

Abstract
Inactivation of NF2/Merlin causes the autosomal dominant cancer predisposition syndrome Familial Neurofibromatosis Type 2 (NF2) and contributes to the development of malignant pleural mesothelioma (MPM). In order to develop a targeted therapy for NF2-mutant tumors, we have exploited the recent realization that Merlin loss drives tumorigenesis by activating the E3 ubiquitin ligase CRL4DCAF1 – thereby inhibiting the Hippo pathway component Lats. Here, we show that MLN4924 – a NEDD8 activating enzyme (NAE) inhibitor – suppresses CRL4DCAF1 and attenuates activation of YAP in NF2-mutant tumor cells. Additionally, MLN4924 sensitizes MPM to traditional chemotherapy, presumably as a result of collateral inhibition of cullin-RING ubiquitin ligases (CRLs) involved in DNA repair. However, even in combination with chemotherapy, MLN4924 does not exhibit significant preclinical activity. Further analysis revealed that depletion of DCAF1 or treatment with MLN4924 does not affect mTOR hyperactivation in NF2-mutant tumor cells, suggesting that loss of Merlin activates mTOR independently of CRL4DCAF1. Intriguingly, combining MLN4924 with the mTOR/PI3K inhibitor GDC-0980 suppresses the growth of NF2-mutant tumor cells in vitro as well as in mouse and patient-derived xenografts. These results provide preclinical rationale for the use of NAE inhibitors in combination with mTOR/PI3K inhibitors in NF2-mutant tumors.
Keywords: NF2/Merlin, CRL4DCAF1, mesothelioma, mTOR, MLN4924
INTRODUCTION
Loss-of-function NF2/Merlin mutations are among the most frequent events driving malignant pleural mesothelioma (MPM), a rare cancer associated with asbestos exposure (1,2). Biallelic NF2 mutations underlie Familial Neurofibromatosis Type 2 (NF2), characterized by debilitating nervous system tumors where treatment relies predominantly on surgeries or radiosurgery (3). The prognosis of MPM is even bleaker as this cancer grows rapidly and is recalcitrant to both radio- and chemotherapy (4). In contrast to some other cancers that can be effectively treated – although not cured – by targeted therapies, NF2-mutant tumors remain a therapeutic challenge owing to the lack of consensus on the molecular mechanisms that underlie their development and the absence of validated targets.
Complicating the outlook, increasing evidence suggests that Merlin is a multifunctional protein that shuttles between the cell cortex and the nucleus in a manner reminiscent but antithetic to that of the cell adhesion and signaling component β-catenin (5,6). At the cell cortex, Merlin interacts with the scaffold and signaling protein Angiomotin to suppress mitogenic signaling through Rac, thereby inhibiting PAK (7). Merlin also regulates mitogenic signaling by suppressing mTORC1 in mesothelial, Schwann, and meningeal cells – tissues most commonly associated with NF2-mutant tumorigenesis (8,9). Although the mechanism by which Merlin suppresses mTOR signaling remains elusive, inactivation of NF2 confers sensitivity to rapalogs in bladder cancer and cetuximab-resistant lung cancer, suggesting that mTORC1 signaling broadly sustains the expansion of Merlin-deficient cancer cells (10,11). Recently, we discovered that the de-phosphorylated conformer of Merlin accumulates in the nucleus and suppresses tumorigenesis by inhibiting the cullin E3 ubiquitin ligase CRL4DCAF1 (5). Depletion of DCAF1 inhibited the hyperproliferation of schwannoma cells isolated from NF2 patients and suppressed the oncogenic potential of Merlin-mutant schwannoma and mesothelioma cell lines (5). Intriguingly, the oncogenic program of gene expression controlled by CRL4DCAF1 includes TEAD target genes, suggesting that Merlin controls Hippo signaling by inhibiting CRL4DCAF1. Following up on this hypothesis, we found that de-repressed CRL4DCAF1 targets Lats1 and 2 for ubiquitylation and inhibition in the nucleus and thus activates YAP-driven transcription and oncogenesis. Analysis of clinical samples indicated that this oncogenic pathway is consistently activated in human NF2-mutant mesothelioma, schwannoma, and meningioma (12). Despite these findings, the mechanisms governing Merlin’s regulation of YAP remains under debate, due in part to potentially non-redundant mechanisms (6).
Currently, multimodal therapy for MPM – including surgery and chemotherapy – extends patient survival by only a few months from the median survival of 1.5 years (4). Therefore, targeting of the oncogenic signaling pathways underlying aggressive NF2 loss-driven tumors – including those comprising a dominant fraction of MPM – would be of great clinical value. It was recently reported that NF2-mutant MPM is preferentially sensitive to a FAK inhibitor in pre-clinical models (13); however, clinical trials using a more selective FAK inhibitor were unable to demonstrate efficacy in NF2-mutant MPM (NCT01870609). Indeed, pre-clinical studies in mice using Nf2 loss-driven xenografts or autochthonous models have failed to completely suppress tumorigenesis using single or combination therapies, further highlighting the need for effective mechanism-based therapeutics (13–18).
Following our identification of CRL4DCAF1 as a primary target of Merlin in the nucleus (5), we sought to obtain proof of principle that pharmacological inhibition of CRL4DCAF1 could be effective in treating NF2-mutant tumors. Cullin E3 ligases, including CRL4DCAF1, are the best-characterized substrates of the ubiquitin-like modifier NEDD8. Neddylation of the cullin subunit at a conserved lysine residue promotes a conformational shift that fully activates ubiquitin ligase-conjugating activity by bringing target substrates into closer proximity with the ubiquitin-charged E2 enzyme (19–21). While multiple factors influence the activity of E3 ligases – including the abundance of adaptor modules – many E3 ligases display decreased ubiquitylation of target proteins upon depletion of active NEDD8 (22). MLN4924 is a first-in-class inhibitor of the NEDD8 activating enzyme (NAE), blocking activation of NEDD8 and thereby depleting the pool of active NEDD8, which can be conjugated to target proteins (23). Importantly, a recent Phase I study of MLN4924 in patients with advanced solid tumors established efficient on-target inhibition of NAE, acceptable dose-limiting toxicities, and anti-tumorigenic activity in some patients (24). Here we show that inhibition of CRL4DCAF1 using MLN4924 sensitizes cells to traditional chemotherapy but displays limited pre-clinical activity even in combination with chemotherapy. However, combined inhibition of CRL4DCAF1 and mTOR/PI3K almost completely suppresses the growth of NF2 loss-driven tumors.
MATERIALS AND METHODS
Animal Studies
Animal studies were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of MSKCC. Xenograft experiments were performed in collaboration with the MSKCC Antitumor Assessment Facility. VAMT, Meso-10, and MSK-LX19 xenografts were implanted in the rear flank of female NOD-scid IL2Rgammanull (NSG) mice obtained from the MSKCC Genomics Core. Drug treatments begun once tumors reached approximately 100 mm3. Tumors were measured by caliper every 3–4 days and mice were sacrificed if tumors reached 1000mm3 or if tumors began to ulcerate.
Apoptosis assay
Meso-33 and VAMT treated with cisplatin and MLN4924 were subjected to a Annexin-V/PI apoptosis assay using the Annexin V:FITC Apoptosis Detection Kit II (BD #556570) according to manufacturer’s instructions. Annexin V- and PI-positive cells were determined using FACS by the MSKCC Flow Cytometry Core Facility using a BD FACSCalibur Cell Analyzer.
Cell culture
All non-primary cell lines were passaged fewer than 10 times between receipt from source and experimentation. Mesothelial or mesothelioma cell lines Meso-9, Meso-10, Meso-33, H-Meso, 211H, H28, H2052, H2452, JMN, and VAMT were obtained from the same stocks as published previously (9) and were obtained between 2003 and 2004. LP9, Met5A, and Meso-37 mesothelioma cell lines were obtained from Dr. Marc Ladanyi (MSKCC) in 2012 (LP9 and Met5a) or 2014 (Meso-37), and were neither tested nor authenticated. Mesothelioma cell lines 211H, H2452, H28, H-Meso, JMN, Meso-9, Meso-10, Meso-37, VAMT, and H2052 were cultured as previously described (9). LP9 and Met5A immortalized mesothelial cells and Merlin-deficient mesothelioma Meso-33 cells were cultured in MCDB 110:199 Earle’s supplemented with EGF (10 ng/ml, Invitrogen #PHG0311), Hydrocortisone (50 μg/ml, CalBioChem #3867), ITS (1%, Invitrogen #I2521), antibiotics (1%, Gemini Bio # 400-101), Fetal Bovine Serum (FBS, 15%, Invitrogen #10437-028), and L-Glutamine (2 mM, Invitrogen #25030-081). LP9 and Met5a were also cultured in RPMI 1640 supplemented with 10% FBS, antibiotics, and L-Glutamine. COS-7 and 293T cells were obtained from ATCC in 2009 and 2015, respectively, and cultured in DMEM-HG supplemented with antibiotics, 10% FBS, and L-Glutamine. FH-912 Nf2+/+ mouse Schwann cells and FC-1801 Nf2−/− schwannoma cells were obtained in 2007 and cultured as previously described (5,25) and were not tested or authenticated. Human schwannoma and Schwann cells were kindly provided by NF2 patients after informed consent for studies approved by the institutional review board at the Plymouth University Peninsula School of Medicine and Dentistry. These Schwann and schwannoma cells were isolated and seeded into Lab-Tek chamber slides coated with poly-L-lysine and mouse laminin at 70–80% confluence essentially as described previously (26). Cells were then starved for 24 hours and treated with DMSO or the indicated drugs in full growth-factor media for 72 hours. Primary Schwann cell and schwannoma proliferation was determined by manual counting of Ki-67+ cells following immunofluorescent staining. Unless otherwise indicated, experiments were performed on cells grown to 70–90% confluency in complete medium.
Compounds
MG132 was obtained from Calbiochem (#474790), solubilized in DMSO, and used at a working concentration of 10–25 μM. MLN4924 was generously provided by Takeda Pharmaceutical Company and solubilized in DMSO for in vitro experiments or 10% Captisol for in vivo experiments. GDC-0980 was generously provided by Genentech and was solubilized in DMSO for in vitro experiments or 0.5% methylcellulose with 0.1% Tween-80 for in vivo experiments. Cisplatin was obtained from the Sloan Kettering Pharmacy and solubilized in saline for in vivo experiments or from Sigma and solubilized in DMF for in vitro experiments. Pemetrexed (Alimta) was obtained from Eli Lilly and solubilized in saline for in vivo experiments.
Lats in vivo ubiquitylation assay
293T cells in 6-well plates were transfected using Lipofectamine 2000 (Invitrogen) with 1 μg of pHis-Myc-Ub and 0.5 μg of pRK5-HA-Lats1, 1 μg of pRK5-Myc-DCAF1, and 0.25 μg pRK5-Myc-Merlin. Cells were treated with 10 μM MG132 for 4 hours and ±MLN4924 at the indicated concentrations before harvest. 24 hours after transfection, cells were scraped into cold PBS and 10% of the sample was lysed in SDS lysis buffer and reserved for immunoblotting of the total lysate. The remaining 90% of each sample was lysed in 1 ml of Guanidinium chloride lysis buffer (6 M Guanidinium-HCL, 0.1 M NaHPO4, 0.01 M Tris/HCL, pH 8.0, 20 mM Imidazole, 10 mM β-mercaptoethanol), sonicated, and centrifuged to. Equal quantities of cleared protein lysates, as extrapolated from the quantified SDS aliquots, were incubated with 100 μl Ni-NTA magnetic beads (Qiagen #36111) for 2 hours at room temperature. After incubation, Ni-NTA magnetic beads were washed once with each of Guanidinium washing buffer (6 M Guanidinium-HCL, 0.1 M NaHPO4, 0.01 M Tris/HCL, pH 8.0, 10 mM β-mercaptoethanol, 0.2% Triton X-100), Urea washing buffer I (8 M Urea, 0.1 M NaHPO4, 0.01 M Tris/HCL, pH 8.0, 10 mM β-mercaptoethanol, 0.2% Triton X-100), Urea washing buffer II (8 M Urea, 0.1 M NaHPO4, 0.01 M Tris/HCL, pH 6.3, 10 mM β-mercaptoethanol, 0.2% Triton X-100) and Urea washing buffer III (8 M Urea, 0.1 M NaHPO4, 0.01 M Tris/HCL, pH 6.3, 10 mM β-mercaptoethanol, 0.1% Triton X-100). Bound proteins were dissociated in 20 μL SDS sample/elution buffer supplemented with 250 mM Imidazole and 5% β-mercaptoethanol and immunoblotted as indicated.
Ubiquitylation assay, immunoprecipitation, and immunoblotting
Ten cm diameter plates of COS-7 or 293T cells were transfected using Lipofectamine 2000 with 3 μg of pRK5-Flag-HA-DCAF1 and 2 μg of pHis-Myc-Ub. When indicated, cells were treated with 25 μM MG132 for 6 hours. Lysates containing 0.5 mg of proteins were subjected to immunoprecipitations and western blotting. To isolate Flag-HA-DCAF1 and associated proteins, extracts were pre-cleared with Mouse IgG-Agarose (Sigma A0919) for one hour and then incubated with anti-FLAG M2 Affinity Gel (Sigma) at 4°C for 2 hours. Immunoprecipitates were washed three or four times with RIPA buffer or RIPA buffer without SDS as indicated, and bound proteins were dissociated in 20 μL 1× SDS sample buffer (25 mM Tris pH 6.8, 4% SDS, 5% Glycerol, bromophenol blue). Eluted proteins were separated on 4–12% Bis-Tris SDS-PAGE gels (Invitrogen) and transferred to nitrocellulose or Immobilon-P membranes (Millipore). Membranes were incubated in blocking buffer (5% skim milk, 0.1% Tween, 10 mM Tris at pH 7.6, 100 mM NaCl) for 0.5–1 hour at room temperature and then with primary antibodies diluted in blocking buffer for another hour at the same temperature or overnight at 4°C. Antibodies and working concentrations are shown in Supplemental Table 1. After three washes, the membranes were incubated with goat anti-Rabbit HRP-conjugated antibody (1:10,000; Santa Cruz sc-2054) or goat anti-mouse HRP-conjugated antibody (1:10,000; Santa Cruz sc-2005) at room temperature for 1 hour and subjected to chemiluminescence using ECL (Pierce #1856136). When indicated, cells were lysed in SDS-boiling buffer (10 mM Tris pH 7.5, 1% SDS, 50 mM NaF, 1 mM NaVO4) or NP-40 lysis buffer (50 mM Tris pH 7.4, 1% NP-40, 150 mM NaCl, 40 mM NaF, 1 mM NaVO4, protease and phosphatase inhibitors) and subjected to SDS-PAGE and immunoblotting. Where indicated, densitometry analysis was performed using ImageJ software.
For extended materials and methods, see Supplemental Materials and Methods.
RESULTS
NEDD8 activating enzyme inhibition abrogates CRL4DCAF1 activity and selectively blocks the proliferation of NF2-mutant schwannoma cells
Since CRLs are differentially sensitive to the effects of NAE inhibition (22), we set out to determine whether NAE inhibition abrogates the ubiquitin conjugating activity of CRL4DCAF1. Preliminary experiments indicated that the fraction of CUL4 associated with DCAF1 is effectively de-neddylated by MLN4924 (Fig. 1A). To examine if neddylation is necessary for the ubiquitin conjugating activity of CRL4DCAF1, we transfected COS-7 cells with Flag-HA-tagged DCAF1 (FH-DCAF1) and Myc-tagged ubiquitin and performed in vivo ubiquitylation assays. Under these conditions, CRL4DCAF1 promotes extensive ubiquitylation of its substrate receptor component, DCAF1, and lower level ubiquitylation of lower molecular weight endogenous substrates (12). Following precipitation of DCAF1, we observed an MLN4924-induced dose-dependent decrease in the precipitation of lower molecular weight Myc-ubiquitylated species, consistent with inhibition of CRL4DCAF1 by MLN4924 (Fig. 1B, arrowhead). Neddylation is likely dispensable for CRL4DCAF1 auto-ubiquitylation or CRL4DCAF1-mediated ubiquitylation of free or ligase-incorporated DCAF1, consistent with prior findings that neddylation is unnecessary for some CRL-mediated ubiquitylation targets (22).
Figure 1. MLN4924 attenuates CRL4DCAF1 activity and reduces the proliferation of NF2-mutant schwannoma cells.
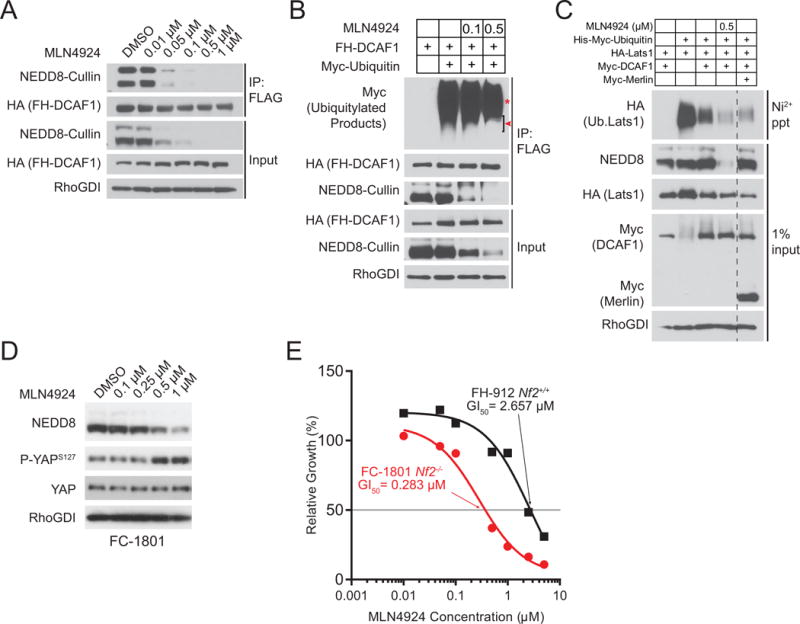
(A) 293T cells transfected with Flag-HA-tagged DCAF1 (FH-DCAF1) were treated with the indicated concentrations of MLN4924 for 4 hours and then lysed in RIPA buffer. Input and FLAG immunoprecipitates were immunoblotted as indicated. (B) COS-7 cells were transfected with FH-DCAF1 and Myc-Ubiquitin as indicated and then treated with MLN4924 at the indicated concentrations for 4 hours. FLAG immunoprecipitates and input were immunoblotted as indicated. Red asterisk indicates putative auto-ubiquitylated DCAF1. Red arrowhead indicates putative CRL4DCAF1 substrates. (C) In vivo Lats1 ubiquitylation assay. 293T cells were transfected as indicated. 24 hours following transfection, cells were treated with MLN4924 (8 hours as indicated) and the proteasome inhibitor MG132 (4 hours, all samples) prior to lysis in a denaturing buffer. Input and Ni-NTA precipitates were immunoblotted as indicated. (D) FC-1801 cells were treated with the indicated concentrations of MLN4924 for 8 hours, lysed, and immunoblotted as indicated. (E) The Nf2-null mouse schwannoma line FC-1801 and the Nf2-wild-type immortalized adult Schwann cell line FH-912 were treated with increasing doses of MLN4924 and subjected to MTT proliferation assay after 72 hours of treatment. Proliferation of MLN4924-treated cells was normalized to respective DMSO-treated controls. The concentrations of MLN4924 required to inhibit cell proliferation by 50% (GI50) are indicated.
CRL4DCAF1-mediated ubiquitylation and inhibition of the Lats tumor suppressor proteins promotes tumorigenesis across multiple NF2-mutant tissues (12). Lats1 and 2 are redundant kinases of the Hippo pathway immediately upstream of a transcriptional module composed of YAP or TAZ. To examine the effect of MLN4924 on the ubiquitylation of pathogenically relevant substrates of CRL4DCAF1, we conducted ubiquitylation assays with recombinant forms of Lats1 and 2. Importantly, MLN4924 administration inhibited Lats1 ubiquitylation as effectively as overexpression of Merlin (Fig. 1C), a manipulation previously shown to inhibit CRL4DCAF1 activity in vitro and in vivo (9). In addition, MLN4924 treatment reduced the oligo-ubiquitylation of Lats2, although to a lesser extent than Merlin expression (Supplemental Fig. S1A, arrowhead). Combined, these data suggest that MLN4924 blocks the inhibitory ubiquitylation of Lats1 and 2, potentially restoring inhibition of YAP.
Based on these in vitro findings, we sought to assess the preclinical efficacy of NAE inhibition in NF2-mutant tumors as a method to inhibit oncogenic CRL4DCAF1 and its ensuing activation of YAP. We elected to first examine the effects of MLN4924 in the Nf2-null mouse schwannoma cells FC-1801, which were derived by infecting primary Nf2Flox/Flox Schwann cells with adenoviral Cre (25). FC-1801 undergo proliferation arrest and lose their tumorigenic potential upon knock-down of DCAF1 as they do upon re-expression of Merlin, consistent with the model that NF2-mutant cells are dependent on CRL4DCAF1 signaling (5). Notably, MLN4924 induced phosphorylation of YAP at Serine 127 – a negative regulatory site – in a dose-dependent fashion in FC-1801 cells (Fig. 1D). In fact, increased phosphorylation of YAP correlated with decreased neddylation of cullins under these conditions. This finding is consistent with the ability of MLN4924 to reduce CRL4DCAF1-mediated inhibition of Lats (Fig. 1C and Supplemental Fig. S1A) and with our previous finding that CRL4DCAF1 is necessary to support oncogenic YAP signaling in this cell line (12).
To determine whether MLN4924 preferentially inhibits the proliferation of cells in which Merlin loss is the driving oncogenic lesion, we compared the anti-proliferative properties of MLN4924 in FC-1801 and the Nf2-WT Schwann cells FH-912, which provide for a genetically matched control for the FC-1801 cells (5). MLN4924 elicited a half-maximal growth inhibitory concentration (GI50) of 0.28 μM in FC-1801 cells and 2.7 μM in the Nf2-WT FH-912 cells, indicating that Nf2-mutant schwannoma cells are approximately ten-fold more sensitive to MLN4924 than their wild type counterparts (Fig. 1E) despite FH-912 displaying a nearly 5-fold higher on-target sensitivity (Supplemental Fig. S1B). Treatment of FC-1801 with MLN4924 for more than 24 hours revealed no change in Lats1 levels, suggesting that inhibition of kinase activity (5) – as opposed to proteasomal degradation – is the predominant route of Lats1 inhibition by CRL4DCAF1 in FC-1801 (Supplemental Fig. S1C, left). To examine the effects of MLN4924 on YAP downstream target genes, we treated Nf2-mutant FC-1801 and Nf2-WT FH-912 cells with MLN4924 for more than 24 hours. Intriguingly, we observed no changes in expression of canonical YAP target genes Ctgf, Cyr61, and Birc5 despite induction of YAP inhibitory phosphorylation in both lines (Supplemental Fig. S1C). We compared transcript levels in both lines and found that FC-1801 expresses nearly identical or reduced levels of these canonical YAP targets as compared to FH-912, suggesting that these genes may not be most relevant to oncogenic CRL4DCAF1 signaling in Schwann cells. Moreover, MLN4924 treatment led to a dose-dependent increase in all transcripts, again suggesting that these specific YAP targets are not relevant to the oncogenic output of dysregulated CRL4DCAF1 in this tissue. Combined, these findings reveal that MLN4924 blocks CRL4DCAF1-mediated ubiquitylation of Lats and activation of YAP and exerts a selective anti-proliferative effect in Merlin-deficient schwannoma cells.
MLN4924 suppresses oncogenic signaling and cell proliferation in NF2-mutant MPM cells
To confirm the identification of CRL4DCAF1 as a therapeutic target in NF2-mutant MPM, we tested the effect of iRNA-mediated depletion of DCAF1 in NF2-mutant Meso-33 and VAMT cells (Supplemental Fig. S2A, top). Acute depletion of DCAF1 with a SMART pool reduced the proliferation of both MPM lines by 50–70% as compared to their control counterparts within 4 days. In addition, tetracycline-inducible knockdown of DCAF1 with two independent shRNAs reduced proliferation of NF2-mutant MPM cells in a manner proportional with the timing and extent of DCAF1 depletion (Supplemental Fig. S2A and S2B). These results confirm the potential therapeutic value of inhibiting CRL4DCAF1 in NF2-mutant MPM.
As predicted, MLN4924 treatment caused a dose-dependent inhibition of cullin neddylation in NF2-mutant Meso-33 cells. Nearly complete inhibition was reached at 0.5 μM, confirming that this compound is capable of efficiently targeting cullin E3 ligases in MPM cells (Fig. 2A). Notably, 0.5 μM MLN4924 induced phosphorylation of YAP at its inhibitory phosphorylation site Serine 127 to a similar extent – if not more efficiently – as compared to DCAF1 depletion (Fig. 2B) (12). To examine MLN4924 effects on downstream YAP signaling, we assessed canonical YAP targets after 36 hours of MLN4924 exposure. Similar to prolonged DCAF1 or YAP knockdown (Fig. 2C, left), extended MLN4924 reduced expression of canonical YAP target genes CTGF, BIRC5, and CYR61 (Fig. 2C, right) despite a relapse of YAP inhibitory phosphorylation. Intriguingly, neither DCAF1 depletion nor MLN4924 administration reduced flux through the mTORC1 pathway as assessed by phosphorylation of S6 ribosomal protein, a key downstream effector of mTORC1, suggesting that loss of Merlin activates mTOR independently of CRL4DCAF1 (Fig. 2A, B, D). Consistent with previous findings, DCAF1 inhibition and MLN4924 treatment induced p27 expression (Fig. 2B), which may contribute to cell cycle arrest (27). Indeed, at a concentration that almost completely blocks cullin neddylation, MLN4924 strongly induced cell cycle progression as demonstrated by suppression of the phosphorylation of Rb and Histone H3 (Fig. 2D). These findings suggest that NAE inhibition in Merlin-deficient cells inhibits oncogenic YAP signaling and suppresses cell cycle progression.
Figure 2. MLN4924 rescues activity of the Lats Hippo pathway kinases and blocks cell cycle progression.
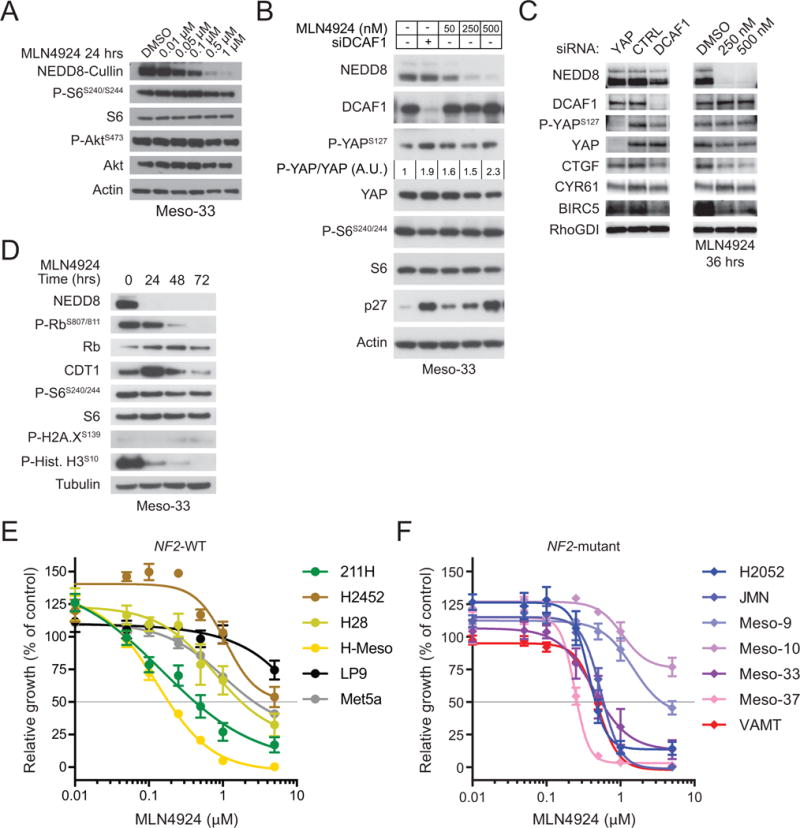
(A) The NF2-mutant mesothelioma line Meso-33 was treated with the indicated concentrations of MLN4924 for 24 hours and immunoblotted as indicated. (B) Meso-33 cells were transfected with control or DCAF1 siRNAs and subsequently treated for 16 hours with DMSO or the indicated concentrations of MLN4924 and lysed 72 hours post-transfection for immunoblotting as indicated. P-YAPS127/YAP fold change based on densitometric analysis is indicated. (C) Meso-33 cells were transfected with indicated siRNAs and harvested after 80 hours (left) or treated with the indicated concentrations of MLN4924 for 36 hours (right) for immunoblotting as indicated. (D) Meso-33 cells were treated with 0.5 μM MLN4924 for the indicated times and immunoblotted as indicated. (E) NF2-positive MPM lines and immortalized but untransformed mesothelial lines (Met5a and LP9) were treated with increasing doses of MLN4924 and subjected to MTT proliferation assay after 72 hours of treatment. Proliferation of MLN4924-treated cells was normalized to respective DMSO-treated controls. Error bars are ±SEM (n=3). (F) NF2-mutant MPM lines were treated as in (E).
To test the hypothesis that MLN4924 preferentially suppresses the proliferation of NF2-mutant mesothelioma cells, we determined the concentration of MLN4924 required to inhibit cell proliferation by 50% (GI50) in a large panel of MPM lines as well as in untransformed Met5a and LP9 mesothelial cells. Whereas the GI50 of MLN4924 was approximately 2 μM or higher in Met5a, LP9, and 2 out of 4 NF2-wild type MPM lines (Fig. 2E), it was equal or lower than 0.55 μM in 5 out of 7 NF2-mutant MPM lines, providing evidence that the large majority of NF2-mutant MPM lines are highly sensitive to MLN4924 (Fig. 2F). MPM tumors and cell lines often have complex genotypes and a large fraction of them exhibit mutations not only in NF2 but also in CDKN2A, CDKN2B, and BAP1. Sensitivity to MLN4924 did not correlate with absence or presence of any of these additional genetic alterations in the MPM lines examined (2). However, the NF2-mutant Meso-9 and Meso-10 lines have not been fully sequenced and it is therefore possible that their relative insensitivity to MLN4924 is due to an unidentified co-occurring genetic alteration. These findings are consistent with the model that NF2-mutant MPM cells are sensitive to MLN4924 because of its ability to inhibit oncogenic signaling through CRL4DCAF1 (5).
MLN4924 cooperates with chemotherapy to significantly reduce Merlin-deficient MPM tumor growth in vivo
MLN4924 inhibits CRL ligases involved in the DNA damage response (DDR) including CRL4DDB2 and CRL4CSA, which function in global genome Nucleotide Excision Repair (NER) and transcription-coupled NER, as well as CRL4CDT2, which suppresses DNA re-replication and apoptosis (28–32). We therefore set out to determine if MLN4924, in addition to inhibiting CRL4DCAF1 and YAP signaling, would also sensitize NF2-mutant tumor cells to DNA-damaging agents. We first examined whether MLN4924 cooperates with cisplatin, a platinum-containing DNA cross-linking drug, because it is used in combination with the folate antimetabolite pemetrexed as first-line chemotherapy for MPM. Treatment of NF2-mutant MPM with MLN4924, cisplatin, or both resulted in DNA damage as shown by Chk1 and Histone H2A.X phosphorylation (Fig. 3A). Cisplatin-induced DNA damage also causes CDT1 degradation, which functions to halt the cell cycle as a result of DNA damage. MLN4924 stabilized CDT1 even in the presence of cisplatin – an effect of MLN4924 that has been shown to induce DNA re-replication and to contribute to further DNA damage and apoptosis (28,29). MLN4924 and cisplatin monotherapy caused comparable levels of apoptosis in Merlin-deficient MPM, however, combining the compounds dramatically increased apoptosis, indicating that MLN4924 sensitizes NF2-mutant MPM cells to chemotherapy (Fig. 3B).
Figure 3. NAE inhibition cooperates with first-line MPM chemotherapy.
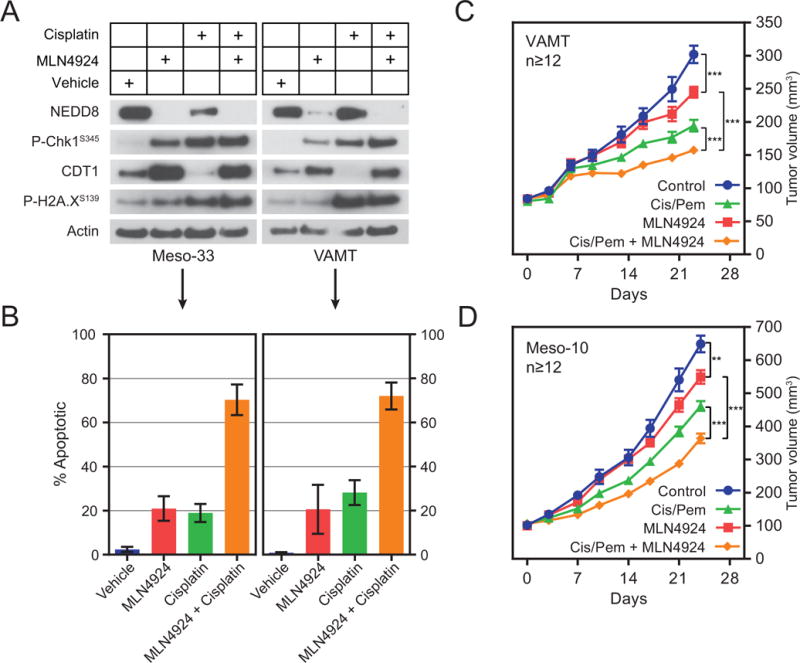
(A) Meso-33 and VAMT were treated with vehicle or the indicated drugs at their respective GI50 concentrations for 24 hours. Cell lysates were immunoblotted as indicated. (B) Meso-33 and VAMT cells were treated with vehicle or the indicated drugs at twice their respective GI50 concentrations for 72 hours and then subjected to an Annexin-5/PI apoptosis assay. Error bars indicate ±SEM (VAMT, n=3; Meso-33, n=2). (C) 15 × 106 VAMT cells and (D) 8 × 106 Meso-10 cells were injected bilaterally in the rear flanks of NSG mice. After xenografts exceeded 100 mm3, mice were treated with vehicle or the indicated drugs. All cisplatin treatments were 3 mg/kg (intraperitoneally, once every 7 days), pemetrexed treatments 100 mg/kg (intraperitoneally, once daily, 5 days on, 2 days off), and MLN4924 treatments 90 mg/kg (subcutaneously, twice per day, three times per week M-W-F). A 7-day holiday was provided between chemotherapy regimens while the MLN4924 was administered continuously. Data are means ± SEM (n≥12). **P < 0.01, ***P < 0.001, unpaired T-test.
To examine the ability of MLN4924 to sensitize NF2-mutant MPM cells to chemotherapy in vivo, we treated NOD-scid IL2Rgammanull (NSG) mice bearing VAMT or Meso-10 xenografts of approximately 100 mm3 size with MLN4924, cisplatin + pemetrexed, or both. Both MLN4924 monotherapy and combination chemotherapy induced a statistically significant reduction in tumor growth (Fig. 3C and D). However, combination therapy with MLN4924 and pemetrexed + cisplatin displayed significantly improved inhibition of tumor growth as compared to either individual treatment modality in both types of tumors (Fig. 3C and D). Individual and combined therapies were well tolerated (Supplemental Fig. S3A and S3B). These preclinical data indicate that combining MLN4924 with first-line chemotherapy may improve the treatment NF2-mutant MPM.
Combined inhibition of NAE and mTOR/PI3K cooperate to block NF2-mutant tumor growth
Although MLN4924 inhibited tumor growth, especially in combination with chemotherapy, it did not completely suppress this event or cause tumor regression, suggesting that NF2 loss-driven tumorigenesis does not depend solely on CRL4DCAF1. Intriguingly, we had observed that treatment with MLN4924 or depletion of DCAF1 does not diminish mTORC1 signaling in NF2-mutant Meso-33 cells (Fig. 2A, B and C), suggesting that this oncogenic signaling pathway may provide a mechanism of resistance to inhibition of CRL4DCAF1. To confirm that DCAF1 promotes oncogenic signaling through regulation of YAP and other targets, but independently of flux through the mTORC1-PI3K signaling pathways, we assessed the expression of these pathways’ downstream effectors following CRL4DCAF1 inhibition. Consistently, we found that DCAF1 depletion dramatically reduces expression of the YAP target gene CTGF but does not affect the phosphorylation of S6 in two NF2-mutant MPM lines (Fig. 4A). Consistent with previous findings (8,9), re-expression of Merlin reduced S6 phosphorylation in FC-1801 schwannoma cells (Fig. 4B). Even in this genetically-defined Nf2-null cell line, DCAF1 depletion and MLN4924 administration did not affect mTORC1 signaling (Fig. 4B), providing further support that Merlin regulates mTORC1 independently of CRL4DCAF1.
Figure 4. MLN4924 combined with mTOR/PI3K inhibition causes complete MPM tumor growth suppression and significantly reduces the growth of NF2-mutant schwannoma.
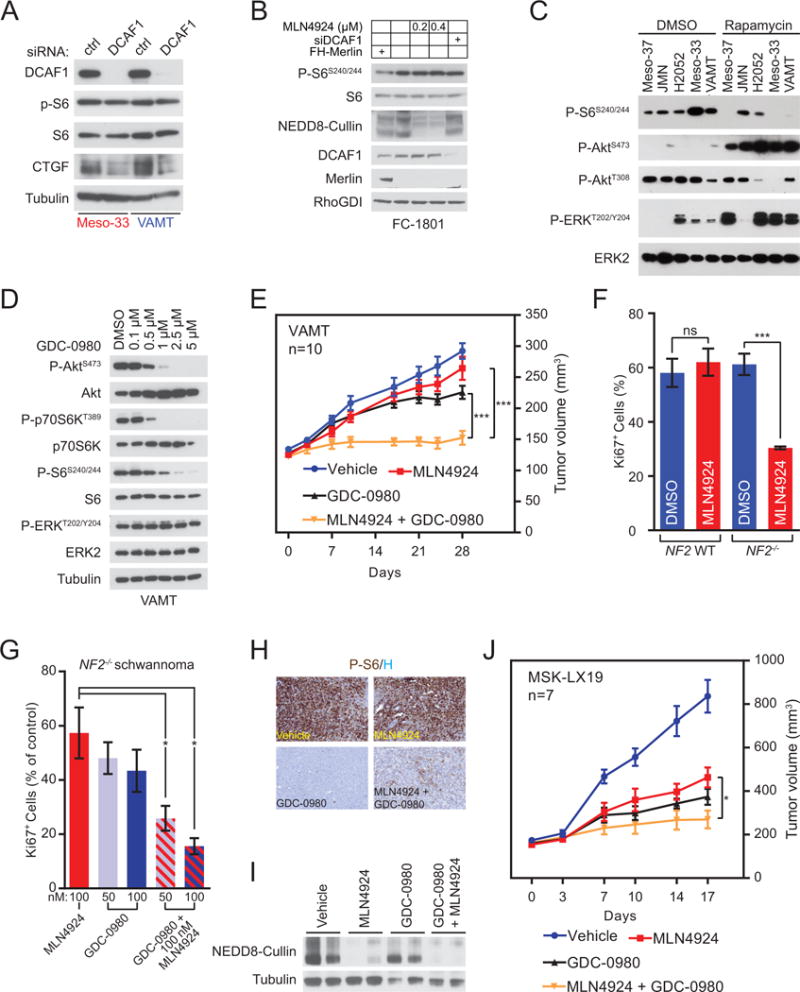
(A) Merlin deficient Meso-33 and VAMT cells were transfected with control or DCAF1 siRNA and harvested after 72 hours for immunoblotting as indicated. (B) FC-1801 cells were transfected with Flag-HA-tagged (FH) Merlin or empty vector and control or DCAF1 siRNAs as indicated. Cells were also treated with the indicated concentration of MLN4924 or DMSO for 4 hours prior to lysis and immunoblotted as indicated. (C) The indicated Merlin-deficient MPM lines were treated with DMSO (left) or 5 nM rapamycin (right) for 4 hours and immunoblotted as indicated. (D) VAMT cells were treated with the indicated concentrations of GDC-0980 for three hours and immunoblotted as indicated. (E) 8–10 × 106 VAMT cells were injected bilaterally in the rear flanks of NSG mice. After xenografts exceeded 100 mm3, mice were treated with vehicle or the indicated inhibitors. MLN4924 treatments were 120 mg/kg (subcutaneously, twice per day, three times per week M-W-F). GDC-0980 treatments were 5 mg/kg (oral, once per day, five times per week). Data are means ± SEM (n=10). ***P < 0.001, unpaired T-test. (F) Primary Schwann cells from normal individuals (NF2 WT, left) and schwannoma cells from NF2 patients (NF2−/−, right) were treated with DMSO or 100 nM MLN4924 for 72 hours. Graph indicates the percentage of proliferating cells based on immunostaining for Ki-67 positivity. Data are means ± SEM (NF2+/+ n=3; NF2−/− n=4). ***P < 0.001, unpaired T-test. (G) Primary NF2−/− human schwannoma cells were treated with DMSO or the indicated concentrations of MLN4924, GDC-0980, or a combination of the two inhibitors as indicated. Ki-67+ cells were counted as in (F) and the graph indicates Ki-67+ relative to DMSO controls. Data are means ± SEM (n=3). *P < 0.05, unpaired T-test. (H and I) On-target mTORC1 and NAE inhibition in MSK-LX19 PDX. NSG mice bearing 200 mm3 MSK-LX19 PDXs were treated once with vehicle or the same doses of the indicated inhibitors as in (E). Tumors were isolated 7 hours after drug administration. (H) Representative images of tumors immunostained for phospho-S6 (P-S6) and counterstained with hematoxylin (H). (I) Lysates from the same tumors as (H) were immunoblotted as indicated to detect neddylated cullins (n=2). (J) Serially transplanted MSK-LX19 tumors were injected unilaterally in the rear flanks of NSG mice. After tumors exceeded 100 mm3, mice were treated as in (E). Data are means ± SEM (n=7). *P < 0.05, unpaired T-test.
Based on these findings, we set out to test the hypothesis that combined inhibition of CRL4DCAF1 and mTORC1 suppresses NF2-mutant MPM growth. We had shown that rapamycin inhibits the proliferation of NF2-mutant MPM lines to a greater extent than NF2-wild type lines (9). In many tumor types, the therapeutic efficacy of rapalogs is limited by the release of mTORC1-mediated negative feedback loops that function to restrain AKT-mTOR signaling (9,33–37). Consistently, we found that treatment with rapamycin induces hyperactivation of both AKT and ERK in NF2-mutant MPM lines (Fig. 4C). GDC-0980, a dual mTOR/PI3K inhibitor, was developed to block mTORC signaling while simultaneously suppressing feedback-regulated pathways (38). Notably, GDC-0980 blocked mTORC1 signaling without activating Akt or ERK in VAMT and Meso-33 (Fig. 4D and Supplemental Fig. S4A). Based on these results, we examined the pre-clinical efficacy of MLN4924 in combination with GDC-0980 in VAMT xenografts. Intriguingly, single agent therapy with GDC-0980 yielded marginally higher – albeit statistically insignificant – efficacy relative to MLN4924 alone (Fig. 4E). However, combination therapy with MLN4924 and GDC-0980 completely suppressed tumor growth (Fig. 4E) and exceeded the efficacy of either single agent alone, standard first-line chemotherapy, and chemotherapy combined with MLN4924 with minimal toxicity (Fig. 3C, 3D and Supplemental Fig. S4E). These findings support the concept that mTORC1 and CRL4DCAF1 are essential and non-redundant oncogenic pathways sustaining NF2-mutant tumor growth, and their combined inhibition may be therapeutically efficacious.
To determine whether combined inhibition of NAE and mTORC1 could be broadly exploited as a rational therapy in NF2 loss-driven tumorigenesis, we evaluated the efficacy of MLN4924 and GDC-0980 in human schwannoma – the hallmark tumor of NF2 patients. Notably, MLN4924 suppressed the proliferation of primary human NF2-mutant schwannoma cells but did not affect that of NF2-WT Schwann cells (Fig. 4F), providing genetic evidence that NF2-mutant tumor cells are selectively sensitive to MLN4924. Importantly, while GDC-0980 monotherapy inhibited NF2-mutant schwannoma to a similar extent as MLN4924, combination therapy significantly reduced the proliferation of these cells in vitro (Fig. 4G), confirming the potential efficacy of dual inhibition of NAE and mTORC1 in NF2-mutant tumors.
Finally, we examined the efficacy of mTOR and NAE inhibition in an NF2-mutant epithelioid MPM patient-derived xenograft (PDX), MSK-LX19, in which we had identified a total loss of expression of Merlin by immunoblotting (Supplemental Fig. S4B). Immunostaining and immunoblotting of small (≤200 mm3) MSK-LX19 tumors grown in NSG mice and treated once with each drug singly and in combination confirmed that MLN4924 inhibits neddylation of cullins but does not appreciably affect mTORC1 signaling (Fig. 4H, 4I and Supplemental Fig. S4C). Conversely, GDC-0980 efficiently blocks mTORC1 signaling in vivo but does not affect neddylation of cullins. Similarly, MLN4924 induced stabilization of p27 while combination therapy significantly increased p27 expression (Supplemental Fig. S4D), suggesting that MLN4924 and GDC-0980 cooperatively induce cell cycle arrest. Whereas MLN4924 or GDC-0980 monotherapy significantly reduced tumor growth (P<0.01 and P<0.001, respectively), MLN4924 and GDC-0980 combination therapy completely suppressed tumor growth (P<0.001) (Fig. 4J). Combined, these data reveal that dual inhibition of CRL4DCAF1 and mTORC1/PI3K abrogates two critical oncogenic pathways sustaining the proliferation of NF2-mutant tumors and exhibit pre-clinical efficacy in both MPM and schwannoma models (Fig. 5).
Figure 5. Signaling pathway and mechanistic effects of MLN4924 and GDC-0980 in NF2/Merlin-mutant tumors.
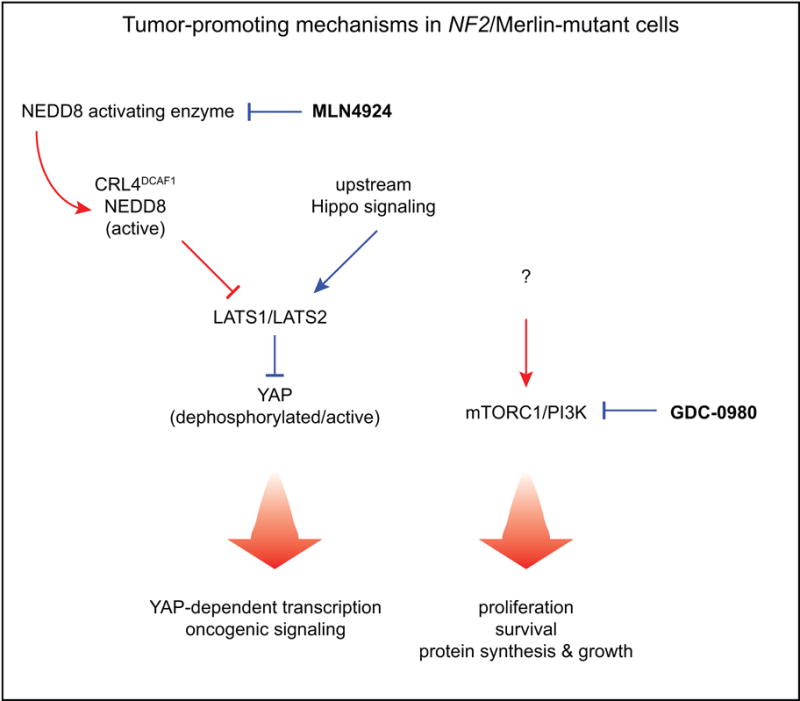
In NF2/Merlin-mutant tumors, dysregulated CRL4DCAF1 and mTORC1 are two predominant oncogenic signaling nodes sustaining tumor growth. MLN4924 inhibits YAP activity in multiple Merlin-mutant tumors and transcription of canonical pro-proliferative and anti-apoptotic YAP targets in mesothelioma. GDC-0980 inhibits mTOR and PI3K, thereby inhibiting downstream mechanisms supporting tumor growth and maintenance including proliferation, survival, protein synthesis, and cell growth. Tumor-promoting mechanisms are indicated in red, tumor-suppressive mechanisms in blue.
DISCUSSION
Incomplete understanding of the signaling circuits dysregulated by inactivation of Merlin has so far prevented the development of rational and effective therapies for NF2 and MPM. Recent studies have revealed that Merlin is a cell adhesion and signaling component, which shuttles between the cell cortex and the nucleus in a manner reminiscent but antithetic to that of β-catenin (5). Intriguingly, loss of Merlin’s inhibition of the nuclear E3 ligase CRL4DCAF1 stabilizes the Hippo pathway component Lats1/2 and hence activates YAP and TAZ, which play redundant roles in sustaining NF2-mutant tumorigenesis (12). Furthermore, loss of Merlin dysregulates additional signaling pathways – including mTORC1 – that may contribute to tumorigenesis (8,9). It remains unclear if Merlin prevents activation of these additional pathways from the cell cortex or in the nucleus through inhibition of CRL4DCAF1.
In this study, we found that inactivation of NAE using MLN4924 inhibits CRL4DCAF1-mediated ubiquitylation of Lats1 and 2, and hence induces phosphorylation and inactivation of YAP. In addition, we found that MLN4924 preferentially inhibits the proliferation of NF2-mutant tumor cells, presumably as a consequence of their reliance on YAP/TAZ signaling for growth. In spite of its ability to curb YAP/TAZ and downstream target genes, MLN4924 did not inhibit the growth of NF2-mutant MPM xenografts as effectively as standard chemotherapy. We therefore set out to exploit the collateral inhibition of CRL ligases intimately regulating the cell cycle and DNA damage response (22). Our results clearly indicate that MLN4924 preferentially sensitizes NF2-mutant tumor cells to chemotherapy, inducing tumor cells exposed to both drugs to undergo apoptosis. Moreover, MLN4924 in combination with pemetrexed + cisplatin suppresses MPM xenograft growth to a much greater extent than either single treatment modality alone. Therefore, MLN4924 inhibits oncogenic signaling through CRL4DCAF1 but also enhances the effect of chemotherapy as a result of collateral inhibition of CRLs involved in DNA repair. These preclinical data provide rationale for combining NAE inhibition with first-line chemotherapy to more effectively treat NF2 loss-driven MPM.
Prior studies have shown that activation of mTORC1 contributes to the hyperproliferation of NF2-mutant MPMs as well as canonical NF2 tumors including meningiomas and schwannomas (8,9). Based on the observation that loss of Merlin activates mTORC1 independently of CRL4DCAF1, we set out to test the effect of combined inhibition of CRL4DCAF1 and mTORC1. Preliminary experiments indicated that rapamycin releases negative feedback loops impinging on both AKT and ERK, leading to potent activation of both pathways in NF2-mutant MPM. This observation may explain, at least in part, the limited efficacy of mTORC1 inhibitors – such as everolimus – in clinical trials for NF2 tumors (3). Effective mTORC1 inhibition would potentially provide a long-term therapy to NF2 patients, which would be otherwise impractical with cytotoxic chemotherapy such as cisplatin + pemetrexed. To circumvent this obstacle, we used combinatorial mTOR/PI3K inhibition using GDC-0980 and verified that this compound blocks mTORC1 signaling without activating AKT or ERK. To block the two key oncogenic pathways underlying NF2-mutant tumorigenesis, we combined GDC-0980 and MLN4924. The combination almost completely suppressed the growth of NF2-mutant MPM cells in vivo and selectively inhibited the proliferation of NF2-mutant primary human schwannoma cells in vitro. Intriguingly, examination of the MSK-LX19 MPM PDX model revealed that these NF2-mutant tumor cells are highly sensitive to MLN4924 or GDC-0980 monotherapy, suggesting that activation of both pathways is required for their outgrowth. Moreover, combination therapy in vivo may induce additive p27 expression (Supplemental Fig. S4D) and thereby cell cycle arrest due to GDC-0980-mediated inhibition of AKT coupled with MLN4924-mediated accumulation of p27. MLN4924 likely induces p27 accumulation via inhibition of both CRL4DCAF1 (27) and the p27 cullin E3 ligase SCFSkp2. Simultaneously, inhibition of mTORC and PI3K may block AKT-mediated phosphorylation of p27, thereby promoting p27 nuclear import and proliferation arrest (39). p27 nuclear import may further protect it from degradation in this context. Combined, our findings illustrate the preclinical efficacy of targeting the CRL4DCAF1-YAP axis and mTORC/PI3K pathway in NF2 loss-driven tumors.
Non-surgical therapies for NF2 tumors and NF2-mutant MPM are relatively ineffective in reducing morbidity and mortality. Our findings provide proof of principle that targeting oncogenic signaling pathways shared amongst NF2-mutant tumors, such as YAP and mTORC1, can radically alter disease progression. We anticipate that state-of-the-art mTORC1/2 inhibitors and forthcoming NAE inhibitors with improved pharmacokinetics/pharmacodynamics will be of great utility in treating NF2 loss-driven tumors.
Supplementary Material
Acknowledgments
We thank Genentech for providing GDC-0980 and Takeda Pharmaceuticals for providing MLN4924. We gratefully acknowledge excellent technical assistance from Viola Allaj and Qi Li. We thank Ralitsa Petrova for scientific discussion and review of the manuscript.
Financial Support: This study was supported by the Mesothelioma Applied Research Foundation – The Law Offices of Peter G Angelos Grant (to F.G. Giancotti), The Children’s Tumor Foundation (to F.G. Giancotti), NIH R01 CA152975 (to F.G. Giancotti), and NIH R01 CA191222-01A1 (to F.G. Giancotti and M. Zauderer).
References
- 1.Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nature genetics. 2016;48:407–16. doi: 10.1038/ng.3520. [DOI] [PubMed] [Google Scholar]
- 2.Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nature genetics. 2011;43:668–72. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blakeley JO, Plotkin SR. Therapeutic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro Oncol. 2016;18:624–38. doi: 10.1093/neuonc/nov200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ladanyi M, Zauderer M, Krug L, Ito T, McMillan R, Bott M, et al. New Strategies in Pleural Mesothelioma: BAP1 and NF2 as novel targets for therapeutic development and risk assessment. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012 doi: 10.1158/1078-0432.CCR-11-2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140:477–90. doi: 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper J, Giancotti FG. Molecular insights into NF2/Merlin tumor suppressor function. FEBS letters. 2014;588:2743–52. doi: 10.1016/j.febslet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila JL, Christian N, et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer cell. 2011;19:527–40. doi: 10.1016/j.ccr.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29:4250–61. doi: 10.1128/MCB.01581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopez-Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol Cell Biol. 2009;29:4235–49. doi: 10.1128/MCB.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012;338:221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pirazzoli V, Nebhan C, Song X, Wurtz A, Walther Z, Cai G, et al. Acquired resistance of EGFR-mutant lung adenocarcinomas to afatinib plus cetuximab is associated with activation of mTORC1. Cell reports. 2014;7:999–1008. doi: 10.1016/j.celrep.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer cell. 2014;26:48–60. doi: 10.1016/j.ccr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shapiro IM, Kolev VN, Vidal CM, Kadariya Y, Ring JE, Wright Q, et al. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Sci Transl Med. 2014;6:237ra68. doi: 10.1126/scitranslmed.3008639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka K, Eskin A, Chareyre F, Jessen WJ, Manent J, Niwa-Kawakita M, et al. Therapeutic potential of HSP90 inhibition for neurofibromatosis type 2. Clin Cancer Res. 2013;19:3856–70. doi: 10.1158/1078-0432.CCR-12-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giovannini M, Bonne NX, Vitte J, Chareyre F, Tanaka K, Adams R, et al. mTORC1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro Oncol. 2014;16:493–504. doi: 10.1093/neuonc/not242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong HK, Lahdenranta J, Kamoun WS, Chan AW, McClatchey AI, Plotkin SR, et al. Anti-vascular endothelial growth factor therapies as a novel therapeutic approach to treating neurofibromatosis-related tumors. Cancer Res. 2010;70:3483–93. doi: 10.1158/0008-5472.CAN-09-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Messerli SM, Prabhakar S, Tang Y, Mahmood U, Giovannini M, Weissleder R, et al. Treatment of schwannomas with an oncolytic recombinant herpes simplex virus in murine models of neurofibromatosis type 2. Hum Gene Ther. 2006;17:20–30. doi: 10.1089/hum.2006.17.20. [DOI] [PubMed] [Google Scholar]
- 18.Nigim F, Esaki S, Hood M, Lelic N, James MF, Ramesh V, et al. A new patient-derived orthotopic malignant meningioma model treated with oncolytic herpes simplex virus. Neuro Oncol. 2016;18:1278–87. doi: 10.1093/neuonc/now031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 20.David MD, Laura AB, Daniel CS, Harold WH, Michal H, Brenda AS. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Molecular cell. 2008 doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett EJ, Rush J, Gygi SP, Harper JW. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell. 2010;143:951–65. doi: 10.1016/j.cell.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 24.Sarantopoulos J, Shapiro GI, Cohen RB, Clark JW, Kauh JS, Weiss GJ, et al. Phase I Study of the Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (TAK-924/MLN4924) in Patients with Advanced Solid Tumors. Clin Cancer Res. 2016;22:847–57. doi: 10.1158/1078-0432.CCR-15-1338. [DOI] [PubMed] [Google Scholar]
- 25.Lallemand D, Manent J, Couvelard A, Watilliaux A, Siena M, Chareyre F, et al. Merlin regulates transmembrane receptor accumulation and signaling at the plasma membrane in primary mouse Schwann cells and in human schwannomas. Oncogene. 2009;28:854–65. doi: 10.1038/onc.2008.427. [DOI] [PubMed] [Google Scholar]
- 26.Kaempchen K, Mielke K, Utermark T, Langmesser S, Hanemann CO. Upregulation of the Rac1/JNK signaling pathway in primary human schwannoma cells. Hum Mol Genet. 2003;12:1211–21. doi: 10.1093/hmg/ddg146. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita K, Ide M, Furukawa KT, Suzuki A, Hirano H, Ohno S. Tumor suppressor protein Lgl mediates G1 cell cycle arrest at high cell density by forming an Lgl-VprBP-DDB1 complex. Mol Biol Cell. 2015;26:2426–38. doi: 10.1091/mbc.E14-10-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei-Wei P, Jian-Jie Z, Chao Y, Ying X, Lian-Jun G, Hai-Yi Z, et al. Ubiquitin E3 ligase CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. The Journal of biological chemistry. 2013:29680–91. doi: 10.1074/jbc.M113.495069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70:10310–20. doi: 10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown JS, Lukashchuk N, Sczaniecka-Clift M, Britton S, le Sage C, Calsou P, et al. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell reports. 2015;11:704–14. doi: 10.1016/j.celrep.2015.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fischer ES, Scrima A, Bohm K, Matsumoto S, Lingaraju GM, Faty M, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–39. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 32.Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Molecular cell. 2006;23:471–82. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 33.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, et al. A negative feedback signaling network underlies oncogene-induced senescence. Cancer cell. 2006;10:459–72. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research. 2006 doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tremblay F, Brûlé S, Um SH, Li Y. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient-and obesity-induced insulin resistance. Proceedings of the …. 2007 doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor …. Molecular cancer therapeutics. 2005 doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 37.Yu Y, Yoon S-OO, Poulogiannis G, Yang Q, Ma XM, Villén J, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science (New York, NY) 2011;332:1322–6. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallin J, Edgar K, Guan J, Berry M, Prior W, Lee L, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Molecular cancer therapeutics. 2011;10:2426–36. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 39.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.