

Ionic scattering factors of atoms that compose biological molecules have been computed by the multi-configuration Dirac–Fock method and parameterized for major curve models in X-ray and electron protein crystallography and single-particle cryo-EM.
Keywords: form factors, electron crystallography, single-particle cryo-EM, structure refinement, X-ray crystallography, imaging, structure determination
Abstract
Ionic scattering factors of atoms that compose biological molecules have been computed by the multi-configuration Dirac–Fock method. These ions are chemically unstable and their scattering factors had not been reported except for O−. Yet these factors are required for the estimation of partial charges in protein molecules and nucleic acids. The electron scattering factors of these ions are particularly important as the electron scattering curves vary considerably between neutral and charged atoms in the spatial-resolution range explored in structural biology. The calculated X-ray and electron scattering factors have then been parameterized for the major scattering curve models used in X-ray and electron protein crystallography and single-particle cryo-EM. The X-ray and electron scattering factors and the fitting parameters are presented for future reference.
1. Introduction
Scattering factors for X-rays and electrons are essential for the analysis of experimental data probed with an X-ray and an electron beam. X-rays are scattered by electrons around the atom, yielding an electron-density map, whereas electrons are scattered by Coulomb potential, yielding a Coulomb-potential map. Hence the scattering factors are directly related to the charged state, particularly the electron scattering factors, which vary considerably between the neutral and charged atoms in the regime when sinθ/λ is smaller than 0.1 Å−1 and do not match until sinθ/λ reaches 0.2–0.3 Å−1 (Fig. 1 ▸). Here θ represents half the scattering angle, λ is the wavelength of the incident X-ray or electrons and the latter values of sinθ/λ correspond to spatial resolutions of 1/(0.2 × 2) – 1/(0.3 × 2) = 2.5 – 1.67 Å. Thus, the charge state essentially affects the structure analysis of proteins and protein complexes including nucleic acids by electron crystallography and single-particle cryo-EM.
Figure 1.

Plots of typical X-ray (upper row) and electron (lower row) scattering factors from the literature [e.g. International Tables for Crystallography (Colliex et al., 2006 ▸)]. The electron scattering factors of O and O− are displayed up to sinθ/λ = 0.25 Å−1 and the others are up to 0.5 Å−1. Electron scattering factors of partially charged O0.3− and O0.6− were calculated from a linear combination of those of the neutral and fully ionized oxygen atoms (Yonekura & Maki-Yonekura, 2016 ▸) and are also overlaid in the lower left graph.
The charge is actually delocalized over several atoms in the protein molecules and nucleic acids, yielding partial charges which can be regarded as effective scattering factors. Partial charges should be assigned to all of the atoms in proteins and protein complexes (see Fig. 2 ▸ for theoretical partial charge distributions in amino acids and a nucleic acid). Although this approach is more qualitative than using form factors representing chemical bonding calculated for typical small molecules (Chang et al., 1999 ▸; Yamashita & Kidera, 2001 ▸; Zhong et al., 2002 ▸), it allows for the information related to charges to be extracted by electron two-dimensional (Mitsuoka et al., 1999 ▸) and three-dimensional (3D) crystallography (Yonekura et al., 2015 ▸; Yonekura & Maki-Yonekura, 2016 ▸) and single-particle cryo-EM (Yonekura & Maki-Yonekura, 2016 ▸). Also, ionic scattering factors could lead to a more accurate structure refinement against cryo-EM data (Yonekura & Maki-Yonekura, 2016 ▸). However, those studies assumed partial charges for a limited number of atoms including O and H of the side chains in titratable residues such as aspartate, glutamate, lysine, arginine and histidine.
Figure 2.

Theoretical charge distributions over atoms in typical amino acids and the template of a nucleic acid. Taken from the crystallographic information files (Brown & McMahon, 2002 ▸) in the PHENIX library (Adams et al., 2010 ▸).
The scattering factors of atoms and ions for X-rays and electrons were calculated (e.g. Doyle & Turner, 1968 ▸; Schmidt & Weiss, 1979 ▸; Rez et al., 1994 ▸; Wang et al., 1996 ▸; Su & Coppens, 1997 ▸; Macchi & Coppens, 2001 ▸) and parameterized previously (e.g. Doyle & Turner, 1968 ▸; Rez et al., 1994 ▸; Waasmaier & Kirfel, 1995 ▸; Peng et al., 1996 ▸; Su & Coppens, 1997 ▸; Peng, 1998 ▸; Macchi & Coppens, 2001 ▸; Colliex et al., 2006 ▸), but there have been no reported scattering factors for chemically unstable ions involved in proteins and protein complexes except for O−. To fill the gap, we calculated the scattering factors of those ions by the multi-configuration Dirac–Fock (MCDF) method in this study. We only selected ions for the atoms, C, N, O, P and S that compose amino acids and nucleic acids, which are the major building blocks of biological molecules. The electron scattering factor of H+ is also presented. We then provide coefficients for the purpose of fitting the calculated scattering factors to the major curve models used in X-ray and electron crystallography and single-particle cryo-EM.
2. Calculations
2.1. Calculation of radial wavefunctions
Radial wavefunctions in the ground state were calculated for C, C+, C−, N, N+, N−, O, O+, O−, O2−, P, P+, P−, P2+, P3+, S, S+, S−, S2+, S2−, S3+ and S4+ by the MCDF method. The GRASP2K package (version 1.1, Jönsson et al., 2013 ▸) was used to perform the extended optimal level calculation for self-consistent fields of wavefunctions. Isolated negative ions are known to be unstable with a short lifetime in vacuum and tend to suffer from the inherent lack of convergence by MCDF calculations. A practical approach to this problem is placing positive charges outside a given radius around the ion (Watson, 1958 ▸; Schmidt & Weiss, 1979 ▸; Rez et al., 1994 ▸), but the resultant scattering curves differ significantly in the range sinθ/λ < 0.4 Å−1, depending on the size of the radius (Schmidt & Weiss, 1979 ▸). Previous studies (Wang et al., 1996 ▸; Macchi & Coppens, 2001 ▸) showed that the Dirac–Fock calculation yielded good solutions for O− and halides with no surrounding positive charges. Based on these studies, we carried out MCDF calculations in the absence of surrounding positive charges; this treatment was straightforward for O−, P− and S− as well as for the neutral atoms and positive ions with the exception of P3+ and S4+. For C− and N−, starting from initial estimates of radial functions for the neutral atom and/or increasing array sizes in the GRASP2K programs yielded reasonable solutions. No stable solutions were obtained using this approach for the closed-shell ions: O2−, S2−, P3+ and S4+.
2.2. Conversion to scattering factors
The scattering factors were calculated from relativistic wavefunctions as described in Su & Coppens (1997 ▸). Briefly, the radial charge density ρ(r) at a radius of r can be calculated as
where Pi(r) and Qi(r) represent the major and minor components, respectively, of the radial wavefunction for the ith orbital, and Nqi is the generalized occupation number for the corresponding orbital. The X-ray scattering factor Fx(s) at s = sinθ/λ can then be converted from ρ(r) by Fourier transform in polar coordinates as
The integral in equation (2) was obtained by the composite-Simpson summation at a fine step of r given by cubic spline interpolation. The scattering factors were computed up to s = 12 Å−1. A python script called scsumrhofft.py (provided in the supporting information) was written for these calculations in equations (1) and (2).
2.3. Parameterization of scattering factors
The ScatCurve package (Yonekura & Maki-Yonekura, 2016 ▸) was used to parameterize the X-ray scattering factors for the major curve models used in X-ray protein crystallography expressed in the form
where n = 4 for the four Gaussians plus a constant model (Colliex et al., 2006 ▸) and n = 5 for the five Gaussians plus a constant model (Waasmaier & Kirfel, 1995 ▸). The program scatcurvefit was modified to achieve a more robust minimization of the difference between the scattering factor and the model.
The X-ray scattering factor F x was converted to the electron scattering factor F el for an atom with Z 0 electrons and Z nuclear charges using the Mott formula:
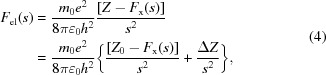 |
where m
0 represents the electron mass, e is the electron charge,  0 is the permittivity of free space, h is Planck’s constant, and (m
0
e
2/8π∊0
h
2) = 0.02393366. ΔZ is the ionic charge and is defined as ΔZ = Z – Z
0. The scattering factor of H+ was also obtained from equation (4) with F
x of H+ = 0 and ΔZ = 1. By using scatcurvefit (Yonekura & Maki-Yonekura, 2016 ▸) again, the electron scattering factors were parameterized for the five Gaussians plus a charge term model (Peng, 1998 ▸) expressed as
0 is the permittivity of free space, h is Planck’s constant, and (m
0
e
2/8π∊0
h
2) = 0.02393366. ΔZ is the ionic charge and is defined as ΔZ = Z – Z
0. The scattering factor of H+ was also obtained from equation (4) with F
x of H+ = 0 and ΔZ = 1. By using scatcurvefit (Yonekura & Maki-Yonekura, 2016 ▸) again, the electron scattering factors were parameterized for the five Gaussians plus a charge term model (Peng, 1998 ▸) expressed as
The calculated scattering factors and fitting to the curve model were evaluated from an R scat factor (Peng, 1998 ▸; Yonekura & Maki-Yonekura, 2016 ▸) by using scatcurvediff and scatcurvefit. R scat is defined as
where F is the scattering amplitude computed in this work and F c is the reference value or fitted value for the curve model.
3. Results and discussion
3.1. Validation of scattering factors
For validation of the X-ray scattering factors obtained from the MCDF calculations, we compared the calculated values for C, N, O, P, S and O− with the reference values, which are tabulated in the International Tables for Crystallography (Colliex et al., 2006 ▸). The tables contain these data up to s = 6 Å−1 for neutral atoms and 1.5 Å−1 for O−. R scat values defined in equation (6) were calculated in these ranges (Table 1 ▸). The slight differences of 0.02–0.01% indicate that the values calculated in this study are consistent with the reference data. Plots of the calculated scattering factors and the reference curves appeared almost identical.
Table 1. R scat factors of X-ray scattering factors against the reference curves.
| Atom | R scat † (%) |
|---|---|
| C | 0.0125 |
| N | 0.0194 |
| O | 0.0223 |
| O− | 0.0223 |
| P | 0.0047 |
| S | 0.0075 |
Defined in equation (6). Calculated between the scattering factors and the reference values in International Tables for Crystallography (Colliex et al., 2006 ▸). Summation over data from sinθ/λ = 0 to 1.5 Å−1 for O− and from 0 to 6 Å−1 for all the others.
Relativistic effects are known to be small for light atoms (e.g. Wang et al., 1996 ▸). Despite this, we included relativistic effects as most of the scattering factors in Colliex et al. (2006 ▸) adopted the relativistic calculation, even for the atoms concerned in this study. The MCDF calculation for those atoms does not require much computing cost on the current PC system. The reference scattering factor for O− was non-relativistically computed (Colliex et al., 2006 ▸) and the difference from our relativistic calculation is very small (Table 1 ▸), confirming a very small contribution from the relativistic term.
The scattering factors of the same atom with and without a charge match well when s = 0.2–0.3 Å−1 (Fig. 1 ▸). The curves of the calculated X-ray and electron scattering factors show these expected appearances, see Figs. 3 ▸ and 4 ▸, respectively. Figs. 3 ▸ and 4 ▸ also indicate that the point, where the curves of different charge states become overlapped, varies for each atom. In contrast, the electron scattering factors of H given in the International Tables for Crystallography (Colliex et al., 2006 ▸) and of H+ derived in Hirai et al. (2007 ▸) do not match even beyond s = 0.5 Å−1 (see Fig. 1 of Yonekura & Maki-Yonekura, 2016 ▸). We calculated the electron scattering factor of H+ from equation (4) by simply setting F x of H+ to 0 and ΔZ = 1. The new curve matches that of neutral H as sinθ/λ increases (Fig. 4 ▸). The calculated X-ray and electron scattering factors, including the new values for H+, are tabulated up to s = 6 Å−1 in Tables S1 and S2, respectively (see supporting information).
Figure 3.

Plots of X-ray scattering factors obtained by our MCDF calculations. The X-ray scattering factors at sinθ/λ = 0 are equal to the number of electrons Z 0. Dashed lines indicate plots of neutral atoms. Magenta and red curves correspond to the scattering factors of O, O+ and O−, and the green curves are those of S2+ and S3+. Plots in the lower row show the same curves as those in the upper row but up to sin θ/λ = 2 Å−1.
Figure 4.

Plots of electron scattering factors converted from the X-ray scattering factors by the Mott formula (equation 4). Broken lines indicate plots for the neutral atoms. Negative ions show negative values in a lower sin θ/λ range. Magenta and red curves correspond to the scattering factors of O, O+ and O−, the cyan and blue curves are those of H, H+ and the green curves are those of S2+ and S3+. The scattering curve of H is derived from International Tables for Crystallography (Colliex et al., 2006 ▸).
We also tried the standard quantum chemistry programs Gaussian 09 (Frisch et al., 2016 ▸) and GAMESS (Schmidt et al., 1993 ▸) for the calculation of the scattering factors, with the hope of obtaining the scattering factors of O2−, S2−, P3+ and S4+, as the MCDF calculation gave no stable solution for these ions. Molecular orbital calculations with ROHF, UHF, MP2,3,4 and CCSD(T) were performed using the various Gaussian-type basis sets such as cc-pVTZ, cc-pV5Z, aug-cc-pVTZ, d-aug-cc-pVTZ and so on. Charge density ρ was then calculated on a sufficiently fine grid and converted to the radial scattering factor. The R scat values, however, were around 0.5–1.0% against the references for neutral atoms and O−, and were much larger than the errors obtained from the MCDF calculations (Table 1 ▸), although the basis sets above were supposed to be available for quantitative calculation. Plots of the scattering factors appear to vary from the reference, particularly in the range of low sinθ/λ. This inconsistency may reflect that the Gaussian-type basis set cannot describe the cusps near the nucleus and inter-electron coalescent points (e.g. Pachucki & Komasa, 2004 ▸). Therefore, we did not adopt these calculations.
3.2. Parameterization of scattering factors
We then parameterized the X-ray scattering factors in Table S1 for the four Gaussians plus a constant (Table 2 ▸) and five Gaussians plus a constant models (Table 3 ▸), and the electron scattering factors in Table S2 for the five Gaussians plus a charge term model (Table 4 ▸). Data up to s = 6 Å−1 were used for all X-ray scattering factors and data from s = 0.04 to 6 Å−1 for the electron scattering factors of the positive ions. Only data from s = 0.04 to 1.5 Å−1 were used for the electron scattering factors of the negative ions, as R scat values between the scattering factors and the fitted curves were getting worse when including data beyond s = 1.5 Å−1. Calculated coefficients and R scat values are summarized in Tables 2 ▸, 3 ▸ and 4 ▸. All the R scat values are less than 0.09% and typically around 0.05–0.005%. The electron scattering factors of the negative ions give slightly worse R scat values, but these values are comparable with those reported for other ions (Peng, 1998 ▸). Equations (4) and (5) are identical for H+ when all a 1–5 = 0, and do not need to be parameterized for the five Gaussians plus a charge term model. Scattering factors of multivalent cations of phosphorus and sulfur may be necessary for some chemicals such as nicotinamide adenine dinucleotide phosphates [see the crystallographic information files (Brown & McMahon, 2002 ▸) in the PHENIX library (Adams et al., 2010 ▸)]. Tables S1, S2 and 2–4 are provided in the supporting information for future reference.
Table 2. Parameterization of X-ray scattering factors of ions for the four Gaussians plus a constant model.
| a 1 | a 2 | a 3 | a 4 | c | R scat † | |
|---|---|---|---|---|---|---|
| Ion | b 1 | b 2 | b 3 | b 4 | (%) | |
| C+ | 6.727E+0 | 1.439E+0 | 2.191E+0 | 9.913E-1 | −6.349E+0 | 0.0154 |
| 4.479E-3 | 6.231E-1 | 1.313E+1 | 2.971E+1 | |||
| C− | 1.598E+0 | 2.161E+0 | 2.360E+0 | 6.549E-1 | 2.238E-1 | 0.0606 |
| 5.843E-1 | 1.346E+1 | 3.831E+1 | 1.405E+2 | |||
| N+ | 1.570E+0 | 2.068E+0 | 1.847E+0 | 2.870E-1 | 2.271E-1 | 0.0025 |
| 4.122E-1 | 8.064E+0 | 1.665E+1 | 3.340E+1 | |||
| N− | 1.569E+0 | 2.640E+0 | 2.795E+0 | 7.386E-1 | 2.551E-1 | 0.0394 |
| 4.393E-1 | 9.202E+0 | 2.624E+1 | 9.062E+1 | |||
| O+ | 1.553E+0 | 2.237E+0 | 2.466E+0 | 5.079E-1 | 2.367E-1 | 0.0015 |
| 3.148E-1 | 5.618E+0 | 1.191E+1 | 2.504E+1 | |||
| O− | 1.535E+0 | 3.038E+0 | 3.280E+0 | 8.555E-1 | 2.886E-1 | 0.0346 |
| 3.459E-1 | 6.556E+0 | 1.876E+1 | 6.328E+1 | |||
| P+ | 1.732E+0 | 6.472E+0 | 3.698E+0 | 9.940E-1 | 1.105E+0 | 0.0065 |
| 5.089E-1 | 1.893E+0 | 2.424E+1 | 5.215E+1 | |||
| P− | 1.990E+0 | 6.243E+0 | 4.686E+0 | 1.928E+0 | 1.146E+0 | 0.0423 |
| 5.903E-1 | 1.967E+0 | 3.046E+1 | 1.031E+2 | |||
| P2+ | 1.825E+0 | 6.368E+0 | 3.076E+0 | 6.076E-1 | 1.124E+0 | 0.0046 |
| 5.431E-1 | 1.924E+0 | 2.114E+1 | 4.185E+1 | |||
| S+ | 1.419E+0 | 6.923E+0 | 4.736E+0 | 1.076E+0 | 8.458E-1 | 0.0115 |
| 2.405E-1 | 1.459E+0 | 2.028E+1 | 4.519E+1 | |||
| S− | 1.505E+0 | 6.721E+0 | 5.614E+0 | 2.130E+0 | 1.024E+0 | 0.0372 |
| 3.506E-1 | 1.507E+0 | 2.411E+1 | 7.728E+1 | |||
| S2+ | 1.442E+0 | 6.787E+0 | 4.206E+0 | 5.939E-1 | 9.715E-1 | 0.0092 |
| 3.088E-1 | 1.488E+0 | 1.846E+1 | 3.950E+1 | |||
| S3+ | 1.611E+0 | 6.550E+0 | 3.434E+0 | 3.284E-1 | 1.078E+0 | 0.0051 |
| 4.062E-1 | 1.543E+0 | 1.637E+1 | 3.422E+1 |
Defined in equation (6). Calculated between the scattering factors and the fitted curves. Summation over data from sinθ/λ = 0 to 2 Å−1.
Table 3. Parameterization of X-ray scattering factors of ions for the five Gaussians plus a constant model.
| a 1 | a 2 | a 3 | a 4 | a 5 | c | R scat † | |
|---|---|---|---|---|---|---|---|
| Ion | b 1 | b 2 | b 3 | b 4 | b 5 | (%) | |
| C+ | 8.431E-2 | 5.957E-1 | 1.145E+0 | 2.224E+0 | 9.528E-1 | −2.457E-3 | 0.0194 |
| 4.787E-2 | 2.273E-1 | 7.042E-1 | 1.328E+1 | 3.012E+1 | |||
| C− | 5.946E-1 | 1.229E+0 | 2.295E+0 | 2.257E+0 | 6.050E-1 | 1.642E-2 | 0.0516 |
| 1.840E-1 | 7.149E-1 | 1.408E+1 | 4.041E+1 | 1.463E+2 | |||
| N+ | 1.670E-1 | 6.515E-1 | 9.987E-1 | 2.749E+0 | 1.427E+0 | 5.943E-3 | 0.0228 |
| 6.426E-2 | 2.094E-1 | 5.482E-1 | 9.105E+0 | 2.215E+1 | |||
| N− | 6.423E-1 | 1.169E+0 | 2.685E+0 | 2.758E+0 | 7.215E-1 | 2.224E-2 | 0.0403 |
| 1.451E-1 | 5.304E-1 | 9.338E+0 | 2.664E+1 | 9.161E+1 | |||
| O+ | 6.042E-1 | 1.171E+0 | 2.398E+0 | 2.376E+0 | 4.253E-1 | 2.527E-2 | 0.0047 |
| 1.078E-1 | 3.764E-1 | 5.797E+0 | 1.247E+1 | 2.613E+1 | |||
| O− | 6.950E-1 | 1.102E+0 | 3.023E+0 | 3.287E+0 | 8.611E-1 | 2.919E-2 | 0.0367 |
| 1.195E-1 | 4.139E-1 | 6.546E+0 | 1.869E+1 | 6.309E+1 | |||
| P+ | 1.496E+0 | 1.992E+0 | 5.676E+0 | 3.739E+0 | 9.422E-1 | 1.546E-1 | 0.0107 |
| 7.120E-2 | 9.188E-1 | 1.987E+0 | 2.446E+1 | 5.304E+1 | |||
| P− | 1.505E+0 | 2.539E+0 | 5.185E+0 | 4.696E+0 | 1.909E+0 | 1.595E-1 | 0.0431 |
| 7.246E-2 | 1.008E+0 | 2.097E+0 | 3.061E+1 | 1.036E+2 | |||
| P2+ | 1.501E+0 | 2.253E+0 | 5.421E+0 | 3.145E+0 | 5.236E-1 | 1.563E-1 | 0.0084 |
| 7.155E-2 | 9.661E-1 | 2.041E+0 | 2.148E+1 | 4.363E+1 | |||
| S+ | 1.477E+0 | 1.268E+0 | 6.299E+0 | 4.788E+0 | 1.011E+0 | 1.569E-1 | 0.0155 |
| 6.175E-2 | 6.681E-1 | 1.521E+0 | 2.047E+1 | 4.613E+1 | |||
| S− | 1.483E+0 | 1.597E+0 | 6.016E+0 | 5.623E+0 | 2.114E+0 | 1.610E-1 | 0.0397 |
| 6.259E-2 | 7.320E-1 | 1.570E+0 | 2.419E+1 | 7.758E+1 | |||
| S2+ | 1.483E+0 | 1.520E+0 | 6.052E+0 | 4.257E+0 | 5.300E-1 | 1.594E-1 | 0.0131 |
| 6.220E-2 | 7.229E-1 | 1.558E+0 | 1.865E+1 | 4.092E+1 | |||
| S3+ | 1.489E+0 | 1.983E+0 | 5.618E+0 | 3.476E+0 | 2.716E-1 | 1.626E-1 | 0.0084 |
| 6.277E-2 | 8.002E-1 | 1.631E+0 | 1.656E+1 | 3.620E+1 |
Defined in equation (6). Calculated between the scattering factors and the fitted curves. Summation over data from sinθ/λ = 0 to 6 Å−1.
Table 4. Parameterization of electron scattering factors of ions for the five Gaussians plus a charge term model.
| a 1 | a 2 | a 3 | a 4 | a 5 | R scat † | |
|---|---|---|---|---|---|---|
| Ion | b 1 | b 2 | b 3 | b 4 | b 5 | (%) |
| C+ | 2.079E-2 | 9.266E-2 | 2.949E-1 | 6.812E-1 | 3.304E-1 | 0.0156 |
| 5.950E-2 | 5.359E-1 | 2.760E+0 | 9.283E+0 | 2.442E+1 | ||
| C- | 2.248E-1 | 8.254E-1 | 1.769E+0 | 1.690E+0 | 6.994E-1 | 0.0810 |
| 5.518E-1 | 4.308E+0 | 1.600E+1 | 5.196E+1 | 1.708E+2 | ||
| N+ | 2.296E-2 | 1.004E-1 | 3.289E-1 | 6.546E-1 | 2.733E-1 | 0.0132 |
| 5.522E-2 | 4.910E-1 | 2.402E+0 | 7.751E+0 | 2.051E+1 | ||
| N− | 2.192E-1 | 7.256E-1 | 1.398E+0 | 1.245E+0 | 4.381E-1 | 0.0430 |
| 4.784E-1 | 3.389E+0 | 1.171E+1 | 3.604E+1 | 1.125E+2 | ||
| O+ | 2.439E-2 | 1.036E-1 | 3.360E-1 | 6.112E-1 | 2.447E-1 | 0.0120 |
| 5.082E-2 | 4.390E-1 | 2.036E+0 | 6.407E+0 | 1.710E+1 | ||
| O− | 2.236E-1 | 6.923E-1 | 1.176E+0 | 9.354E-1 | 2.821E-1 | 0.0218 |
| 4.372E-1 | 2.918E+0 | 9.670E+0 | 2.868E+1 | 8.489E+1 | ||
| P+ | 6.191E-2 | 3.154E-1 | 6.324E-1 | 1.661E+0 | 1.027E+0 | 0.0447 |
| 6.525E-2 | 6.224E-1 | 3.009E+0 | 1.258E+1 | 3.411E+1 | ||
| P− | 4.590E-1 | 1.002E+0 | 2.724E+0 | 3.228E+0 | 1.336E+0 | 0.0728 |
| 5.019E-1 | 3.545E+0 | 1.445E+1 | 4.313E+1 | 1.303E+2 | ||
| P2+ | 4.997E-2 | 2.497E-1 | 4.690E-1 | 1.099E+0 | 6.144E-1 | 0.0233 |
| 5.712E-2 | 5.372E-1 | 2.398E+0 | 1.051E+1 | 2.704E+1 | ||
| S+ | 6.232E-2 | 3.129E-1 | 6.541E-1 | 1.742E+0 | 9.377E-1 | 0.0391 |
| 6.149E-2 | 5.785E-1 | 2.848E+0 | 1.107E+1 | 2.978E+1 | ||
| S− | 4.496E-1 | 9.810E-1 | 2.598E+0 | 2.717E+0 | 8.614E-1 | 0.0543 |
| 4.656E-1 | 3.259E+0 | 1.233E+1 | 3.583E+1 | 1.055E+2 | ||
| S2+ | 5.029E-2 | 2.440E-1 | 4.671E-1 | 1.196E+0 | 7.100E-1 | 0.0230 |
| 5.364E-2 | 4.908E-1 | 2.186E+0 | 8.911E+0 | 2.266E+1 | ||
| S3+ | 3.991E-2 | 1.787E-1 | 3.465E-1 | 7.425E-1 | 5.600E-1 | 0.0124 |
| 4.652E-2 | 4.001E-1 | 1.626E+0 | 6.936E+0 | 1.724E+1 |
Defined in equation (6). Calculated between the scattering factors and the fitted curves. Summation over data from sinθ/λ = 0.04 to 6 Å−1 for the positive ions and from 0.04 to 1.5 Å−1 for the negative ions.
3.3. Radiation damage
Density losses in the negatively charged side chains of aspartate and glutamate were often observed in recent single-particle reconstructions (e.g. Bartesaghi et al., 2014 ▸; Fromm et al., 2015 ▸; Hryc et al., 2017 ▸) and are likely to be interpreted as radiation damage (Fioravanti et al., 2007 ▸) even when reconstructed from data sets collected with a low accumulated electron dose. Part of the density loss should be attributed to negatively charged atoms, and this can be checked if a map calculated without lower-resolution data recovers the corresponding density (Yonekura et al., 2015 ▸). In contrast, an intense beam such as an X-ray-free electron laser causes ionization damage to samples and this may affect the diffraction patterns (Hau-Riege, 2007 ▸). The scattering factors of these radicals losing some electrons may help us to treat the data properly (Hau-Riege, 2007 ▸).
4. Conclusions
We have provided the X-ray and electron scattering factors of C+, C−, N+, N−, O+, O−, P+, P−, P2+, S+, S−, S2+ and S3+, the electron scattering factor of H+ and the coefficients for the four Gaussians plus a constant, the five Gaussians plus a constant and the five Gaussians plus a charge term models. The scattering factors of partially charged atoms and the fitting parameters for a curve model can be obtained by a linear combination of those for neutral and fully ionized atoms using scatcurvecomb (as in Yonekura & Maki-Yonekura, 2016 ▸). We are now testing the calculated values in this study for structure refinement against data obtained by electron 3D crystallography and single-particle cryo-EM.
Supplementary Material
Supplementary Tables. DOI: 10.1107/S2052252518005237/fq5001sup1.pdf
Supplementary code.. DOI: 10.1107/S2052252518005237/fq5001sup2.zip
Acknowledgments
We thank Toshiyuki Azuma for comments on features of divalent anions. Calculations with Gaussian 09 were performed in the HOKUSAI GreatWave HPC system, RIKEN.
Funding Statement
This work was funded by the Japan Science and Technology Agency SENTAN program grant SENTAN to Koji Yonekura. Japan Society for the Promotion of Science Grant-in-Aid grants 24657111 and 16H04757.
References
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed]
- Bartesaghi, A., Matthies, D., Banerjee, S., Merk, A. & Subramaniam, S. (2014). Proc. Natl Acad. Sci. USA, 111, 11709–11714. [DOI] [PMC free article] [PubMed]
- Brown, I. D. & McMahon, B. (2002). Acta Cryst. B58, 317–324. [DOI] [PubMed]
- Chang, S., Head-Gordon, T., Glaeser, R. M. & Downing, K. H. (1999). Acta Cryst A55, 305–313. [DOI] [PubMed]
- Colliex, C., Cowley, J. M., Dudarev, S. L., Fink, M., Gjønnes, J., Hilderbrandt, R., Howie, A., Lynch, D. F., Peng, L. M., Ren, G., Ross, A. W., Smith, V. H., Spence, J. C. H., Steeds, J. W., Wang, J., Whelan, M. J. & Zvyagin, B. B. (2006). Electron diffraction. In International Tables for Crystallography, Vol. C, edited by E. Prince, pp. 263–281. Dordrecht: Kluwer.
- Doyle, P. A. & Turner, P. S. (1968). Acta Cryst. A24, 390–397.
- Fioravanti, E., Vellieux, F. M. D., Amara, P., Madern, D. & Weik, M. (2007). J. Synchrotron Rad. 14, 84–91. [DOI] [PubMed]
- Frisch, M. J. et al. (2016). Gaussian 09, Revision A.02, Gaussian Inc., Wallingford, CT, USA.
- Fromm, S. A., Bharat, T. A., Jakobi, A. J., Hagen, W. J. & Sachse, C. (2015). J. Struct. Biol. 189, 87–97. [DOI] [PMC free article] [PubMed]
- Hau-Riege, S. P. (2007). Phys. Rev. A, 76, 042511.
- Hirai, T., Mitsuoka, K., Kidera, A. & Fujiyoshi, Y. (2007). J. Electron Microsc. 56, 131–140. [DOI] [PubMed]
- Hryc, C. F., Chen, D. H., Afonine, P. V., Jakana, J., Wang, Z., Haase-Pettingell, C., Jiang, W., Adams, P. D., King, J. A., Schmid, M. F. & Chiu, W. (2017). Proc. Natl Acad. Sci. USA, 114, 3103–3108. [DOI] [PMC free article] [PubMed]
- Jönsson, P., Gaigalas, G., Bieroń, J., Froese Fischer, C. & Grant, I. P. (2013). Comput. Phys. Commun. 184, 2197–2203.
- Macchi, P. & Coppens, P. (2001). Acta Cryst. A57, 656–662. [DOI] [PubMed]
- Mitsuoka, K., Hirai, T., Murata, K., Miyazawa, A., Kidera, A., Kimura, Y. & Fujiyoshi, Y. (1999). J. Mol. Biol. 286, 861–882. [DOI] [PubMed]
- Pachucki, K. & Komasa, J. (2004). Chem. Phys. Lett. 389, 209–211.
- Peng, L.-M. (1998). Acta Cryst. A54, 481–485.
- Peng, L.-M., Ren, G., Dudarev, S. L. & Whelan, M. J. (1996). Acta Cryst. A52, 257–276.
- Rez, D., Rez, P. & Grant, I. (1994). Acta Cryst. A50, 481–497.
- Schmidt, M. W., Baldridge, K. K., Boatz, J. A., Elbert, S. T., Gordon, M. S., Jensen, J. H., Koseki, S., Matsunaga, N., Nguyen, K. A., Su, S., Windus, T. L., Dupuis, M. & Montgomery, J. A. (1993). J. Comput. Chem. 14, 1347–1363.
- Schmidt, P. C. & Weiss, A. (1979). Z. Naturforsch. 34, 1471–1481.
- Su, Z. & Coppens, P. (1997). Acta Cryst. A53, 749–762.
- Waasmaier, D. & Kirfel, A. (1995). Acta Cryst. A51, 416–431.
- Wang, J., Smith, V. H., Bunge, C. F. & Jáuregui, R. (1996). Acta Cryst. A52, 649–658.
- Watson, R. E. (1958). Phys. Rev. 111, 1108–1110.
- Yamashita, H. & Kidera, A. (2001). Acta Cryst. A57, 518–525. [DOI] [PubMed]
- Yonekura, K. & Maki-Yonekura, S. (2016). J. Appl. Cryst. 49, 1517–1523.
- Yonekura, K., Kato, K., Ogasawara, M., Tomita, M. & Toyoshima, C. (2015). Proc. Natl Acad. Sci. USA, 112, 3368–3373. [DOI] [PMC free article] [PubMed]
- Zhong, S., Dadarlat, V. M., Glaeser, R. M., Head-Gordon, T. & Downing, K. H. (2002). Acta Cryst. A58, 162–170. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables. DOI: 10.1107/S2052252518005237/fq5001sup1.pdf
Supplementary code.. DOI: 10.1107/S2052252518005237/fq5001sup2.zip