

Abstract
Nitazoxanide (NTZ) is an FDA-approved anti-protozoal drug that inhibits several bacteria and viruses as well. However, its effect on poxviruses is unknown. Therefore, we investigated the impact of NTZ on vaccinia virus (VACV). We found that NTZ inhibits VACV production with an EC50 of ~2μM, a potency comparable to that reported for several other viruses. The inhibitory block occurs early during the viral life cycle, prior to viral DNA replication. The mechanism of viral inhibition is likely not due to activation of intracellular innate immune pathways, such as protein kinase R (PKR) or interferon signaling, contrary to what has been suggested to mediate the effects of NTZ against some other viruses. Rather, our finding that addition of exogenous palmitate partially rescues VACV production from the inhibitory effect of NTZ suggests that NTZ impedes adaptations in cellular metabolism that are needed for efficient completion of the VACV replication cycle.
Keywords: Vaccinia virus, nitazoxanide, protein kinase R, metabolism, palmitate
Introduction
The thiazolide nitazoxanide (NTZ) has extraordinarily broad antimicrobial activity (Shakya et al., 2017). In the United States, it is FDA-approved for the treatment of cryptosporidiosis and giardiasis and it has been used extensively throughout the world for these and other parasitic infections. Because it appears to be quite safe and also has activity against bacteria and viruses, it is being evaluated in multiple clinical trials for a variety of other infectious and even some noninfectious diseases (clinicaltrials.gov, 2017; Koszalka et al., 2017; Nikolova et al., 2014). Understanding how NTZ functions will be important for optimizing these efforts to repurpose it for new clinical applications.
NTZ has been reported to have activity in cell culture against many medically important viruses. In addition to a RNA viruses such as influenza virus, hepatitis C virus, and human immunodeficiency virus type I (Rossignol and Keeffe, 2008; Rossignol et al., 2009; Trabattoni et al., 2016), it is known to inhibit a few DNA viruses including human cytomegalovirus (HCMV) (Mercorelli et al., 2016). This breadth of activity is striking since these viruses utilize a wide range of replication strategies. Various studies aiming to identify the NTZ mechanism(s) have identified effects on innate immune signaling, which might explain its broad antiviral effects (Petersen et al., 2016b; Trabattoni et al., 2016). However, other studies have implicated virus-specific effects of NTZ, such as its ability to inhibit transcriptional activation by the HCMV IE2 protein (Mercorelli et al., 2016) and to block the maturation of the influenza A hemagglutinin protein (Rossignol et al., 2009). Thus, whether NTZ acts through multiple different virus-specific mechanisms or one common underlying one remains unclear.
The impact of NTZ on poxviruses has not been reported. Poxviruses are important as pathogens, as vaccine vectors, and as models of host-virus interactions. Variola, the cause of smallpox, has been eradicated from circulation in humans, but the potential for intentional or inadvertent re-introduction remains a concern (Ferguson et al., 2003; Koblentz, 2017). Other poxviruses, including monkeypox, are relatively uncommon but occasionally cause life-threatening disease in humans (McCollum and Damon, 2014). Some others, such as molluscum contagiosum virus, are very common but cause only mild disease. In addition to its value in the smallpox elimination campaign, vaccinia virus (VACV) has been used experimentally as a vector to deliver other antigens, as well as for oncolytic therapy (Chan and McFadden, 2014). However, VACV can cause severe disease in immunocompromised patients and even in individuals with the common skin condition eczema (Bray and Wright, 2003; Petersen et al., 2016a). Although vaccinia immune globulin, cidofovir and a few experimental compounds have activity against poxviruses, development or identification of new safe drugs that inhibit poxviruses could have clinical applications and facilitate development and testing of new applications of VACV.
Here we report our investigations of the impact of NTZ on the VACV replicative cycle. We found that NTZ inhibits VACV production, further extending its spectrum activity to Poxviridae family. We show that several factors and pathways that have been suggested to mediate the antiviral effects of NTZ appear not to mediate its impact on VACV. Rather, our finding that addition of palmitate but not several other metabolites, partially reverses the antiviral effects of NTZ suggests that the drug blocks specific metabolic adaptations needed for efficient viral reproduction.
Methods
Cells, viruses
Primary human foreskin fibroblasts (HF) and BSC40 cells were propagated and maintained at 37°C in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% NuSerum (Corning) and penicillin-streptomycin (100 U/ml). HF with CRISPR/Cas9-mediated knock out of STING (HF STING−/−) or PKR (PKR−/−) have been described (Child et al., 2018; Gray et al., 2016).
WT VACV, VC2 containing a lacZ cassette (VC2+lacZ) and VACV lacking its PKR antagonist E3L (VACVΔE3L, kindly provided by Bertram Jacobs, Arizona State University) have been described (Beattie et al., 1995; Child et al., 2012). HF were infected at a high (3 pfu/cell) or low (0.1 pfu/cell) MOI for 1 hour, after which the inoculum was removed and replaced with medium containing vehicle alone (0.1% DMSO) or NTZ immediately or at later times (Fig. 1D), diluted from a 10 mM stock made in DMSO. Viral production was measured 24–48 h post infection (hpi) either by measuring β-galactosidase (β-gal) activity using a fluorometric substrate cleavage assay (Child et al., 2006), or by titering virus in cell lysates, made by three freeze-thaw cycles, on BSC40 cells. Other drugs and metabolites were added at 1 hpi along with 0.1% DMSO or NTZ.
Figure 1. NTZ inhibits VACV production.

HF were infected with VACV at the indicated MOIs and treated with varying concentrations of NTZ or 10 μM TZ at 1 hpi (A–C) or with 10 μM NTZ added at the times indicated in (D). Viral production was quantified by measuring β-gal activity at (A) 48 hpi or (D) 24 hpi or by titering cell-associated virus at (B) 48 hpi and (C) 24 hpi (mean ± SD, N=3). (E) The EC50 was calculated using Prism 7 (MOI 0.1). Depicted is the median EC50 from 3 independent experiments.
For glutamine starvation experiments, monolayers were infected with VACV (MOI = 0.1) in glutamine-free DMEM (Gibco) containing 2% NuSerum or in normal DMEM for 1h. After removing the inoculum, the monolayers were refed with the appropriate medium containing either 0.1% DMSO, 10 μM NTZ, 7mM dimethyl-α-ketoglutarate (αKG, Aldrich Chemistry), or a combination of NTZ and αKG. Viral production was measured at 48 hpi by β-gal activity assays.
Chemical reagents
NTZ, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), antimycin A, 2,4-DNP, oligomycin, and sodium palmitate were obtained from Sigma. Z-VAD-FMK was obtained from Selleckchem. Tizoxanide (TZ) was obtained from MedChem Express. Cidofovir was obtained from Gilead Sciences Inc. DL-mevalonolactone was obtained from TCI Chemicals. Sodium palmitate was conjugated to fatty acid free BSA (Sigma) according to the Seahorse Bioscience protocol.
Immunoblot analyses
Cell lysates were made by washing the monolayers with phosphate-buffered saline (PBS) followed by incubation in 2% sodium dodecyl sulfate (SDS). Following sonication, the proteins were separated by SDS-PAGE, transferred to a membrane (Immobilon®-P), and then probed with antibodies using the Western Star chemiluminescent detection system (Applied Biosystems) using the following antibodies: VACV D8 and A14 (obtained from Yan Xiang, University of Texas, San Antonio (Meng et al., 2011)), PKR D7F7 (#12297, Cell Signaling Technology), P-PKR E120 (ab32036, Abcam) actin (A2066, Sigma-Aldrich), IRF3 (D83B9, Cell Signaling Technology), H3 (ab1791, Abcam), STING (ab92605, Abcam).
Viral genome replication and transcription
VACV DNA replication was assessed by real-time PCR analyses of DNA purified from HF infected with VACV (MOI = 3) and treated with 0.1% DMSO, 10 μM cidofovir (CDV), or 10 μM NTZ. The cells were washed once with PBS, and lysed in 100 μl TE buffer containing 1% SDS, then incubated in 150 μg/ml proteinase K, 1% SDS, 200 mM NaCl and 0.3 μL β-mercaptoethanol at 55°C for 30 min. Following phenol:chloroform extraction, the DNA was ethanol precipitated and resuspended in TE. 10 ng of each sample was analyzed by real-time PCR with primers that bind to the E3L gene (forward - 5′-CGTCAGCCATAGCATCAGCA-3′, and reverse 5′-TGACAACGGAGGCGGATAAG-3). The relative genome replication was calculated as the fold change between samples collected at 1hpi and 24hpi.
The same primers were used to analyze expression of E3L RNA. Whole cell RNA was purified from mock infected and infected cells (MOI = 3) using TRiZOL (Ambion, Inc.), then reverse transcribed using the Applied Biosystems High Capacity cDNA Reverse Transcription kit, and quantified by real time PCR. The relative abundance of E3L RNA, normalized to cellular GAPDH (primers: forward 5′-gaaggtcggagtcaacggattt -3′ and reverse 5′-gaatttgccatgggtggaat -3′) was calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Nuclear-cytoplasmic fractionation
HF in 6 well plates were mock infected or infected with VACV (MOI = 3) and treated with 0.1% DMSO or 10μM NTZ 1 hpi. At 4 hpi, the monolayers were washed twice with PBS, and lysed by incubation in 300 μl of PBS containing 1mM dithiothreitol and 1% NP-40 for 10 min at room temperature. The cytoplasmic fractions were separated by pelleting of the nuclei at 4,000×g for 10 min at 4°C. The nuclear pellets were washed once with PBS, and then lysed in 35μl 2% SDS and sonicated. HCMV that had been UV irradiated in a Stratalinker 1800 (Stratagene) for 6 auto-cross-link cycles, was used as a control for IRF3 translocation to the nucleus. Equal volumes were analyzed by immunoblot assays as described above.
Radiolabeling
HF were infected with VACV (MOI = 3) and then treated with 0.1% DMSO or 10 μM NTZ. 1 hour prior to harvest, the medium was replaced with methionine-free DMEM (Gibco) supplemented with 2% NuSerum and 100 μCi/ml 35S-methionine (Perkin Elmer Health Sciences, Inc.). After washing the monolayers with PBS, the cells were lysed in 2% SDS. Following sonication of the lysates, equal volumes were separated by SDS-PAGE. The gels were stained with Coomassie blue, which showed similar total protein loading in each lane, dried, and analyzed by autoradiography.
Statistical analysis
Unpaired two-tailed t tests were performed using Prism 7 software (GraphPad).
Results
NTZ inhibits vaccinia virus production
To determine whether NTZ inhibits VACV, we used a recombinant virus that expresses lacZ, under control of the P11 late promoter, which allows β-galactosidase (β-gal) activity assays to measure viral production. We first infected human fibroblasts (HF) at a low MOI (0.1 pfu/cell), and at 1 hpi, removed the inoculum and added medium with varying concentrations of NTZ (Fig. 1A). NTZ inhibited production of new virus in a dose-dependent manner, increasing from a ~1 log reduction at a concentration of 5 μM, to more than 2 logs at 20 μM (Fig. 1A). The active metabolite of NTZ, tizoxanide (TZ) (Rossignol, 2014), resulted in a comparable level of inhibition. We detected a similar dose-response curve by measuring viral titers (Fig. 1B), confirming that β-gal activity measurements provide a reliable measure of viral production as we have shown in several previous studies (Carpentier et al., 2016; Child et al., 2004; Child et al., 2002) (Fig. 1B). NTZ also inhibited VACV production in HeLa cells, BSC40 cells, and guinea pig fibroblasts (data not shown), suggesting that its activity is not cell type- or host species-specific. NTZ inhibited VACV production after a high MOI (3 pfu/cell) infection of HF (Fig. 1C) and even when added as late as 6 h post infection (Fig. 1D) supporting the conclusion that it acts after viral entry. Dose-response curves made from 3 independent experiments, revealed EC50 values ranging from 0.7μM to 2.6μM with a median of 1.95μM (Fig. 1E). Thus, NTZ is a potent inhibitor of VACV replicative cycle.
NTZ inhibits the VACV replicative cycle at early times post infection
To investigate the mechanism by which NTZ inhibits VACV, we first explored when during the viral lifecycle NTZ acts. By qRT-PCR, we detected transcription of the E3L early gene at 1 hpi and it increased ~20-fold by 4 hpi in the absence of NTZ (Fig. 2A). We observed a small and nonsignificant decrease in E3L transcript accumulation by 4 hpi when NTZ was added at 1 hpi. These result support the conclusion that NTZ functions after VACV has entered the cells and has started to express its genes.
Figure 2. NTZ inhibits the VACV replicative cycle after entry.

HF were infected with VACV at an MOI of 3 and mock treated or treated with 10 μM NTZ at 1h pi. (A) E3L transcript abundance, relative the amount present at 1 hpi, was quantified by qRT-PCR as described in the Methods. (B) Proteins were labelled by incubating cells with 100 μCi/ml of S35-methionine for 1h before collecting cell lysates and were analysed by SDS-PAGE and autoradiography. (C) Cell lysates were collected at the indicate times timepoints and the accumulation of late VACV proteins D8 and A14 was monitored via immunoblot assay. (D) The fold increase in VACV DNA at 24 hpi, compared to amount present at 1 hpi, was measured by qPCR. (* p < 0.05; ** p < 0.005; ns, not significant)
We monitored de novo protein synthesis by metabolic labeling with [35S]-methionine at 8 and 24 hpi. After viral entry, VACV shuts off host protein synthesis as early as 6 hpi in cells such as BS-C-1, but progression through the replicative cycles appears to be slower in HF (Burgess and Mohr, 2015; Liu et al., 2014). This shutoff is mediated in part by the action of the late protein D10 and to a lesser extent by the early protein D9, both of which increase the degradation of host mRNAs, effectively prioritizing the synthesis of its own viral proteins (Parrish and Moss, 2006; Shors et al., 1999). In untreated infected HF, we detected shut off of host protein synthesis at 24 hpi, but not at 8 hpi. Treatment with NTZ prevented the shut off of host protein synthesis (Fig. 2B). NTZ did not diminish protein synthesis in uninfected cells at either time point.
We also evaluated the impact of NTZ on late viral protein synthesis by examining infected cell lysates for representative viral proteins (Fig. 2C). Immunoblot assays revealed that NTZ markedly reduced accumulation of the VACV envelope proteins D8 and A14 at 24 hpi. Together with the noted decrease in expression of β-gal, which is regulated in this virus by the P11 late promoter (Fig. 1), these data suggest that NTZ blocks VACV prior to the late phase of the replication cycle.
As viral DNA replication is necessary for the expression of intermediate and late VACV genes, inhibition of viral DNA replication may explain the inhibitory effects of NTZ on late viral protein production. Therefore, we assessed the impact of NTZ on viral genome replication by real time PCR (Fig. 2D). In untreated cells, we detected a ~1000-fold increase in genome copy number at 24 hpi compared to 1 hpi. NTZ reduced this genome amplification by more than 10-fold, even more than the control VACV inhibitor cidofovir. These results provide additional support for the conclusion that NTZ acts at an early phase of the replication cycle.
NTZ does not function by activating protein kinase R (PKR)
Previous reports have shown that NTZ can activate PKR, leading to eIF2α phosphorylation and inhibition of translation, potentially explaining its broad antiviral effects (Ashiru et al., 2014; Elazar et al., 2009). However, our radiolabeling results did not show any evidence that NTZ reduced protein synthesis in either uninfected or infected cells (Fig. 2B), suggesting that PKR was not activated under these conditions.
To investigate the interaction of NTZ and the PKR pathway more directly, we evaluated the total abundance and phosphorylation state of both PKR and eIF2α, in the presence or absence of NTZ (Fig. 3A). NTZ did not alter the level of PKR phosphorylation in mock infected cells or in VACV infected cells at 8 or 24 hpi, compared to the untreated controls (Fig. 3A). Phosphorylation of eIF2α was likewise unaffected. As a control, we included cells infected with VACVΔE3L, a mutant lacking the PKR antagonist E3L, which is well known to activate the PKR pathway and thus increase the levels of phospho-PKR and phospho-eIF2α (Child et al., 2002; Langland and Jacobs, 2002), as was evident in our blots.
Figure 3. NTZ does not act via PKR.

(A) Lysates of HF infected with VACV or VACVΔE3Lat an MOI of 3 were analysed by immunoblots for PKR, P-PKR, eIF2α, P-eIF2α, and actin accumulation. (B) Viral production was monitored at 48 hpi of HF or HF-PKR−/− (MOI = 0.1) with VACV after addition of no drug (0.1% DMSO) or 10 μM NTZ at 1 hpi (mean ± SD, N=3; ns, not significant)
We also evaluated the antiviral activity of NTZ in PKR−/− HF in which PKR expression was eliminated by CRISPR/Cas9 genome editing. NTZ was as effective at inhibiting VACV production in PKR−/− as in wild-type HF (Fig 3B). We also found the NTZ was similarly effective at inhibiting VACV in PKR−/− HeLa cells (data not shown). Thus, NTZ does not affect the PKR pathway with or without VACV infection and PKR is not necessary for NTZ’s VACV-inhibitory activity.
NTZ does not act by STING or IRF3 signaling
Although NTZ did not increase PKR expression in our cells (Fig. 3), it has been reported to induce IFN expression and a subset of other interferon-regulated genes in vivo and in isolated PBMCs (Petersen et al., 2016a; Trabattoni et al., 2016). NTZ has also been shown to cause mitochondrial stress by uncoupling the inner mitochondrial membrane (Amireddy et al., 2017). Therefore, we considered the possibility that NTZ might act by causing leakage of mitochondrial DNA (mtDNA) into the cytosol, where it could activate the cGAS/STING pathway (Liu et al., 2016; Rongvaux et al., 2014). Moreover, since VACV is a DNA virus that replicates in the cytoplasm, its genome could potentially act synergistically with cytosolic mtDNA leakage to activate cGAS/STING. We therefore compared the antiviral impact of NTZ in wild type vs. STING−/− HF (Fig. 4A). NTZ inhibited VACV to a similar extent in both cells, leading us to conclude that this pathway is not necessary for NTZ function.
Figure 4. NTZ does not act by cGAS/STING or IRF3 signaling.

(A) HF or HF STING−/− were infected with VACV (MOI = 0.1) and mock treated or treated with 10 μM NTZ at 1 hpi. Viral production was determined at 48 hpi by β-gal expression (ns, not significant). (B) HF were mock infected or infected with VACV (MOI = 3) and mock treated or treated with 10 μM NTZ at 1 hpi. UV-inactivated HCMV was used as a control for IRF3 translocation to the nucleus. Nuclear-cytosolic fractionation was performed at 4 hpi, and IRF3 distribution was determined by immunoblot assays. H3 was used as a control for the nuclear fraction, and STING expression for the cytosolic fraction.
In addition to cGAS/STING, several other cellular sensors signal through pathways that converge on IRF3 (Chen and Jiang, 2013). Phosphorylation and dimerization of IRF3, followed by translocation to the nucleus leads to an upregulation of type I IFN and interferon-stimulated genes (ISGs). To investigate whether IRF3 activation mediates the NTZ effects, we determined the localization of IRF3 in NTZ treated HF, with and without infection (Fig. 4B). We did not detect any IRF3 relocalization to the nucleus in NTZ treated cells regardless of VACV infection, while a substantial portion of the cellular IRF3 did relocalize to the nuclear fraction after infection with UV-inactivated HCMV, as has been reported (DeFilippis et al., 2010). The distribution of STING in the cytoplasmic fraction and histone H3 in the nucleus confirmed the success of the fractionation protocol. Thus, NTZ appears not to exert its anti-VACV effects through IRF3.
Role of mitochondrial functions in the NTZ anti-VACV mechanism
VACV alters cellular metabolism during infection. For example, the VACV replicative cycle requires exogenous glutamine, whereas glucose deficiency has no impact (Fontaine et al., 2014; Greseth and Traktman, 2014). The impaired VACV production in glutamine-deficient medium can be rescued by supplementation with alpha-ketoglutarate (α-KG). Therefore, we hypothesized that inhibition of glutamine metabolism by NTZ during infection might play a role in its ability to suppress VACV production. We explored this possibility by infecting HF with VACV in glutamine-free or normal medium, with no drug or NTZ, and in the presence or absence of α-KG (Fig. 5A). In glutamine-free medium, α-KG partially rescued VACV infection, as has been reported (Fontaine et al., 2014; Greseth and Traktman, 2014). However, α-KG did not rescue of viral production from inhibition by NTZ. In the presence of glutamine, α-KG alone inhibited viral production and caused complete inhibition when used in combination with NTZ. Although we do not know why α-KG inhibited VACV when glutamine was present, its failure to reverse the antiviral effect of NTZ suggests that the drug does not act by blocking glutamine utilization.
Figure 5. α-KG supplementation does not rescue VACV production after treatment with NTZ.

(A) HF were infected with VACV (MOI = 0.1) in DMEM either containing or lacking glutamine (Gln), and treated at 1hpi with either 10 μM NTZ or 7mM α-KG, or a combination of both in the respective media. Viral production was determined at 48 hpi by β-gal expression. (B) HF were infected with VACV (MOI = 0.1) and treated with 10 μM NTZ, 20 μM Z-VAD-FMK (zVAD), 10 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), 1 μM antimycin A (AM), 2.5 μM oligomycin (OM), 400 μM 2,4-dinitrophenol (DNP), or a combination of 10 μM NTZ and 20 μM zVAD. Viral production was measured at 48 hpi by β-gal expression.
Because NTZ is a mitochondrial uncoupler (Amireddy et al., 2017), we next considered whether energy depletion would mimic the antiviral effects of NTZ. Oligomycin (OM), which inhibits the mitochondrial F1FO-ATP synthase, had no effect on VACV production (Fig. 5B). Similarly, antimycin A, which inhibits cytochrome c reductase in the electron transport chain, only slightly reduced VACV production. Since both drugs inhibit mitochondrial ATP production, it seems unlikely that ATP depletion by mitochondrial uncoupling explains the NTZ effect. Additionally, although treatment with the protonophore FCCP inhibited VACV production, another OXPHOS uncoupler, 2,4-DNP did not have a large effect, suggesting that loss of inner mitochondrial membrane potential is not sufficient to account of the NTZ antiviral effects.
Cell death by apoptosis and pyroptosis can be triggered by mitochondrial factors. Although NTZ does not cause any clear cytopathology suggestive of apoptosis or pyroptosis in uninfected HF, it is possible that these pathways are being triggered by NTZ in infected cells and lead to an antiviral state. However, our finding that the pan-caspase inhibitor zVAD did not reduce the antiviral effect of NTZ argues against these mechanisms (Fig. 5B).
Palmitate partially rescues anti-viral effect of NTZ on VACV
In bacteria and parasites, NTZ has been shown to inhibit pyruvate:ferredoxin oxidoreductase (PFOR), a thiamine-dependent enzyme that produces acetyl-CoA from pyruvate (Kennedy et al., 2016). Although vertebrate cells do not have PFOR, they have a functionally similar enzyme, pyruvate dehydrogenase (PDH) that also uses thiamine pyrophosphate (TPP) as a cofactor to produce acetyl-CoA, which is needed in a variety of metabolic pathways including fatty acid and cholesterol synthesis. Since palmitate is a precursor for complex fatty acid synthesis, and can also be converted to acetyl-CoA via β-oxidation and thereby potentially obviate the dependence on PDH, we tested whether palmitate could suppress the NTZ anti-viral effect.
Interestingly, we found that addition of palmitate significantly lessened inhibition of VACV production by NTZ (Fig. 6). Although we cannot exclude the possibility that palmitate alters the pharmacology of NTZ, these results suggest that NTZ may be interfering with fatty acid metabolism in a way that is circumvented in part by adding palmitate. Moreover, addition of mevalonate, an intermediate in the cholesterol biosynthesis pathway, further rescued viral production.
Figure 6. Palmitate rescues viral production after NTZ treatment.
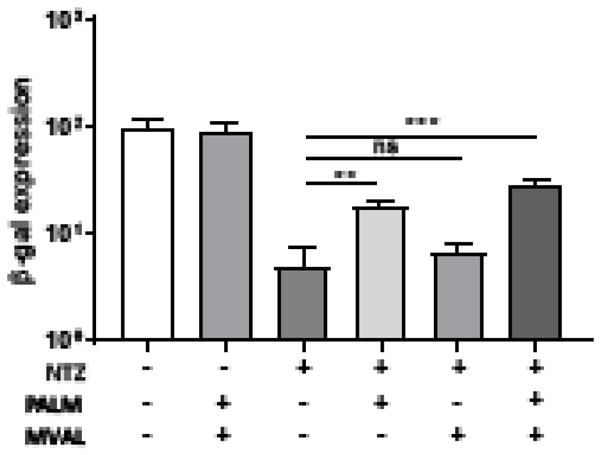
HF were infected with VACV (MOI = 0.1) and mock treated or treated with 10μM NTZ at 1 hpi along with 100 μM palmitate (PALM) or mevalonate (MVAL) or both. Viral production was measured at 48 hpi by β-gal expression. (**p < 0.005, ***p < 0.001)
Discussion
To our knowledge, this is the first report demonstrating that NTZ inhibits a member of the Poxviridae family. The potency of inhibition of VACV by NTZ (EC50 = ~2 μM) is similar to previously reported values for other viruses including as HCMV (EC50 = 3.2 μM) (Mercorelli et al., 2016), influenza A (1.0 – 3.3 μM) (Rossignol et al., 2009) and HCV (EC50 = 0.25 μM) (Korba et al., 2008). In fact, the potency of NTZ is even quite similar to what has been reported for bacteria and parasites (Lam et al., 2012; Laura et al., 2015). We found that TZ, the active metabolite of NTZ that is detected in plasma (Rossignol, 2014), is comparably active against VACV (Fig. 1A–C). Pharmacologic data from humans suggest that standard dosing with 500 mg of NTZ is sufficient to achieve TZ plasma levels above 10 μM within 1 hour after treatment, and maintain levels above that threshold until approximately 5 hours post treatment, with a peak concentration of ~35 μM (Balderas-Acata, 2011). Moreover, TZ plasma levels only dropped below the EC50 10 hours post treatment, suggesting that standard dosing should be sufficient to sustain an effective antiviral concentration during treatment. Another study reported a lower peak level, 6.4 μM, after a single 500 mg dose (Stockis et al., 1996). However, treatment with a higher single dose, 4 g, resulted in peak plasma concentrations close to 200 μM in patients and appeared to be safe (Stockis et al., 2002). The concentration remained above 30 μM for over 12 hours, further supporting the possibility of treating viral infections with NTZ. Thus, NTZ has potential utility in controlling poxviral infections.
Although the similarity of the dose responses across multiple infection systems seems most consistent with NTZ acting on a single cellular process, there are publications suggesting it targets specific virus factors. In the case of HCMV, Mercorelli et al. found that NTZ inhibited viral production and we have confirmed this effect (data not shown) (Mercorelli et al., 2016). These authors went on to provide evidence that NTZ inhibited the transcriptional activation properties of the HCMV IE2 protein. However, those experiments relied on IE2 expression by transduction of adenovirus vectors, followed by NTZ treatment for 72 hours before assaying the effect on an IE2 target gene. The reduced expression of IE2-target genes under these conditions could be due to NTZ inhibiting IE2 protein function as the authors suggest, but the experiments did not rule out the possibility that NTZ might be inhibiting IE2 activity by inhibiting its expression from the adenoviral vector. Further studies will be needed to distinguish between these possibilities.
Another example of NTZ targeting specific viral targets has been reported for influenza A. Unlike during HCMV infection, TZ was shown to act at a post-translational step, selectively inhibiting the maturation of the viral hemagglutinin by blocking the terminal glycosylation of HA after synthesis in the ER, subsequently hindering its progression to the Golgi and further insertion into the plasma membrane (Rossignol et al., 2009). Although these results suggested that NTZ inhibited a specific step in the viral life cycle of influenza, the dependency of glycosylation on metabolic intermediates including acetyl-CoA (Wellen and Thompson, 2012) may indicate that a more general effect of NTZ on the host cell metabolism may be the cause of the defect in HA maturation. NTZ has also been shown to cause the mobilization of Ca2+ from the ER into the cytosol, perturbing N-linked glycosylation and trafficking of the BVDV E2 glycoprotein from the ER to the Golgi (Ashiru et al., 2014), supporting the notion that NTZ may not be inhibiting influenza by a virus-specific mechanism.
The more parsimonious explanation for the broad antiviral effects of NTZ is that it alters a host cell pathway that is required by many viruses. The viruses that are now known to be sensitive to NTZ include RNA and DNA viruses, ones that replicate in the nucleus and in the cytoplasm, and ones that are enveloped as well as non-enveloped. All viruses share a reliance on the host cell protein synthetic machinery. Thus, the idea that NTZ might activate PKR in infected cells was an appealing one. However, we found that the PKR pathway was not activated by NTZ (Fig. 3A). Consistent with our results, Rossignol et al. found that TZ did not inhibit translation or affect eIF2α phosphorylation in uninfected or influenza A infected cells (Rossignol et al., 2009). Moreover, we found that NTZ still exerts its antiviral effects on VACV (Fig. 3B) and on HCMV (data not shown) in PKR−/− cells. Thus, at least in several viral systems, PKR does not mediate the NTZ effects.
Another possibility we considered was that NTZ might activate the interferon system and thereby inhibit multiple different viruses through various ISGs. VACV is unusual among DNA viruses in that it replicates in the cytoplasm. Coupled with the observation that NTZ causes mitochondrial stress, we considered the hypothesis that the combined effects of VACV DNA with mitochondrial DNA that could leak out of the mitochondria might activate the cGAS–STING pathway. However, NTZ remained effective even in STING−/− cells, ruling out that possibility. While others have detected an effect of NTZ on expression of a subset of ISGs (Trabattoni et al., 2016), we found no evidence that a major mediator of the IFN pathway, IRF3, is activated by NTZ (Fig. 4). Together with the fact that NTZ inhibits bacteria and protozoa in various culture systems in which there is no interferon, these results suggest that the interferon pathway activation cannot explain the broad antimicrobial effects of NTZ.
Since NTZ uncouples mitochondrial electron transport, we considered the possibility that mitochondrial effects, aside from DNA leakage, might mediate the antiviral responses. The failure of the pan-caspase inhibitor z-VAD to block the NTZ antiviral effects argues against pyroptosis or apoptosis, both of which can be mediated by mitochondrial factors, being a key element of the NTZ mechanism. Since NTZ uncouples mitochondrial electron transport from ATP production, we also investigated whether energy depletion might be important. However, another OXPHOS uncoupler, dinitrophenol (DNP), the F1Fo-ATP synthase inhibitor, oligomycin, and a respiratory chain complex III inhibitor, antimycin A, (Fig. 6) did not inhibit viral production to a large extent, thus arguing that energy limitation is not critical for the NTZ antiviral effect.
Vaccinia virus production is dependent on the metabolic switch of host cells from glucose oxidation to glutamine anaplerosis to support biosynthetic activities (Fontaine et al., 2014). Uncoupled respiration can likely lead to a relative depletion of acetyl-CoA and other TCA cycle intermediates resulting in insufficient substrates necessary to support added demands imposed by viral replicative cycle (Balcke et al., 2011) Consistent with this idea, we found that addition of palmitate, which can produce acetyl-CoA by β-oxidation, but not alpha-ketoglutarate did partially reverse the inhibitory effect of NTZ (Fig. 6).
Interestingly, Greseth and Trakman discovered that palmitate also partially rescued the VACV production from the effects of several fatty acid biosynthesis pathway inhibitors (Greseth and Traktman, 2014). Blocking mitochondrial import of palmitate with the CPT1 inhibitor, etomoxir, maintained the negative effects on viral production and this result was interpreted as a requirement for palmitate in energy production. This mechanism might also explain the antiviral activity of NTZ on other viruses, including HCMV and HCV, that alter fatty acid metabolism (Huang et al., 2013; Nasheri et al., 2013; Oem et al., 2008; Park et al., 2009; Spencer et al., 2011; Yang et al., 2008). On the other hand, our finding that other mitochondrial inhibitors had little or no effect on VACV production argues against the notion that NTZ acts by depleting energy production and suggests a possible imbalance in TCA cycle metabolites. As well, we observed that NTZ blocked the VACV-induced shut off of protein synthesis and markedly inhibited viral DNA and protein synthesis, which was not the case in studies using fatty acid synthesis and CPT1 inhibitors. Variation in the experimental conditions used, including cell types, might account for some of the differences. Regardless, the common observation in both studies that palmitate partially rescues VACV production is intriguing.
Our results add VACV to the list of viruses that are susceptible to NTZ. VACV is unusual in that it is a DNA virus that replicates in the cytoplasm and thus has quite different host cell requirements and potentially different susceptibilities to host defenses than other viruses that have been studied. Our investigation of the mechanism of anti-VACV action differ from some reports in other systems that have suggested that NTZ acts by triggering one or more host defense system, especially the PKR pathway. Although the mechanism or mechanisms by which NTZ exerts such a broad antiviral effect remains unclear, the partial rescue of its inhibitory effects by palmitate suggests an effect on the production of acetyl-CoA. Further investigation of the metabolic impact of NTZ in uninfected and infected cells should help to more clearly elucidate the mechanism.
Highlights.
Nitazoxanide inhibits vaccinia virus replication.
The inhibition occurs after entry but before late gene expression.
The mechanism is likely due to nitazoxanide interfering with metabolic adaptions needed for efficient virus replication
Acknowledgments
We thank Yan Xiang (University of Texas Health Science Center at San Antonio) and Bertram Jacobs (Arizona State University), Michael Lagunoff (University of Washington) and Daniel Stetson (University of Washington) for reagents. We thank Stephanie Child, Avraham Bayer, Nicolle Esparo and Lauren Gentles for technical assistance.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health NIH grant under award number RO1AI027762 (A.P.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amireddy N, Puttapaka SN, Vinnakota RL, Ravuri HG, Thonda S, Kalivendi SV. The Unintended Mitochondrial Uncoupling Effects of the FDA Approved Anti-Helminth Drug Nitazoxanide Mitigates Experimental Parkinsonism in Mice. J Biol Chem. 2017 doi: 10.1074/jbc.M117.791863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashiru O, Howe JD, Butters TD. Nitazoxanide, an antiviral thiazolide, depletes ATP-sensitive intracellular Ca(2+) stores. Virology. 2014;462–463:135–148. doi: 10.1016/j.virol.2014.05.015. [DOI] [PubMed] [Google Scholar]
- Balcke GU, Kolle SN, Kamp H, Bethan B, Looser R, Wagner S, Landsiedel R, van Ravenzwaay B. Linking energy metabolism to dysfunctions in mitochondrial respiration--a metabolomics in vitro approach. Toxicol Lett. 2011;203:200–209. doi: 10.1016/j.toxlet.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Balderas-Acata JIRRB, Patricio Estaban, Perez-Becerril Fabiola, Espinosa-Martinez Clara, Victoria Burke-Fraga Victoria, Gonzalez-de la Parra Mario. Bioavailability of two oral-suspension formulations of a single dose of nitazoxanide 500 mg: An open-label, randomized-sequence, two-period crossover, comparison in healthy fasted Mexican adult volunteers. Bioequivalence & Bioavailability. 2011;3:43–47. [Google Scholar]
- Beattie E, Denzler KL, Tartaglia J, Perkus ME, Paoletti E, Jacobs BL. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol. 1995;69:499–505. doi: 10.1128/jvi.69.1.499-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M, Wright ME. Progressive vaccinia. Clin Infect Dis. 2003;36:766–774. doi: 10.1086/374244. [DOI] [PubMed] [Google Scholar]
- Burgess HM, Mohr I. Cellular 5′-3′ mRNA exonuclease Xrn1 controls double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe. 2015;17:332–344. doi: 10.1016/j.chom.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier KS, Esparo NM, Child SJ, Geballe AP. A Single Amino Acid Dictates Protein Kinase R Susceptibility to Unrelated Viral Antagonists. PLoS Pathog. 2016;12:e1005966. doi: 10.1371/journal.ppat.1005966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WM, McFadden G. Oncolytic Poxviruses. Annu Rev Virol. 2014;1:119–141. doi: 10.1146/annurev-virology-031413-085442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Jiang Z. The essential adaptors of innate immune signaling. Protein Cell. 2013;4:27–39. doi: 10.1007/s13238-012-2063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Brennan G, Braggin JE, Geballe AP. Species specificity of protein kinase r antagonism by cytomegalovirus TRS1 genes. J Virol. 2012;86:3880–3889. doi: 10.1128/JVI.06158-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Hakki M, De Niro KL, Geballe AP. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol. 2004;78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Hanson LK, Brown CE, Janzen DM, Geballe AP. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J Virol. 2006;80:10173–10180. doi: 10.1128/JVI.00905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Hickson SE, Bayer A, Malouli D, Fruh K, Geballe AP. Antagonism of the Protein Kinase R Pathway in Human Cells by Rhesus Cytomegalovirus. J Virol. 2018:92. doi: 10.1128/JVI.01793-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Jarrahian S, Harper VM, Geballe AP. Complementation of vaccinia virus lacking the double-stranded RNA-binding protein gene E3L by human cytomegalovirus. J Virol. 2002;76:4912–4918. doi: 10.1128/JVI.76.10.4912-4918.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- clinicaltrials.gov. [Date accessed: 29 November 2017];U.S. National Library of Medicine. [Google Scholar]
- DeFilippis VR, Sali T, Alvarado D, White L, Bresnahan W, Fruh KJ. Activation of the interferon response by human cytomegalovirus occurs via cytoplasmic double-stranded DNA but not glycoprotein B. J Virol. 2010;84:8913–8925. doi: 10.1128/JVI.00169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar M, Liu M, McKenna SA, Liu P, Gehrig EA, Puglisi JD, Rossignol JF, Glenn JS. The anti-hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2alpha via protein kinase activated by double-stranded RNA activation. Gastroenterology. 2009;137:1827–1835. doi: 10.1053/j.gastro.2009.07.056. [DOI] [PubMed] [Google Scholar]
- Ferguson NM, Keeling MJ, Edmunds WJ, Gani R, Grenfell BT, Anderson RM, Leach S. Planning for smallpox outbreaks. Nature. 2003;425:681–685. doi: 10.1038/nature02007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine KA, Camarda R, Lagunoff M. Vaccinia virus requires glutamine but not glucose for efficient replication. J Virol. 2014;88:4366–4374. doi: 10.1128/JVI.03134-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, Stetson DB. The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity. 2016;45:255–266. doi: 10.1016/j.immuni.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greseth MD, Traktman P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 2014;10:e1004021. doi: 10.1371/journal.ppat.1004021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Tseng CP, Liao MH, Lu SC, Yeh WZ, Sakamoto N, Chen CM, Cheng JC. Hepatitis C virus replication is modulated by the interaction of nonstructural protein NS5B and fatty acid synthase. J Virol. 2013;87:4994–5004. doi: 10.1128/JVI.02526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AJ, Bruce AM, Gineste C, Ballard TE, Olekhnovich IN, Macdonald TL, Hoffman PS. Synthesis and Antimicrobial Evaluation of Amixicile-Based Inhibitors of the Pyruvate-Ferredoxin Oxidoreductases of Anaerobic Bacteria and Epsilonproteobacteria. Antimicrob Agents Chemother. 2016;60:3980–3987. doi: 10.1128/AAC.00670-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblentz GD. The De Novo Synthesis of Horsepox Virus: Implications for Biosecurity and Recommendations for Preventing the Reemergence of Smallpox. Health Secur. 2017 doi: 10.1089/hs.2017.0061. [DOI] [PubMed] [Google Scholar]
- Korba BE, Montero AB, Farrar K, Gaye K, Mukerjee S, Ayers MS, Rossignol JF. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008;77:56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Koszalka P, Tilmanis D, Hurt AC. Influenza antivirals currently in late-phase clinical trial. Influenza Other Respir Viruses. 2017;11:240–246. doi: 10.1111/irv.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KK, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, Anderson HJ, Andersen RJ, Av-Gay Y, Roberge M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8:e1002691. doi: 10.1371/journal.ppat.1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland JO, Jacobs BL. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology. 2002;299:133–141. doi: 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- Laura C, Celina E, Sergio SB, Guillermo D, Carlos L, Luis A. Combined flubendazole-nitazoxanide treatment of cystic echinococcosis: Pharmacokinetic and efficacy assessment in mice. Acta Trop. 2015;148:89–96. doi: 10.1016/j.actatropica.2015.04.019. [DOI] [PubMed] [Google Scholar]
- Liu S, Feng M, Guan W. Mitochondrial DNA sensing by STING signaling participates in inflammation, cancer and beyond. Int J Cancer. 2016;139:736–741. doi: 10.1002/ijc.30074. [DOI] [PubMed] [Google Scholar]
- Liu SW, Wyatt LS, Orandle MS, Minai M, Moss B. The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice. J Virol. 2014;88:202–211. doi: 10.1128/JVI.02426-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McCollum AM, Damon IK. Human monkeypox. Clin Infect Dis. 2014;58:260–267. doi: 10.1093/cid/cit703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Zhong Y, Embry A, Yan B, Lu S, Zhong G, Xiang Y. Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology. 2011;409:271–279. doi: 10.1016/j.virol.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercorelli B, Luganini A, Nannetti G, Tabarrini O, Palu G, Gribaudo G, Loregian A. Drug Repurposing Approach Identifies Inhibitors of the Prototypic Viral Transcription Factor IE2 that Block Human Cytomegalovirus Replication. Cell Chem Biol. 2016;23:340–351. doi: 10.1016/j.chembiol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- Nasheri N, Joyce M, Rouleau Y, Yang P, Yao S, Tyrrell DL, Pezacki JP. Modulation of fatty acid synthase enzyme activity and expression during hepatitis C virus replication. Chem Biol. 2013;20:570–582. doi: 10.1016/j.chembiol.2013.03.014. [DOI] [PubMed] [Google Scholar]
- Nikolova K, Gluud C, Grevstad B, Jakobsen JC. Nitazoxanide for chronic hepatitis C. Cochrane Database Syst Rev. 2014:CD009182. doi: 10.1002/14651858.CD009182.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oem JK, Jackel-Cram C, Li YP, Zhou Y, Zhong J, Shimano H, Babiuk LA, Liu Q. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J Gen Virol. 2008;89:1225–1230. doi: 10.1099/vir.0.83491-0. [DOI] [PubMed] [Google Scholar]
- Park CY, Jun HJ, Wakita T, Cheong JH, Hwang SB. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J Biol Chem. 2009;284:9237–9246. doi: 10.1074/jbc.M808773200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Moss B. Characterization of a vaccinia virus mutant with a deletion of the D10R gene encoding a putative negative regulator of gene expression. J Virol. 2006;80:553–561. doi: 10.1128/JVI.80.2.553-561.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen BW, Harms TJ, Reynolds MG, Harrison LH. Use of Vaccinia Virus Smallpox Vaccine in Laboratory and Health Care Personnel at Risk for Occupational Exposure to Orthopoxviruses - Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2015. MMWR Morb Mortal Wkly Rep. 2016a;65:257–262. doi: 10.15585/mmwr.mm6510a2. [DOI] [PubMed] [Google Scholar]
- Petersen T, Lee YJ, Osinusi A, Amorosa VK, Wang C, Kang M, Matining R, Zhang X, Dou D, Umbleja T, Kottilil S, Peters MG. Interferon Stimulated Gene Expression in HIV/HCV Coinfected Patients Treated with Nitazoxanide/Peginterferon-Alfa-2a and Ribavirin. AIDS Res Hum Retroviruses. 2016b;32:660–667. doi: 10.1089/aid.2015.0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159:1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol JF. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res. 2014;110:94–103. doi: 10.1016/j.antiviral.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol JF, Keeffe EB. Thiazolides: a new class of drugs for the treatment of chronic hepatitis B and C. Future Microbiol. 2008;3:539–545. doi: 10.2217/17460913.3.5.539. [DOI] [PubMed] [Google Scholar]
- Rossignol JF, La Frazia S, Chiappa L, Ciucci A, Santoro MG. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level. J Biol Chem. 2009;284:29798–29808. doi: 10.1074/jbc.M109.029470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya A, Bhat HR, Ghosh SK. Update on Nitazoxanide: A Multifunctional Chemotherapeutic Agent. Curr Drug Discov Technol. 2017 doi: 10.2174/1570163814666170727130003. [DOI] [PubMed] [Google Scholar]
- Shors T, Keck JG, Moss B. Down regulation of gene expression by the vaccinia virus D10 protein. J Virol. 1999;73:791–796. doi: 10.1128/jvi.73.1.791-796.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Schafer XL, Moorman NJ, Munger J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J Virol. 2011;85:5814–5824. doi: 10.1128/JVI.02630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockis A, Allemon AM, De Bruyn S, Gengler C. Nitazoxanide pharmacokinetics and tolerability in man using single ascending oral doses. Int J Clin Pharmacol Ther. 2002;40:213–220. doi: 10.5414/cpp40213. [DOI] [PubMed] [Google Scholar]
- Stockis A, Deroubaix X, Lins R, Jeanbaptiste B, Calderon P, Rossignol JF. Pharmacokinetics of nitazoxanide after single oral dose administration in 6 healthy volunteers. Int J Clin Pharmacol Ther. 1996;34:349–351. [PubMed] [Google Scholar]
- Trabattoni D, Gnudi F, Ibba SV, Saulle I, Agostini S, Masetti M, Biasin M, Rossignol JF, Clerici M. Thiazolides Elicit Anti-Viral Innate Immunity and Reduce HIV Replication. Sci Rep. 2016;6:27148. doi: 10.1038/srep27148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13:270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, Conrads TP, Wang T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology. 2008;48:1396–1403. doi: 10.1002/hep.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]