

Abstract
The advent of high throughput epigenome mapping technologies has ushered in a new era of multi-omics where powerful tools can now delineate and record different layers of genomic output. Integrating various components of the epigenome from these multi-omics measurements allows the interrogation of cellular heterogeneity in addition to the discovery of molecular connectivity maps between the genome and its functional output. Mapping of chromatin accessibility dynamics and higher order chromatin structure has enabled new levels of understanding of cell fate decisions, identity, and function in normal development, physiology, and disease. We provide a perspective on the progress of the epigenomics field and applications, and anticipate an even greater revolution in our understanding of the human epigenome for years to come.
Keywords: epigenetics, gene regulation, genomics, chromatin architecture
Subject Terms: Genetics, Epigenetics
Cells in multicellular organisms are genetically homogeneous but structurally and functionally heterogeneous owing to the differential expression of genes. Classic Mendelian inheritance of phenotypic traits results from allelic differences caused by mutations of the DNA sequence. In contrast, other genetic phenomena, such as X chromosome inactivation during early embryo development in female mammals, position-effect variegation in flies, and chromosomal imprinting, exhibit non-Mendelian inheritance patterns. Conrad Waddington introduced the term “epigenetic landscape” to describe “the interactions of genes with their environment, which bring the phenotype into being”1. Epigenetics is the study of reversible, heritable changes in gene expression that do not involve changes to the underlying DNA sequence—a change in phenotype without a change in genotype. Epigenetic mechanisms are mediated by either chemical modifications of the DNA itself or by modifications of proteins such as chromatin that are closely associated with DNA. Some of the best characterized epigenetic modifications thought to initiate and sustain epigenetic changes include DNA methylation, chromatin remodeling, histone modification, and non-coding RNA (ncRNA)-associated mechanisms2. Earlier studies showed that heterochromatin and euchromatin are associated with distinct DNA methylation and histone modification patterns that correlate with particular states of gene activity, leading to the concept of an ‘epigenetic code’ that determines the chromatin state and, consequently, gene expression3. In mammals, epigenetic regulation is crucial for a variety of different processes such as development, cell differentiation, and proliferation4. A thorough understanding of the regulatory networks and epigenetic mechanisms that underlie context-specific gene expression programs and cellular phenotypes remains a critical scientific goal with broad implications for human health.
Fundamentally, epigenomics, as the name suggests, is the study of the effects of chromatin structure—including higher order chromatin folding and attachment to the nuclear matrix, packaging of DNA around nucleosomes, covalent modifications of histone tails (acetylation, methylation, phosphorylation, ubiquitination), and DNA methylation—on the genetic material of a cell, known as the epigenome. This rapidly expanding field of study is analogous to genomics and proteomics, the study of the genome and proteome of a cell, respectively. The epigenome can differ from cell type to cell type, and in each individual cell it can potentially modulate gene expression in a number of ways—by organizing the nuclear architecture of the chromosomes, inhibiting or facilitating transcription factor access to DNA, and mediating gene expression. Some have referred to these multifaceted aspects of the epigenome as representing a second dimension of the genomic sequence that is pivotal for maintaining cell-type-specific gene expression patterns5. The logic behind performing epigenetic analysis on a global level is that inferences can then be made about epigenetic modifications which may not otherwise be possible through analysis of specific loci. The term “epigenomics” differs from “epigenetics” in that the former does not necessarily imply gene memory; in practice epigenomic is used to describe comprehensive analyses of chromatin constituents or of gene regulation. Epigenetics, on the other hand, is often thought to encompass three major types of memory that utilize related mechanisms over different time scales6: 1) mitotically heritable transcriptional states established during development (cellular memory), 2) mitotically heritable changes in the responsiveness of organisms to environmental stimuli due to previous experiences where genes can experience a more robust secondary transcriptional response, and 3) meiotically heritable changes in gene expression and physiology of organisms in response to experiences in the previous generations—i.e., where parental experience impacts behavior of the offspring.
The arrival of the first practical massively parallel ‘next generation’ sequencing (NGS) platform in the mid-2000’s marked the beginning of a revolution in genomic research7,8. The ability to sequence vast quantities of DNA enables entire genomes or specific targeted genomic regions from many samples to be sequenced accurately and at high depth, which has led to the continued development and refinement of a wide range of applications8. The epigenetics community was among the first to capitalize on this development, combining NGS with established methods to capture epigenetically modified genomic regions9. A key advantage of NGS platforms is their ability to provide a comprehensive and unbiased view of the epigenome, freeing investigators from content-limited microarray platforms9,10. As in the other genomics fields, epigenomics relies heavily on bioinformatics. Central to being able to unravel the mechanisms underlying the workings of the epigenome at the molecular level is access to robust, reproducible, and streamlined technologies that generate data that can be immediately integrated into existing –omic databases. The goal is to create an ultimate picture of the epigenome integrating DNA methylation, chromatin dynamics and accessibility, and expression. Some of the more established technology platforms of DNA methylation, chromatin profiling, and expression profiling have been extensively reviewed elsewhere5. Here we discuss a few of the more recent cutting edge -omic technologic developments created to facilitate the molecular biologists’ ability to better interrogate the epigenome, and describe some illustrative examples where application of these methods have provided novel and unexpected insights into the molecular mechanisms of phenotypic plasticity in development and disease.
Higher-order chromosomal structure profiling platforms
It has long been hypothesized that communication between widely spaced genomic elements is facilitated through the spatial organization of chromosomes that brings genes and their regulatory elements together in close proximity11–13. Eukaryotic genomes are tightly folded and packaged in a highly organized manner to accommodate the spatial constraints of the nucleus while allowing regulatory factors access to the underlying sequences to affect transcriptional control. The dynamic “folding” of chromatin and chromosome architecture regulates patterns of cellular gene expression during differentiation and development, or in response to environmental signals. Understanding how chromosomes fold can provide insight into the complex relationships between chromatin structure, gene activity, and the functional state of the cell11,14.
Over the last two decades, numerous studies have assessed the spatial proximity and nuclear organization of specific genomic loci, initially through microscopy techniques such as fluorescent in situ hybridization (FISH), and more recently by chromosome conformation capture (3C15). These methods have provided strong evidence that long-range chromosomal interactions are widespread, suggesting a high level of communication between dispersed elements in the genome16,17. Specifically, 3C is a molecular technique that uses formaldehyde cross-linking and locus-specific PCR to detect physical contacts between genomic loci15. Significant effort over the past few years has focused on obtaining comprehensive mapping of chromosomal interactions. Several adaptations of 3C have been developed that allow large-scale detection of genomic interactions by using microarrays or high-throughput sequencing technologies. The 4C method (3C-on chip, or circular 3C) allows identification of regions throughout the genome that are physically close to a single locus of interest; similarly, 5C (3C–carbon copy) is not anchored on a single locus and is used for mapping dense interaction networks throughout large chromosomal regions of interest. Hi-C, first introduced in 2009, brings these analyses to the -omic level by enabling probing of three three-dimensional architecture of whole genomes through coupling proximity-based ligation with massively parallel sequencing18. An increasing number of studies leveraging the power of Hi-C has provided new insight into the global organization of the genome. For example, chromosomes are now believed to be partitioned into megabase-scale topologically associating domains (TADs19–21) and smaller, nested subTADs22,23. Conceptually, the TADs/subTADs are thought to represent particular areas of the genome where all pairs of loci interact more frequently with one another than their surrounding regions. The TADs/subTADs can also form higher order “A” and “B” compartments of active and inactive chromatin, respectively18,23. Significantly, at each level in the chromatin folding hierarchy, the folding patterns exhibit a complex connection to genome function and dysfunction in models of normal development and disease24. The advent of Hi-C has established a burgeoning field for studying chromatin interactomes and regulation networks in 3D, and has been transformative in our ability to understand the architecture of the genome at high resolution.
Chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) is a variation of Hi-C that features an immunoprecipitation step to map long-range DNA interactions25, producing a directed view of long-range contacts associated with a protein factor of interest. In this method, DNA-protein complexes are crosslinked and fragmented. Specific antibodies are then used to immunoprecipitate proteins of interest, while specific linkers are ligated to the DNA fragments, which ligate when in proximity. Subsequent deep sequencing provides base-pair resolution of ligated fragments, providing a genome-wide unbiased and de novo discovery of long-range chromatin interactions. Hi-C and ChIA-PET provide a nice balance of resolution and reasonable coverage in the eukaryotic genome to map long-range interactions.
Genomic analyses of protein-directed chromosomal architecture
Variations on the Hi-C theme have been made in an attempt to achieve enhanced specificity in mapping the relationship between protein binding and the 3-dimensional genome; however, because Hi-C interrogates all possible proximity ligations genome-wide, deep sequencing is required to fully identify chromatin architectural features. Enrichment strategies have been developed to target factor-directed interactions via ChIA-PET (described above) and locus-specific interactions via Capture-C and related methods26. One such technique is HiChIP, an rapid, efficient, and technically simplified method for mapping factor-directed chromatin conformation27 Long-range DNA contacts are first established in situ in the nucleus; chromatin immunoprecipitation (ChIP) is then performed on the contact library, directly capturing long-range interactions associated with a protein of interest. High throughput sequencing then identifies two distantly located segments of the genome from one fragment, indicating that the factor of interest was associated with the long-range interaction. An advantage of HiChIP is the dramatically lower cell number necessary to produce high-confidence contact maps27, which will facilitate future studies of chromatin conformation in systems previously unmeasurable by conventional strategies.
A genomic window into chromatin accessibility
Major insights into the epigenetic information encoded within chromatin have come from high-throughput, genome-wide methods for assaying chromatin accessibility (so called “open chromatin”28), nucleosome positioning29, and transcription factor (TF) occupancy30. The drawback with existing methods is that they all require millions (often hundreds of millions) of cells as starting material, involve complex and time-consuming sample preparation protocols, and cannot easily interrogate the interplay of chromatin accessibility, nucleosome positioning, and TF binding simultaneously in the same sample. Traditionally, enzymes such as micrococcal nuclease and DNase I have been used to preferentially cleave nucleosome-depleted DNA sequences to measure chromatin sensitivity31 to identify active regulatory sequences in the genome. Genome-wide tools have been developed leveraging the activity of DNase I32,33 and a prokaryotic transposase enzyme, Tn5, that preferentially integrates into active, open chromatin elements in vivo34. ATAC-seq (assay for transposase-accessible chromatin using sequencing) takes advantage of hyperactive Tn5 transposase with loaded in vitro adaptors for high-throughput DNA sequencing to provide comparable information about unfixed eukaryotic accessible chromatin to that given by the new DNase-seq methods34. What sets ATAC-seq apart from the other molecular tools is its simple protocol and low cell number requirement34, opening up the possibility of assessing chromatin accessibility in samples for which large numbers of cells is not feasible.
Modulation of chromosomal architecture at will
While work over the last two decades have demonstrated a correlation between chromatin topology and the underlying cellular gene activity, the critical question of whether dynamic changes in chromosome folding is a cause or consequence of genome function is still unresolved35,36. It would appear from the wealth of accumulated whole genome 3-dimensonal data that chromosomal architecture is a dynamic yet highly organized structure, presumably built progressively from stabilization of functional contacts between genes and their respective regulatory elements. However, direct hypothesis testing regarding how chromatin loops are organized and what their function(s) are has been difficult due to the fact that most of the high dimensional data has been obtained at a population-average level, and that disruption of TAD insulation does not appear to impact higher-order genomic compartmentalization37. What has been missing in the field was a heterologous reagent that can be readily programmed to connect any two endogenous DNA segments to facilitate DNA loop engineering, and to molecularly dissect the elements contributing to chromosomal structural borders and/or key architectural loops.
A major advance that begins to address the causal link between chromatin structure and function came with the development of CLOuD9 (chromatin loop reorganization using CRISPR–dCas9), a method to reversibly establish new chromatin loops38. Taking advantage of the powerful genome editing technology Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), CLOuD9 uses orthogonal Cas9 species fused to a reversible dimerization domain to target and subsequently bring together any two chromosomal loci in the genome to affect gene expression. An appropriate analogy is that of protein structure—once the structure for a particular protein is resolved, the key residues contributing to the protein’s function can be readily identified and engineered; similarly, as knowledge of the formation and maintenance of specific chromatin loops increases, comparable structure-informed reverse genetic engineering will allow one to manipulate the genome, with myriad applications. Similar reagents39 are being developed that will allow creation of de novo DNA looping in a variety of cell types, aiding understanding of endogenous loops and enabling creation of new regulatory connections. More importantly, mechanistic links can now be assigned to what were previously only correlations between chromatin conformations and transcriptional regulation (Figure 1A).
Figure 1.

(A) Measurement of long-range contact of DNA elements highlighted by single-cell ATAC-seq (scATACT-seq). Structured cis-variability across single epigenomes highlighted by single-cell ATAC-seq (scATACT-seq). Pearson correlation coefficient representing chromosome compartment signal of interaction frequency from a population chromatin conformation capture assay (left panel) or scATAC-seq (middle panel) from chromosome 1. Data in white represents masked regions due to highly repetitive regions. (right panel, upper box) Permuted cis-correlation map for chromosome 1. (right panel, lower box) Representative region depicting long-range covariability. From Buenrostro et al., 2015.
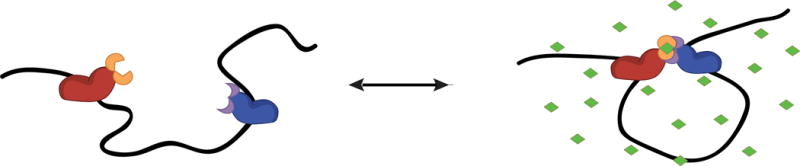
(B) Schematic of CLOuD9 as a reversible method for manipulating chromosomal loops. Addition of abscisic acid (ABA, green) brings two complementary CLOuD9 constructs (CLOuD9 S. pyogenes (CSP), CLOuD9 S. aureus (CSA), red and blue, respectively) into proximity, remodeling chromatin structure. Removal of ABA restores the endogenous chromatin conformation. From Morgan et al., 2017.
Epigenetic editing with CRISPR
Several effective and precise tools have been developed that enable site-specific manipulation of DNA methylation40–43, directly addressing the relationship between DNA methylation and gene expression while bringing to light a novel approach that can selectively and heritably alter gene expression. The technologies all take advantage of deactivated Cas9 (dCas9) nuclease fused to either the catalytic domain of the DNA methyltransferase (DNMT3A) or Ten-Eleven Translocation methylcytosine dioxygenase1 (TET1), and demonstrate specific DNA methylation activity for the targeted region and heritable effects through mitotic divisions. Together, these tools should allow more mechanistic studies of DNA methylation and its role in guiding molecular processes that determine cellular fate.
From bulk populations to single-cell epigenomics
One of the most exciting and powerful recent developments in epigenomics is the application of technologies allowing analyses at the single-cell level44,45. Because epigenetic information is encoded in multiple forms, ranging from covalent modifications on DNA, chromatin accessibility and compaction, post-translational modifications of histones, and higher-order chromosomal conformation, each layer of epigenetic information requires a separate molecular approach to profile it. For many biological questions, observations of epigenetic regulatory systems at the single-cell level will likely elucidate intercellular differences that will lead to a better understanding of the underlying mechanisms compared with bulk analysis46. For example, advances in whole-genome and whole-transcriptome amplification have permitted the sequencing of the minute amounts of DNA and RNA present in single cells, offering a window into the extent and nature of genomic and transcriptomic heterogeneity found in both normal development and disease47. For instance, a recent single-cell transcriptional profiles of the murine non-myocyte cardiac cellular landscape using single-cell RNA sequencing (scRNA-seq48). Detailed molecular analyses of the scRNA-seq data revealed the diversity of the cardiac cellulome and facilitated development of techniques to isolate understudied cardiac cell populations, such as mural cells and glia, offering insights into the structure and function of the mammalian cardiac cellulome and providing an important resource in cardiac cell biology. Indeed, single-cell approaches stand poised to revolutionize our capacity to understand the range and magnitude of epigenomic diversity that occur during the lifetime of an individual organism. In addition, combined single-cell methods are also rapidly emerging that allow analyses of epigenetic–transcriptional correlations at different time scales, thereby enabling detailed investigations of how epigenetic states are associated with phenotype and allowing for discovery of new layers of molecular connectivity between the genome and its functional output44,45.
Methods to interrogate 3-dimensional chromosomal structure and chromatin accessibility in individual cells have been developed over the last few years, such that these features are now assayable at the single-cell level44,45. As discussed above, Hi-C enables measurement of the proximity of genomic loci in 3-dimensional space. Variations and optimizations have been performed to increase throughput and resolution to the single cell level, a method referred to as single-cell Hi-C (scHi-C49–52). Single-cell Hi-C has allowed visualization and reconstruction of the 3-dimensional organization of every chromosome in individual haploid cells53 and revealed how data from population Hi-C can obscure the dynamic reorganization of chromosome compartments during the cell cycle51. The resolution of scHi-C methods is expected to continue to improve such that eventually it will be possible to accurately map contacts between specific promoters and their enhancers. Nonetheless, scHi-C has bridged current gaps between genomics and microscopy analyses of chromosomes, demonstrating how modular organization underlies dynamic chromosome structure, and linking these structures with genome activity patterns50,51.
Similarly, the resolution of ATAC-seq has been improved with the development of single cell ATAC-seq (scATAC-seq54). Taking advantage of microfluidics to process single cells while introducing cell-identifying barcodes as part of the tagging process, scATAC-seq is a robust method that allows parallel processing of a large number of samples to reveal the landscape and principles of mammalian DNA regulatory variation. In addition to providing insights into cell-to-cell variation, scATAC-seq allows identification of specific trans-factor and cis-elements associated with the variance in cell-type-specific accessibility. Interestingly, scATAC-seq elucidates the pattern of accessibility variation in cis across the genome that recapitulates chromosome compartments de novo, thereby linking single-cell accessibility variation to 3-dimensional genome organization54 (Figure 1B).
These single cell epigenomic approaches will ultimately allow a full understanding of genome regulation that involves integrating three different layers of data: one-dimensional data regarding the state of local chromatin (such as patterns of protein binding along chromosomes and the accessibility of chromatin), three-dimensional data describing the population-averaged folding of chromatin inside cells, and single-cell observations of three-dimensional spatial co-localization of genetic loci and trans factors that reveal information about their dynamics and frequency of co-localization. However, despite these advances, there remain significant challenges and limitations that apply to these single-cell epigenome methods. Currently, important bottlenecks include the limited capture rate, low mappability rates, and high levels of PCR duplicates45. Improved computational tools will also be required to process, integrate, and visualize connections between the different molecular layers within and between cells.
Applications of epigenomics tools to disease
The advent of the epigenomic tools over the past few years has brought forth a more holistic view of the interplay between the genome and a very active epigenome, forming an causal link between the underlying genome, the regulatory epigenome, and the functional consequences stemming from perturbations in both. Importantly, these tools are being leveraged to uncover mechanisms of complex diseases. Such integrative approaches may provide insights into the causal regulatory mechanisms of disease for purposes of early-stage detection as well as therapeutic development.
One such example is coronary artery disease (CAD). Meta-analyses of genome-wide association studies in humans have identified hundreds of loci associated with CAD and myocardial infarction susceptibility55,56. However, the mechanisms and functions of many of these loci have remained unclear due to the fact that a large number of the variants reside in non-coding regions. Quertermous and colleagues applied an integrative approach to investigate some of these causal regulatory variants in CAD using genomic, epigenomic, and transcriptomic analyses with targeted experimental follow-up at selected candidate loci57. The authors hypothesized that understanding the epigenetic gene regulatory mechanisms in primary cultured human coronary artery smooth muscle cells (HCASMCs) will provide greater insights into these disease variants as well as the underlying biology of the vessel wall. To begin to dissect the epigenomic changes in CAD, they performed ATAC-seq on stimulated HCASMCs, normal, and atherosclerotic human coronary artery tissue, to generate chromatin accessibility profiles. These were then integrated with ChIP-seq data to define HCASMC-enriched cis-regulatory regions. Publicly available annotations were incorporated to identify several representative loci, and expression quantitative trait loci (eQTL) analyses were done on cohorts of normal and atherosclerotic arteries to validate the endogenous functions of these variants in the appropriate disease environment. Overall, 64 candidate regulatory variants in stimulated HCASMCs and 26 in coronary arteries ex vivo were identified; of these candidate variants, the functionality of seven were confirmed via allele-specific binding, enhancer traps and allelic expression imbalance57.
lncRNAs in cardiac development and disease
Long noncoding RNAs (lncRNAs) are among the several families of noncoding RNAs that have emerged as powerful regulators of cellular and tissue function58,59. Initially considered the “dark matter of the genome,” lncRNAs are now regarded as critical epigenetic regulators of gene expression60. The regulation of cardiac pathways by lncRNAs is still poorly understood. Numerous studies have revealed that lncRNAs have important roles in healthy and diseased hearts by transcriptome profiling in cardiac tissues and identifying hundreds of differentially expressed lncRNAs. Several integrative approaches have linked lncRNAs with specific biological functions, such as modulating chromatin states, regulating transition of chromatin state during cardiomyocyte differentiation, and affecting physiological traits implicated in cardiac remodeling61. Other lncRNAs, such as MHRT62, dictate cardiac chromatin signatures through binding of the chromatin repressor complex. Other lncRNA functions include regulation of cardiomyocyte metabolism, hypertrophy, differentiation, and proliferation63. Whether lncRNAs participate in the regulation of inflammation and fibrosis, both of which represent hallmarks of cardiac remodeling, remains to be determined. Of course, not all differentially expressed lncRNAs will turn out to be functionally important, but their unique association with chromatin states and enhancers suggests fundamental signaling roles. Due to space limitations we refer interested readers to recent reviews on lncRNAs64,65 and microRNAs66,67 in cardiovascular research, as both have been extensively studied as mechanistic regulators of cardiovascular development and disease and potential therapeutic targets.
Elucidating the principles governing the enhancer connectome to identify targets of disease-associated DNA elements
The inability to casually link intergenic mutations to their presumed target genes has limited a better molecular understanding of human diseases. This is a particularly glaring gap as the majority of inherited risk factors for common diseases reside in intergenic enhancers and noncoding features in DNA68. Taking advantage of the HiChIP technology27, Mumbach and colleagues set out to define the high-resolution landscape of enhancer–promoter regulation in primary human cells. They generated high-resolution contact maps of active enhancers and target genes in primary human T cell subtypes and coronary artery smooth muscle cells through a series of H3K27ac HiChIP experiments69. Perhaps not surprisingly, enhancer–promoter contacts were found to be highly dynamic in related cell types and to often involve genomic elements with shared accessibility. Functions were assigned to autoimmune and cardiovascular disease risk variants from genome-wide association study (GWAS)-identified SNPs; the SNPs were also subsequently linked to their putative target genes (Figure 2). These target genes were further validated through modulation of the linked enhancers by CRISPR gain and loss of function assays, and through correlation with expression quantitative trait loci and allele-specific enhancer loops in primary cells from patients. Importantly, because HiChIP demonstrated that the majority of disease-associated enhancers contact their targets beyond the nearest gene(s) in the linear genome, these data also expand the number of potential target genes for autoimmune and cardiovascular diseases. Moreover, these results solidify the idea that mapping chromosomal structural conformation in primary cells can lead to the identification of novel, often hidden, regulatory connections underlying gene function in human disease.
Figure 2. HiChIP identifies allele-specific loops in coronary smooth muscle of coronary artery disease-associated single nucleotide polymorphisms.
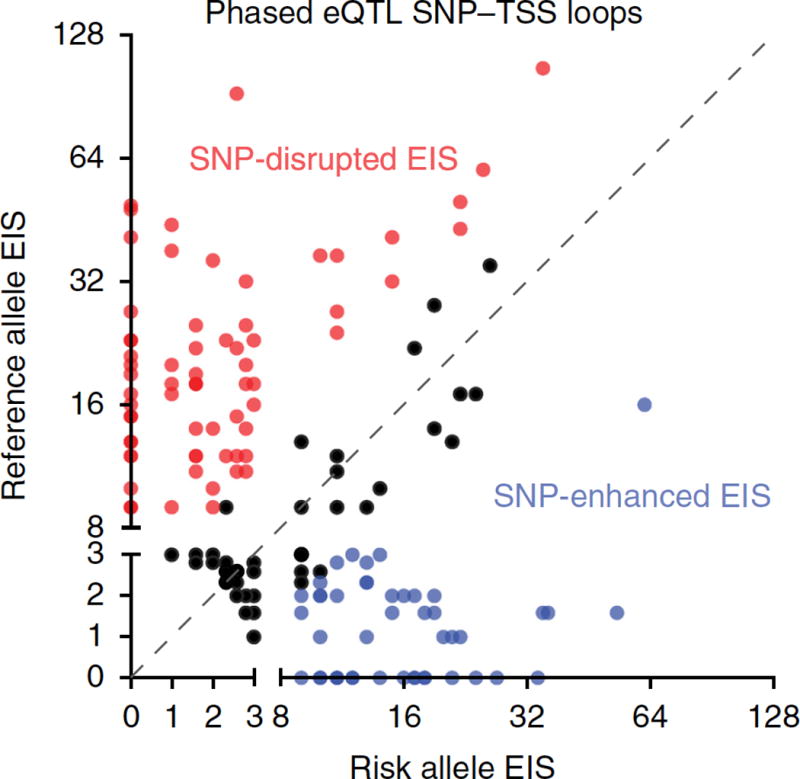
Genome phasing information in human coronary artery smooth muscle cells (HCASMCs) was used to measure enhancer–promoter interactions at allele-specific CAD-associated SNPs, allowing the examination of functional consequences of risk variants compared to their alternative alleles for a set of CAD-associated SNP–target genes. Many risk alleles disrupted enhancer–target gene interactions (red), but a subset of pathogenic SNPs increased enhancer–target gene interactions (blue). From Mumbach et al., 2017.
Hi-C applications to disease
Mounting evidence suggests that the unique genome configurations of loops and chromosome territories may be linked to the establishment and/or maintenance of human disease phenotypes70–72. For instance, the spatial proximity of chromosomes directly influences the probability of translocation between specific loci73. In fact, 3D domain disruption of TAD and sub-TAD boundaries via mutations, deletions, or genetic rearrangements has been linked to aberrant activation of genes via ectopic looping of enhancers in limb malformation syndromes70 and cancer71. Mutations affecting proper function of architectural proteins such as cohesin and the structural properties of the nuclear lamina give rise to diseases known as cohesinopathies74 and laminopathies75, respectively. Additionally, how the genome folds in 3D space facilitates connection of distal SNPs with their target genes was demonstrated when a functional link between a mutation in an intron of the fat mass and obesity associated (FTO) gene and an evolutionarily conserved aberrant enhancer-promoter contacts to IRX3, a novel determinant of body mass and composition, was established76. Recently, Hi-C was used to investigate the mechanisms of epigenomic function in adult cardiac myocytes, using a murine model of pressure overload–induced hypertrophy77. Chromatin capture was used to determine the structure of the cardiac myocyte epigenome, providing a high-resolution resource of the endogenous chromatin architecture in cardiac myocytes while specifically delineating the global changes in chromatin interactions during heart failure. The role of the chromatin structural protein CCCTC-binding factor (CTCF) was also examined by using an in vivo loss-of-function model, revealing its potential role in remodeling long-range interactions of cardiac enhancers and changes in larger scale genome accessibility. These findings demonstrate the promise of epigenomic technologies as a novel means of exploring the 3-dimensional features of the cardiac nucleus, and support epigenomic plasticity as a common feature of cardiac pathophysiology induced by distinct stimuli.
Thus, aberrant 3D chromosomal structure represents a new dimension through which an understanding of sporadic and familial disease states can be made, leading to unraveling of novel therapeutic interventions based on preventing or rewiring pathological 3D contacts. Together, these epigenomic studies support an emerging model in which genome folding and misfolding are critically linked to the onset and progression of a broad range of human diseases. Knowledge of the dynamics of genomic interactions in disease may enable new strategies for therapeutic intervention. Novel technologies to manipulate the epigenome and the 3D chromatin, such as the ability to create novel chromatin contacts and loops at will (CLOuD938), should allow heretofore unavailable investigations into the mechanisms and sequelae of both cis- and trans-chromosomal interactions to be performed.
Epigenomics as a conduit to personalized “regulomes”
As a whole, epigenetic variation can yield information on cellular states and developmental histories in ways that genotype information cannot. Furthermore, in contrast to fixed genome sequences, epigenetic patterns are plastic, and regulated gene expression plays key roles in nearly every developmental program and disease state. This type of temporally sensitive and dynamic information feedback between perturbation and outcome is essential to tailor precise medical treatments for individual patients. Manipulating aberrant, disease-causing epigenetic marks would thus appear to hold considerable therapeutic promise.
The rapid development of high-throughput technologies and computational frameworks have allowed researchers to examine biological systems in unprecedented detail. The ability to study biological phenomena at the -omics levels in turn can be expected to lead to significant advances in personalized and precision medicine. Patients can be treated according to their own molecular characteristics. Individual -omes as well as the integrated profiles of multiple –omic information are expected to be valuable for health monitoring, preventative measures, and precision medicine, transforming medical care from traditional symptom-oriented diagnosis and treatment of diseases toward disease prevention and early diagnostics. Furthermore, regulome analysis may directly investigate chromatin or TF pathways that are direct drug targets.
Some groups have already begun to take advantage of these sensitive genomic technologies. For example, ATAC-seq was utilized to better visualize the personal regulome from a standard blood draw, the most common source of human samples for clinical diagnostics78, where the authors provided foundational data and methods to compare and visualize differences in personal regulomes. The number, location, and potential sources of in vivo variation in chromatin accessibility on a genome-wide scale were identified and analyzed, opening up the possibility that potential variation in chromatin accessibility in the population may be a key to understanding and managing healthy and diseased states. Finally, comparisons of regulome variation in healthy versus diseased patients documented the feasibility of using the personal regulome approach to investigate disease bio-markers and mechanisms78.
Moreover, the newer single cell technologies have been shown to be compatible with the small sample sizes of human biopsies and clinical workflows. A drawback to current therapeutics is their non-specific effects. Development of locus-specific epigenetic modifiers, used in conjunction with epigenetic biomarkers of response, will enable truly precision interventions. Using these epignomic tools to monitor personal regulomes in health and disease offers many exciting possibilities.
Conclusion
What more needs to be done to understand the complete epigenome? For the most part, we are still collecting in the initial phases of discovering the components. Just as the full sequence of a genome has greatly facilitated progress in genetics, a clearer understanding for epigenetics will likely come when all the parts are known. It is encouraging to see the great strides that have been made in the last decade, and the anticipated advances and challenges in systems biology-powered personalized medicine that will define the future of personalized health care that is now becoming unraveled.
Acknowledgments
Sources of funding: The authors are funded by NIH and HHMI.
Nonstandard Abbreviations and Acronyms
- FISH
fluorescent in situ hybridization
- NGS
next generation’ sequencing
- 3C
chromosome conformation capture
- TADs
topologically associating domains
- ChIA-PET
Chromatin interaction analysis by paired-end tag sequencing
- CLOuD9
chromatin loop reorganization using CRISPR–dCas9
Footnotes
Disclosures: None.
References
- 1.Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41:10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- 2.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Fischle W, Cheung W, Jacobs S, Khorasanizadeh S, Allis CD. Beyond the double helix: writing and reading the histone code. Novartis Found Symp. 2004;259:3–17. discussion 17–21– 163–9. [PubMed] [Google Scholar]
- 4.Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, Bennett DA, Houmard JA, Muoio DM, Onder TT, Camahort R, Cowan CA, Meissner A, Epstein CB, Shoresh N, Bernstein BE. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;155:39–55. doi: 10.1016/j.cell.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Urso A, Brickner JH. Epigenetic transcriptional memory. Curr Genet. 2016;63:435–439. doi: 10.1007/s00294-016-0661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 8.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 9.Hurd PJ, Nelson CJ. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief Funct Genomic Proteomic. 2009;8:174–183. doi: 10.1093/bfgp/elp013. [DOI] [PubMed] [Google Scholar]
- 10.Meaburn E, Schulz R. Next generation sequencing in epigenetics: insights and challenges. Semin Cell Dev Biol. 2012;23:192–199. doi: 10.1016/j.semcdb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Sexton T, Cavalli G. The role of chromosome domains in shaping the functional genome. Cell. 2015;160:1049–1059. doi: 10.1016/j.cell.2015.02.040. [DOI] [PubMed] [Google Scholar]
- 12.Dekker J. Gene regulation in the third dimension. Science. 2008;319:1793–1794. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pombo A, Dillon N. Three-dimensional genome architecture: players and mechanisms. Nature Publishing Group. 2015;16:245–257. doi: 10.1038/nrm3965. [DOI] [PubMed] [Google Scholar]
- 14.Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:661–678. doi: 10.1038/nrg.2016.112. [DOI] [PubMed] [Google Scholar]
- 15.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 16.Miele A, Dekker J. Long-range chromosomal interactions and gene regulation. Mol Biosyst. 2008;4:1046–1057. doi: 10.1039/b803580f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krivega I, Dean A. Enhancer and promoter interactions—long distance calls. Current Opinion in Genetics & Development. 2012;22:79–85. doi: 10.1016/j.gde.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012:1–5. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-Dimensional Folding and Functional Organization Principles of the Drosophila Genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 2014:1–16. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norton HK, Phillips-Cremins JE. Crossed wires: 3D genome misfolding in human disease. The Journal of Cell Biology. 2017;216:3441–3452. doi: 10.1083/jcb.201611001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fullwood MJ, Ruan Y. ChIP-based methods for the identification of long-range chromatin interactions. J Cell Biochem. 2009;107:30–39. doi: 10.1002/jcb.22116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dekker J, Misteli T. Long-Range Chromatin Interactions. Cold Spring Harb Perspect Biol. 2015;7:a019356. doi: 10.1101/cshperspect.a019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Meth. 2016;13:919–922. doi: 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee B-K, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan K-K, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, Abyzov A, Addleman N, Bhardwaj N, Boyle AP, Cayting P, Charos A, Chen DZ, Cheng Y, Clarke D, Eastman C, Euskirchen G, Frietze S, Fu Y, Gertz J, Grubert F, Harmanci A, Jain P, Kasowski M, Lacroute P, Leng J, Lian J, Monahan H, O’Geen H, Ouyang Z, Partridge EC, Patacsil D, Pauli F, Raha D, Ramirez L, Reddy TE, Reed B, Shi M, Slifer T, Wang J, Wu L, Yang X, Yip KY, Zilberman-Schapira G, Batzoglou S, Sidow A, Farnham PJ, Myers RM, Weissman SM, Snyder M. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raj A, McVicker G. The genome shows its sensitive side. Nat Meth. 2014;11:39–40. doi: 10.1038/nmeth.2770. [DOI] [PubMed] [Google Scholar]
- 32.He HH, Meyer CA, Hu SS, Chen M-W, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, Liu XS, Brown M. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Meth. 2014;11:73–78. doi: 10.1038/nmeth.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vierstra J, Wang H, John S, Sandstrom R, Stamatoyannopoulos JA. Coupling transcription factor occupancy to nucleosome architecture with DNase-FLASH. Nat Meth. 2014;11:66–72. doi: 10.1038/nmeth.2713. [DOI] [PubMed] [Google Scholar]
- 34.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Meth. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cavalli GG, Misteli TT. Functional implications of genome topology. Nat Struct Mol Biol. 2013;20:290–299. doi: 10.1038/nsmb.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- 37.Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell. 2017;169:930–944.e22. doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan SL, Mariano NC, Bermudez A, Arruda NL, Wu F, Luo Y, Shankar G, Jia L, Chen H, Hu J-F, Hoffman AR, Huang C-C, Pitteri SJ, Wang KC. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat Commun. 2017;8:15993. doi: 10.1038/ncomms15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hao N, Shearwin KE, Dodd IB. Programmable DNA looping using engineered bivalent dCas9 complexes. Nat Commun. 2017;8:1628. doi: 10.1038/s41467-017-01873-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vojta A, Dobrinić P, Tadić V, Bočkor L, Korać P, Julg B, Klasić M, Zoldoš V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016:gkw159. doi: 10.1093/nar/gkw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7:46545–46556. doi: 10.18632/oncotarget.10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDonald JI, Celik H, Rois LE, Fishberger G, Fowler T, Rees R, Kramer A, Martens A, Edwards JR, Challen GA. Reprogrammable CRISPR/Cas9-based system for inducing site-specific DNA methylation. Biol Open. 2016;5:866–874. doi: 10.1242/bio.019067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167:233–235.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark SJ, Lee HJ, Smallwood SA, Kelsey G, Reik W. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016;17:72. doi: 10.1186/s13059-016-0944-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelsey G, Stegle O, Reik W. Single-cell epigenomics: Recording the past and predicting the future. Science. 2017;358:69–75. doi: 10.1126/science.aan6826. [DOI] [PubMed] [Google Scholar]
- 46.Macaulay IC, Voet T. Single cell genomics: advances and future perspectives. PLoS Genet. 2014;10:e1004126. doi: 10.1371/journal.pgen.1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Macaulay IC, Ponting CP, Voet T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet. 2017;33:155–168. doi: 10.1016/j.tig.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, Pinto AR. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018;22:600–610. doi: 10.1016/j.celrep.2017.12.072. [DOI] [PubMed] [Google Scholar]
- 49.Flyamer IM, Gassler J, Imakaev M, Brandão HB, Ulianov SV, Abdennur N, Razin SV, Mirny LA, Tachibana-Konwalski K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 2017;544:110–114. doi: 10.1038/nature21711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagano T, Lubling Y, Várnai C, Dudley C, Leung W, Baran Y, Mendelson Cohen N, Wingett S, Fraser P, Tanay A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017;547:61–67. doi: 10.1038/nature23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, Shendure J. Massively multiplex single-cell Hi-C. Nat Meth. 2017;14:263–266. doi: 10.1038/nmeth.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, Cramard J, Faure AJ, Ralser M, Blanco E, Morey L, Sansó M, Palayret MGS, Lehner B, Di Croce L, Wutz A, Hendrich B, Klenerman D, Laue ED. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 2017;544:59–64. doi: 10.1038/nature21429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.CARDIoGRAMplusC4D Consortium. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, König IR, Cazier J-B, Johansson Å, Hall AS, Lee J-Y, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikäinen L-P, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do R, DIAGRAM Consortium, CARDIOGENICS Consortium. Doney ASF, Mokhtari El N, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han B-G, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki M-L, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Müller-Nurasyid M, MuTHER Consortium. Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schäfer A, Sivananthan M, Song C, Stewart AFR, Tan S-T, Thorgeirsson G, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AFR, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Cardiogenics. Carlquist JF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NEE, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason V, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJP, Khaw K-T, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, McKeown PP, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Mühleisen TW, Muhlestein JB, Münzel T, Musunuru K, Nahrstaedt J, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller CL, Pjanic M, Wang T, Nguyen T, Cohain A, Lee JD, Perisic L, Hedin U, Kundu RK, Majmudar D, Kim JB, Wang O, Betsholtz C, Ruusalepp A, Franzén O, Assimes TL, Montgomery SB, Schadt EE, Björkegren JLM, Quertermous T. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat Commun. 2016;7:12092. doi: 10.1038/ncomms12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 59.Wang KCK, Chang HYH. Molecular mechanisms of long noncoding RNAs. Molecular Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rinn JL, Chang HY. Genome Regulation by Long Noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ounzain S, Micheletti R, Beckmann T, Schroen B, Alexanian M, Pezzuto I, Crippa S, Nemir M, Sarre A, Johnson R, Dauvillier J, Burdet F, Ibberson M, Guigo R, Xenarios I, Heymans S, Pedrazzini T. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J. 2015;36:353–68a. doi: 10.1093/eurheartj/ehu180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han P, Li W, Lin C-H, Yang J, Shang C, Nuernberg ST, Jin KK, Xu W, Lin C-Y, Lin C-J, Xiong Y, Chien H, Zhou B, Ashley E, Bernstein D, Chen P-S, Chen H-SV, Quertermous T, Chang C-P. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang C-P, Dorn GW, Thum T, Heymans S, Cardiolinc network Long noncoding RNAs in cardiac development and ageing. Nature Publishing Group. 2015;12:415–425. doi: 10.1038/nrcardio.2015.55. [DOI] [PubMed] [Google Scholar]
- 64.Haemmig S, Simion V, Yang D, Deng Y, Feinberg MW. Long noncoding RNAs in cardiovascular disease, diagnosis, and therapy. Curr Opin Cardiol. 2017;32:776–783. doi: 10.1097/HCO.0000000000000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Viereck J, Thum T. Long Noncoding RNAs in Pathological Cardiac Remodeling. Circ Res. 2017;120:262–264. doi: 10.1161/CIRCRESAHA.116.310174. [DOI] [PubMed] [Google Scholar]
- 66.Olson EN. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Science Translational Medicine. 2014;6:239ps3. doi: 10.1126/scitranslmed.3009008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Viereck J, Thum T. Circulating Noncoding RNAs as Biomarkers of Cardiovascular Disease and Injury. Circ Res. 2017;120:381–399. doi: 10.1161/CIRCRESAHA.116.308434. [DOI] [PubMed] [Google Scholar]
- 68.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, Wei Y, Nguyen T, Greenside PG, Corces MR, Tycko J, Simeonov DR, Suliman N, Li R, Xu J, Flynn RA, Kundaje A, Khavari PA, Marson A, Corn JE, Quertermous T, Greenleaf WJ, Chang HY. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat Genet. 2017;49:1602–1612. doi: 10.1038/ng.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–114. doi: 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hnisz D, Weintraub AS, Day DS, Valton A-L, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, Reddy J, Borges-Rivera D, Lee TI, Jaenisch R, Porteus MH, Dekker J, Young RA. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y, McCord RP, Ho Y-J, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu J, Krantz ID. Cohesin and human disease. Annu Rev Genomics Hum Genet. 2008;9:303–320. doi: 10.1146/annurev.genom.9.081307.164211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mattout A, Cabianca DS, Gasser SM. Chromatin states and nuclear organization in development–a view from the nuclear lamina. Genome Biol. 2015;16:174. doi: 10.1186/s13059-015-0747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, Lee JH, Puviindran V, Tam D, Shen M, Son JE, Vakili NA, Sung H-K, Naranjo S, Acemel RD, Manzanares M, Nagy A, Cox NJ, Hui C-C, Gomez-Skarmeta JL, Nobrega MA. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosa-Garrido M, Chapski DJ, Schmitt AD, Kimball TH, Karbassi E, Monte E, Balderas E, Pellegrini M, Shih T-T, Soehalim E, Liem D, Ping P, Galjart NJ, Ren S, Wang Y, Ren B, Vondriska TM. High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure. Circulation. 2017;136:1613–1625. doi: 10.1161/CIRCULATIONAHA.117.029430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qu K, Zaba LC, Giresi PG, Li R, Longmire M, Kim YH, Greenleaf WJ, Chang HY. Individuality and Variation of Personal Regulomes in Primary Human T Cells. Cell Systems. 2015;1:51–61. doi: 10.1016/j.cels.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]