed] [Google Scholar]
- 74.Poyet P, Labrie F. Comparison of the antiandrogenic/androgenic activities of flutamide, cyproterone acetate and megestrol acetate. Mol Cell Endocrinol. 1985;42(3):283–288. doi: 10.1016/0303-7207(85)90059-0. [DOI] [PubMed] [Google Scholar]
- 75.Xiong B, et al. Precise cerebral vascular atlas in stereotaxic coordinates of whole mouse brain. Front Neuroanat. 2017;11:128. doi: 10.3389/fnana.2017.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Myers M, Britt KL, Wreford NG, Ebling FJ, Kerr JB. Methods for quantifying follicular numbers within the mouse ovary. Reproduction. 2004;127(5):569–580. doi: 10.1530/rep.1.00095. [DOI] [PubMed] [Google Scholar]
- 77.Steyn FJ, Wan Y, Clarkson J, Veldhuis JD, Herbison AE, Chen C. Development of a methodology for and assessment of pulsatile luteinizing hormone secretion in juvenile and adult male mice. Endocrinology. 2013;154(12):4939–4945. doi: 10.1210/en.2013-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Czieselsky K, et al. Pulse and Surge Profiles of Luteinizing Hormone Secretion in the Mouse. Endocrinology. 2016;157(12):4794–4802. doi: 10.1210/en.2016-1351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.