

Abstract
Inhibition of WEE1 is emerging as a promising chemosensitization strategy in many cancers including acute leukemia. Our lab and others have demonstrated that a small-molecule inhibitor of WEE1, AZD1775, sensitizes acute leukemia cells to cytarabine; however, a mechanism of combinatorial activity has remained elusive. Thus, we sought to determine the relative contribution of WEE1 targets CDK1 and CDK2 to the combinatorial activity of AZD1775 and cytarabine. To accomplish this, we expressed “WEE1 resistant” CDK1 (CDK1-AF) and CDK2 (CDK2-AF) constructs in a T-ALL cell line. Expression of CDK1/2-AF together, but neither alone, enhanced the anti-proliferative effects, DNA damage and apoptosis induced by cytarabine. Furthermore, pharmacologic inhibition of CDK1 alone or CDK1 and CDK2 together reduced the combinatorial activity of AZD1775 and cytarabine. Thus, increased activity of both CDK1 and CDK2 in response to WEE1 inhibition is necessary for the combinatorial activity of AZD1775 and cytarabine. This suggests the role of WEE1 in cells with accumulated DNA damage extends beyond regulation of CDK1 and the G2/M checkpoint and highlights the importance of WEE1 in mediating progression through the cell cycle.
Keywords: WEE1, AZD1775, cytarabine, leukemia, CDK1, CDK2, DNA damage, apoptosis
Introduction
Advances such as risk-directed chemotherapy have substantially improved survival in AML and ALL (Grimwade and Hills, 2009). However, novel therapies are needed for patients who relapse or cannot tolerate standard induction treatment. One strategy to improve outcomes is addition of targeted agents that enhance the efficacy of chemotherapies currently used to treat these diseases. WEE1 is emerging as a promising target in cells treated with genotoxic chemotherapies. WEE1 is a tyrosine kinase that inhibits CDK1 and CDK2 by phosphorylation at tyrosine 15. In response to DNA damage, WEE1 activity is upregulated to promote cell cycle arrest at the intra-S or G2/M checkpoint (Matheson et al., 2016; Perry and Kornbluth, 2007). This promotes DNA damage repair prior to entry into mitosis and allows cells to avoid DNA damage-induced apoptosis or propagation of mutations to daughter cells.
Our lab previously demonstrated that AZD1775, a small-molecule inhibitor of WEE1, enhances the anti-proliferative effects of cytarabine (Ara-C) in AML and T-ALL cells by abrogating the S-phase arrest induced by Ara-C and increasing DNA damage (Ford et al., 2015; Porter et al., 2012). This work, along with the work of others, provided support for a clinical trial examining the efficacy of AZD1775 combined with Ara-C in AML (NCT02666950). As this combination progresses to clinic trials, a complete understanding the mechanism of the combinatorial activity of AZD1775 and Ara-C will be necessary to identify patients most likely to benefit from treatment. While a number of studies have examined alterations in the cell cycle in response to AZD1775 and anti-metabolite chemotherapies such as Ara-C, the relative contribution of WEE1 targets CDK1 and CDK2 has not been studied. Thus, we sought to understand the contribution of CDK1 and CDK2 to the combinatorial activity of AZD1775 and Ara-C. We hypothesized that increased activity of CDK1 and/or CDK2 is required for the combinatorial activity of AZD1775 and Ara-C. Using genetic models of “WEE1 resistant” CDK1 and CDK2, we demonstrate that increased activity of both CDK1 and CDK2 is required to enhance the anti-proliferative effects of Ara-C in a T-ALL cell line. This is confirmed by pharmacologic data demonstrating inhibition of CDK1 alone or CDK1 and CDK2 together can reduce the combinatorial effect of AZD1775 and Ara-C. Together, these data highlight the unique ability of WEE1 to regulate two CDKs that function in different phases of the cell cycle.
Materials and Methods
Cell Culture
Jurkat and MV4;11 cells were generous gifts from the laboratories of Drs. Douglas Graham and James DeGregori. Cell lines were DNA fingerprinted by multiplex PCR using the Profiler Plus or Identifier Kits (ABI) as previously described (Van Linden et al., 2013), and periodically tested for Mycoplasma by PCR. Cells were cultured in RPMI with 10% FBS and penicillin/streptomycin at 37°C in humidified air supplemented with 5% CO2 and maintained in culture for no longer than 2 months.
Lentivirus Preparation
CDK1/2-AF constructs were generously provided by Dr. David O. Morgan and cloned into response vectors of the Lenti-X Tet-On 3G Inducible System (Clontech Laboratories, Inc.) Virus-containing media was prepared according to manufacturer's protocol.
Chemicals, Antibodies, and Reagents
AZD1775 was provided by AstraZeneca (Wilmington, DE). Cytarabine, doxycycline, and puromycin were purchased from Sigma-Aldrich and diluted in water. RO-3306 and roscovitine were purchased from EMD Millipore (Billerica, MA) and diluted in DMSO. Antibodies against HA-tag, GAPDH, PARP, γH2AX, tubulin, and actin were purchased from Cell Signaling Technology (Danvers, MA).
Flow Cytometry
Cell viability was determined with the Guava EasyCytePlus (Millipore, Billerica, MA) by measuring cell counts with propdium iodide exclusion.
Statistical Analysis
Data analysis and graphing was performed using Graphpad Prism 5 (GraphPad Software, La Jolla, CA). Unless otherwise indicated, graphs represent the mean from a minimum of three biological replicate experiments, and error bars portray the standard error of the mean. Oneway ANOVA was used to compare 3 or more samples with a single variable. The Bonferroni correction was applied to determine significance between any two conditions. Non-linear regression was used to generate dose-response curves and determine IC50 values.
Results
Expression of WEE1-Resistant CDK1 and CDK2 Enhances the Anti-Proliferative Effects of Cytarabine
Inhibition of WEE1 reduces inhibitory phosphorylation of CDK1 and CDK2 in response to DNA damage. This prevents cell cycle arrest at the intra-S or G2/M checkpoint and results in an increase in apoptosis (Hirai et al., 2009; Porter et al., 2012). However, it is not known whether increased activity of one or both CDKs is required to enhance the efficacy of DNA damaging agents. We sought to determine the necessity of CDK1/2 activity in the context of Ara-C treatment by expressing ‘WEE1-resistant’ CDK constructs in the T-ALL cell line Jurkat. These mutant CDK constructs substitute alanine for threonine at position 14 and phenylalanine for tyrosine at position 15 yielding CDK constructs that cannot be inhibited by phosphorylation (CDK1/2-AF). Previous reports have confirmed expression of these constructs results in increased CDK activity (Jin et al., 1996). The CDK1/2-AF constructs were cloned into doxycycline-inducible vectors, and expression of the HA-tagged proteins was observed when cells were treated with doxycycline (Figure 1a). To test the effects of CDK1/2-AF expression on cell viability in cytarabine-treated cells, Jurkat cells transduced with vector control, CDK1-AF, and/or CDK2-AF plasmids were treated with Ara-C with and without doxycycline. Expression of CDK1-AF or CDK2-AF alone did not sensitize cells to cytarabine, but we observed significant sensitization when both CDK1-AF and CDK2-AF were expressed (Figure 1b). Next, these cell lines were treated with Ara-C in concentrations ranging from 0.5 nM to 40 nM. Doxycycline-induced expression of CDK1-AF or CDK2-AF alone did not shift the dose-response curve of Ara-C. However, a shift of the dose-response curve was observed in cells expressing both CDK1-AF and CDK2-AF. The IC50 value of Ara-C was 6.09 nM when expression of the CDK constructs was not induced, and this decreased to 2.86 nM when both constructs were expressed (Figure 1c). Thus, increased activity of both CDK1 and CDK2 enhances the anti-proliferative effects of Ara-C.
Figure 1. Expression of CDK1-AF and CDK2-AF sensitizes T-ALL cells to cytarabine.

A. Jurkat cells transduced with empty vector (pLVX), CDK1-AF, or CDK2-AF constructs were give no treatment (NT) or doxycycline (1 ug/mL) for 48 hr after which protein lysates were subjected to western blotting with antibodies specific to HA-tag and GAPDH. B. Jurkat cells transduced with empty vector (pLVX), CDK1-AF, and/or CDK2-AF were treated with Ara-C (10 nM) and/or doxycycline (1 ug/mL) for 72 hr. Viable cell counts are normalized to cells receiving no treatment (NT). Results are shown as mean ± SEM from three independent experiments. *, P < 0.05. **, P < 0.01. ***, P < 0.001. C. Jurkat cells transduced with empty vector (pLVX), CDK1-AF, and/or CDK2-AF were treated with doxycycline (1 ug/mL) and Ara-C (0.5-40 nM) for 72 hr. Viable cell counts are normalized to cells receiving no Ara-C treatment (NT). Results are shown as mean ± SEM from three independent experiments. IC50 values for each cell line with and without doxycycline are displayed below the corresponding graph.
Expression of WEE1-Resistant CDK1 and CDK2 Increases DNA Damage and Apoptosis in Cytarabine-Treated Cells
Our lab and others have demonstrated that AZD1775 enhances DNA damage induction and apoptosis in cells treated with Ara-C (Ford et al., 2015; Porter et al., 2012; Tibes et al., 2012). Therefore, we questioned whether increased activity of CDK1 and CDK2 was responsible for these events. Jurkat cells expressing CDK1-AF and/or CDK2-AF were treated with Ara-C and/or doxycycline as well as Ara-C and AZD1775 as a positive control. Expression of CDK1-AF or CDK2-AF alone did not increase apoptosis as evidenced by a lack of cleaved PARP. However, expression of both constructs produced cleaved PARP comparable to levels observed in cells treated with Ara-C and AZD1775 (Figure 2). We observed a slight increase in γH2AX in cells expressing either CDK1-AF or CDK2-AF suggesting that increased activity of either CDK can induce some DNA damage in cells exposed to Ara-C. However, more DNA damage occurred when activity of both CDK1 and CDK2 increased. Surprisingly, expression of both CDK constructs induced less DNA damage than AZD1775 in cytarabine-treated cells (Figure 2). Thus, increased activity of both CDK1 and CDK2 increases DNA damage and apoptosis, although DNA damage induction is greater in cells treated with AZD1775.
Figure 2. Expression of CDK1-AF and CDK2-AF Increases DNA Damage and Apoptosis in Cytarabine-Treated Cells.
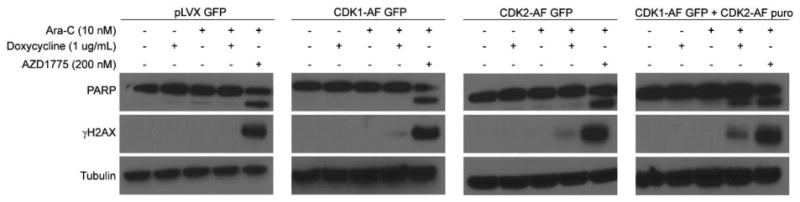
Jurkat cells transduced with empty vector (pLVX), CDK1-AF, and/or CDK2-AF constructs were treated with the indicated doses of Ara-C, doxycycline, and/or AZD1775 for 24 hr. Protein lysates were subjected to western blotting with antibodies specific to PARP, γH2AX, and tubulin.
Pharmacologic Inhibition of CDK1 and CDK2 Reduces the Combinatorial Activity of AZD1775 and Cytarabine
We then asked whether pharmacologic inhibition of CDK1 alone or both CDK1 and CDK2 could reduce the combinatorial activity of AZD1775 and Ara-C. RO-3306 is an ATP-competitive inhibitor of CDK1. The drug is 10-fold more selective for CDK1 than for CDK2 and 50-fold more selective for CDK1 than for CDK4 and CDK6. (Vassilev et al., 2006). AZD1775 treatment significantly enhanced the anti-proliferative effect of Ara-C in Jurkat cells, but the effect of the combination was reduced when cells were treated with RO-3306 (Figure 3a). This is consistent with the findings in CDK1/2-AF-expressing Jurkat cells which suggests increased activity of both CDK1 and CDK2 is necessary for the combinatorial activity of AZD1775 and Ara-C. Next, cells were treated with roscovitine, an ATP-competitive inhibitor of CDK1, CDK2, CDK5, and CDK7 (Cicenas et al., 2015). Inhibition of CDK1 and CDK2 with roscovitine reduced the combinatorial effect of AZD1775 and Ara-C in Jurkat cells as well as in the AML cell line, MV;411 (Figure 3b,c). Roscovitine treatment also reduced apoptosis as evidenced by reduced cleaved PARP in cells treated with AZD1775 and Ara-C (Figure 3d). Together, these data confirm increased activity of CDK1 and CDK2 is required for the combinatorial activity of AZD1775 and Ara-C.
Figure 3. Inhibition of CDK1 or CDK1 and CDK2 Reduces the Combinatorial Effect of AZD1775 and Cytarabine.
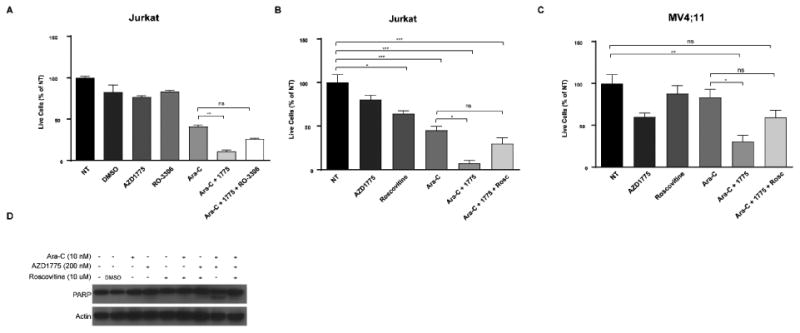
A. Relative numbers of viable Jurkat cells treated with DMSO (vehicle control), AZD1775 (200 nM), RO-3306 (2.5 uM), and/or Ara-C (10 nM) for 72 hr. Viable cell counts are normalized to cells receiving no treatment (NT). Results are shown as mean ± SEM from three independent experiments. **, P < 0.01. B and C. Relative numbers of viable Jurkat (B) or MV4;11 (C) cells treated with DMSO (vehicle control), AZD1775 (200 nM), roscovitine (10 uM), and/or Ara-C (10 nM) for 72 hr. Viable cell counts are normalized to cells receiving no treatment (NT). Results are shown as mean ± SEM from three independent experiments. *, P < 0.05. **, P < 0.01. ***, P < 0.001. D. Jurkat cells were treated with the indicated doses of roscovitine, AZD1775, and/or Ara-C for 24 hr after which protein lysates were subjected to western blotting with antibodies specific to PARP and actin.
Discussion
Inhibition of WEE1 sensitizes cancer cells to many DNA damaging agents; however, the mechanisms of sensitization are not fully understood. Our group previously reported that AZD1775 sensitized AML and T-ALL cells to Ara-C by abrogating S-phase arrest and promoting apoptosis over DNA damage repair (Ford et al., 2015; Porter et al., 2012). This work, combined with data from others, provided justification for a clinical trial examining the effects of AZD1775 in combination with Ara-C in patients with AML (NCT02666950). An enhanced understanding of the mechanism of combinatorial activity between AZD1775 and Ara-C will assist in identifying patients most likely to benefit from this combination and in anticipating mechanisms of resistance.
In this study, we have demonstrated that expression of WEE1 resistant CDK1 and CDK2 reduces proliferation and increases DNA damage and apoptosis in cytarabine-treated cells. Furthermore, inhibition of CDK1 alone or CDK1 and CDK2 together increased viability in cells treated with AZD1775 and Ara-C indicating that increased activity of CDK1 and CDK2 is required for combinatorial activity. Many studies have suggested that inhibition of WEE1 sensitizes cells to DNA damaging agents by preventing cell cycle arrest at the G2/M checkpoint and promoting premature mitotic entry (Hirai et al., 2009; Li et al., 2002; Wang et al., 2001). However, the requirement for increased activity of CDK2, as well as CDK1, suggests that forced activation of CDK1 and disruption of G2/M-phase arrest is not sufficient to sensitize T-ALL cells to Ara-C. This is consistent with previous findings from our lab which demonstrated that AZD1775 abrogates S-phase arrest in cells treated with Ara-C but does not induce premature mitosis (Van Linden et al., 2013). Therefore, it is possible that increased activity of CDK1 and CDK2 sensitizes cells to Ara-C by disrupting the S-phase arrest induced by Ara-C or by altering cell cycle dynamics in multiple phases.
In addition to disrupting DNA damage-induced cell cycle arrest, inhibition of WEE1 also impairs homologous recombination (HR), increases replication origin firing, and reduces cellular nucleotide pools (Beck et al., 2012; Krajewska et al., 2013; Pfister et al., 2015; Garcia et al., 2017). Incorporation of Ara-C into DNA during replication results in DNA single-strand breaks and stalled replication forks (Ewald et al., 2008), and stalled replication forks are frequently repaired by HR (Iraqui et al., 2012). Indeed, HR is the major survival mechanism in AML cells treated with sapacitabine, a nucleoside analogue derivative of Ara-C (Liu et al., 2010). As inhibition of WEE1 impairs HR (Krajewska et al., 2013), AZD1775 could sensitize cells to Ara-C by preventing HR-mediated DNA damage repair. Additionally, hyperactivation of CDK1 and CDK2 in response to WEE1 inhibition results in firing of an increased number of origins of replication as well as decreased nucleotide synthesis (Beck et al., 2012; Pfister et al., 2015). Increased replication origin firing could lead to Ara-C incorporation into DNA at an increased number of sites while reduced nucleotide concentrations might increase the Ara-C to deoxycytidine triphosphate ratio leading to increased incorporation of the nucleoside analog. Either, or both, of these events could promote an increase in DNA damage induction in cells treated with Ara-C.
Of note, we observed less DNA damage induction as measured by γH2AX in cells expressing CDK1-AF and CDK2-AF treated with Ara-C compared with cells treated with AZD1775 and Ara-C. Therefore, AZD1775 could enhance DNA damage induction in cytarabine-treated cells through mechanisms independent of CDK1 and CDK2 such as off-target effects of AZD1775 or altered signaling through histone H2B, another substrate of WEE1 (Mahajan et al., 2012). While we observed differences in DNA damage induction in cytarabine-treated cells expressing CDK1-AF and CDK2-AF compared to cells treated with AZD1775, we did not observe a substantial difference in apoptosis induction. Thus, increased activity of CDK1 and CDK2 could sensitize cells to Ara-C through mechanisms independent of DNA damage induction such as altering apoptotic signaling cascades which both CDK1 and CDK2 are known to do (Chu et al., 2012; Konishi et al., 2002; Megyesi et al., 2016).
In conclusion, this study demonstrates that increased activity of WEE1 substrates CDK1 and CDK2 is required for the combinatorial activity of AZD1775 and Ara-C in a T-ALL cell line. While additional work is required to determine how CDK1 and CDK2 cooperate to sensitize cells to Ara-C, this report highlights the unique ability of WEE1 to regulate two CDKs necessary for cell cycle progression.
Highlights.
Neither active CDK1 or CDK2 is sufficient to sensitize leukemia cells to ARA-C
Activation of both CDK1 and CDK2 is necessary sensitize leukemia cells to ARA-C
Activation of CDK1 and CDK2 increases DNA damage and apoptosis induced by ARA-C
Inhibition of CDK1 or CDK1 and CDK2 reduces the effect of WEE1 inhibition and ARA-C
Acknowledgments
This work was supported by grants from the National Institutes of Health to C.C. Porter (CA172385), the University of Colorado Cancer Center (CA046934; Theodorescu), and the CU Medical Scientist Training Program (GM008497; Gutierrez-Hartmann).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beck H, Nahse-Kumpf V, Larsen MS, O'Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuasen RG, Sorensen CS. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu R, Terrano DT, Chambers TC. Cdk1/cyclin B plays a key role in mitotic arrest-induced apoptosis by phosphorylation of Mcl-1, promoting its degradation and freeing Bak from sequestration. Biochemical pharmacology. 2012;83:199–206. doi: 10.1016/j.bcp.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I, Stankevicius V, Kaupinis A, Valius M. Roscovitine in cancer and other diseases. Annals of Translational Medicine. 2015;3:135. doi: 10.3978/j.issn.2305-5839.2015.03.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine Dephosphorylation of H2AX Modulates Apoptosis and Survival Decisions. Nature. 2009;458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald B, Sampath D, Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene. 2008;27:6522–6537. doi: 10.1038/onc.2008.316. [DOI] [PubMed] [Google Scholar]
- Ford JB, Baturin D, Burleson TM, Van Linden AA, Kim YM, Porter CC. AZD1775 sensitizes T cell acute lymphoblatic leukemia cells to cytarabine by promoting apoptosis over DNA repair. Oncotarget. 2015;6:28001–28010. doi: 10.18632/oncotarget.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwade D, Hills RK. Independent prognostic factors for AML outcome. Hematology/ the American Society of Hematology Education Program. 2009:385–395. doi: 10.1182/asheducation-2009.1.385. [DOI] [PubMed] [Google Scholar]
- Garcia TB, Snedeker JC, Baturin D, Gardner L, Fosmire SP, Zhou C, Jordan CT, Venkataraman S, Vibhakar R, Porter CC. A small molecule inhibitor of WEE1, AZD1775, syndergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017 Jun 27; doi: 10.1158/1535-7163.MCT-16-0660. pii:molcanther.0660.2016. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- Iraqui I, Chekkal Y, Jmari N, Pietrobon V, Freon K, Costes A, Lambert SA. Recovery of arrested replication forks by homologous recombination is error-prone. PLoS Genet. 2012;8:e1002976. doi: 10.1371/journal.pgen.1002976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J Cell Biol. 1996;134:963–970. doi: 10.1083/jcb.134.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A. Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell. 2002;9:1005–1016. doi: 10.1016/s1097-2765(02)00524-5. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Heijink AM, Bisselink YJWM, Seinstra RI, Sillje HHW, de Vries EGE, van Vugt MATM. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2013;32:3001–3008. doi: 10.1038/onc.2012.296. [DOI] [PubMed] [Google Scholar]
- Li J, Wang Y, Sun Y, Lawrence TS. Wild-type TP53 inhibits G(2)-phase checkpoint abrogation and radiosensitization induced by PD0166285, a WEE1 kinase inhibitor. Radiat Res. 2002;157:322–330. doi: 10.1667/0033-7587(2002)157[0322:wttigp]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang Y, Benaissa S, Matsuda A, Kantarjian H, Estrov Z, Plunkett W. Homologous recombination as a resistance mechanism to replication-induced double-strand breaks caused by the antileukemia agent CNDAC. Blood. 2010;116:1737–1746. doi: 10.1182/blood-2009-05-220376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Fang B, Koomen JM, Mahajan NP. H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat Struct Mol Biol. 2012;19:930–937. doi: 10.1038/nsmb.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci. 2016;37:872–881. doi: 10.1016/j.tips.2016.06.006. [DOI] [PubMed] [Google Scholar]
- Megyesi J, Tarcsafalvi A, Seng N, Hodeify R, Price PM. Cdk2 phosphorylation of Bcl-xL after stress converts it to a pro-apoptotic protein mimicking Bax/Bak. Cell Death Discov. 2016;2 doi: 10.1038/cddiscovery.2015.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JA, Kornbluth S. Cdc25 and Wee1: analogous opposites? Cell Div. 2007;2:12. doi: 10.1186/1747-1028-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, Mavrommati I, Pai CC, Zalmas LP, Drobnitzky N, et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell. 2015;28:557–568. doi: 10.1016/j.ccell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, Zaberezhnyy V, Patel PR, Gao D, Tan AC, DeGregori J. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012;26:1266–1276. doi: 10.1038/leu.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Bogenberger JM, Chaudhuri L, Hagelstrom RT, Chow D, Buechel ME, Gonzales IM, Demuth T, Slack J, Mesa RA, et al. RNAi screening of the kinome with cytarabine in leukemias. Blood. 2012;119:2863–2872. doi: 10.1182/blood-2011-07-367557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, Reigan P, Porter CC. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther. 2013;12:2675–2684. doi: 10.1158/1535-7163.MCT-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61:8211–8217. [PubMed] [Google Scholar]
- Zhou L, Cai X, Han X, Xu N, Chang DC. CDK1 switches mitotic arrest to apoptosis by phosphorylating Bcl-2/Bax family proteins during treatment with microtubule interfering agents. Cell Biol Int. 2014;38:737–746. doi: 10.1002/cbin.10259. [DOI] [PubMed] [Google Scholar]