

Abstract
Unlike breast cancer that is positive for estrogen receptor-α (ERα), there are no targeted therapies for triple negative breast cancer (TNBC). ERα is silenced in TNBC through epigenetic changes including DNA methylation and histone acetylation. Restoring ERα expression in TNBC may sensitize patients to endocrine therapy. Expression of c-Src and ERα are inversely correlated in breast cancer suggesting that c-Src inhibition may lead to re-expression of ERα in TNBC. KX-01 is a peptide substrate-targeted Src/pretubulin inhibitor in clinical trials for solid tumors. KX-01 (1 mg/kg body weight-BID) inhibited growth of tamoxifen-resistant MDA-MB-231 and MDA-MB-157 TNBC xenografts in NUDE mice that was correlated with Src kinase inhibition. KX-01 also increased ERα mRNA and protein, as well as increased the ERα targets progesterone receptor (PR), pS2 (TFF1), cyclin D1 (CCND1) and c-myc (MYC) in MDA-MB-231 and MDA-MB-468 but not MDA-MB-157 xenografts. MDA-MB-231 and MDA-MB-468 tumors exhibited reduction in mesenchymal markers (vimentin, β-catenin) and increase in epithelial marker (E-cadherin) suggesting mesenchymal-to-epithelial transition (MET). KX-01 sensitized MDA-MB-231 and MDA-MB-468 tumors to tamoxifen growth inhibition and tamoxifen repression of the ERα targets pS2, cyclin D1 and c-myc. Chromatin immunoprecipitation (ChIP) of the ERα promoter in KX-01 treated tumors demonstrated enrichment of active transcription marks (acetyl-H3, acetyl-H3Lys9), dissociation of HDAC1, and recruitment of RNA polymerase II. Methylation-specific PCR and bisulfite sequencing demonstrated no alteration in ERα promoter methylation by KX-01. These data demonstrate that in addition to Src kinase inhibition, peptidomimetic KX-01 restores ERα expression in TNBC through changes in histone acetylation that sensitize tumors to tamoxifen.
Implications: Src kinase/pretubulin inhibitor KX-01 restores functional ERα expression in ERα- breast tumors, a novel treatment strategy to treat triple-negative breast cancer.
INTRODUCTION
Breast cancer is the most common cancer in women and the cause of substantial morbidity and mortality. Estrogen receptor (ERα) expression in tumors is a marker for better prognosis and a predictor for response to endocrine therapy (1,2). However, approximately one-third of breast cancers do not express ERα and these patients are generally associated with poor prognosis and worse clinical outcomes (3,4). A subset of ERα negative tumors termed Triple Negative Breast Cancer (TNBC) lacks expression of ERα and progesterone receptor (PR), and does not overexpress the membrane receptor HER2. Patients with TNBC have several clinical characteristics that make them difficult to treat including rapid risk of recurrence at 1-3 years, increased mortality in the first 5 years, and rapid progression from distant recurrence to death (5,6).
TNBC patients are not candidates for targeted therapies directed against ERα or HER2 and therapy is limited to cytotoxic chemotherapy and radiation therapy that is associated with significant toxicities. The opportunity to re-express ERα in TNBC patients to sensitize tumors to less toxic endocrine therapy agents represents a promising therapeutic strategy, although there are currently no agents that achieve this result in the clinic. In experimental systems, certain histone deacetylase (HDAC) inhibitors and DNA methyltransferase (DNMT) inhibitors resulted in re-expression of ERα in ERα-negative breast cancer cells and sensitization of cells to endocrine therapy agents (7–9). These studies provided the basis for current clinical trials with HDAC inhibitors panobinostat, entinostat and DNMT inhibitor 5-azacytidine (10) to re-express ERα and sensitize tumors to ERα modulators such as tamoxifen.
ERα is silenced in TNBC through epigenetic changes including DNA methylation and altered histone acetylation (11,12) and possibly additional signaling pathways that silence ER expression. c-Src is an oncogenic non-receptor tyrosine kinase overexpressed in TNBC and identified as a therapeutic target for TNBC (13,14). ERα and Src expression are inversely correlated in human primary breast cancers (15). Inhibition of Src may provide a mechanism for re-expression of ERα in TNBC.
Peptidomimetics represent a novel class of drugs that interact with the peptide substrate sites of proteins. KX-01 is the ‘first in class’ peptidomimetic non-ATP kinase inhibitor that targets the substrate binding site of Src and inhibits its kinase activity and downstream targets (16,17). Additionally a second mechanism of action for KX-01 at higher doses was identified as inhibition of tubulin polymerization (18,19). KX-01 has completed phase I clinical testing for solid tumors (NCT00658970) and has completed a phase II trial for prostate cancer (NCT01074138) (20). A phase 1b trial for acute myeloid leukemia is in progress (21) and a phase 1b/IIa clinical trial for KX-01 in combination with paclitaxel was initiated in patients with solid tumors including breast cancer (22).
Previous studies from this laboratory demonstrated the efficacy of KX-01 as a single agent and in combination with tamoxifen for ERα positive breast cancer (17), and in combination with paclitaxel or doxorubicin for TNBC (18). The efficacy of KX-01 in slowing tumor growth was correlated with significant inhibition of Src kinase in the tumors. During these studies, it was found that KX-01 restored ERα protein expression in TNBC xenografts. These data provided the basis to test whether orally bioavailable, clinical peptidomimetic KX-01 could be valuable as an endocrine therapy sensitization agent in TNBC. The present study was undertaken to determine whether KX-01 could restore tamoxifen sensitivity to TNBC, and to understand the mechanisms for the re-expression of ERα.
Materials and methods
Cell culture and reagents
ERα/PR/Her2-negative MDA-MB- 231, MDA-MB- 468 breast cancer cell lines and the ERα/PR-positive MCF-7 breast cancer cell line were obtained from American Type Culture Collection (ATCC). Cells were grown in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 1% penicillin/streptomycin (Invitrogen). To generate an MDA-MB-468 luc+ cell line, MDA-MB-468 cells were transduced with lentiviral particles expressing the firefly luciferase gene and RFP. Transduced cells were then selected for antibiotic resistance (G418; Invitrogen) and surviving colonies were screened for bioluminescence in complete media supplemented with 150 μg/ml D-luciferin (Gold Bio, USA) by in vitro imaging using the IVIS XRMS small animal imaging system (Perkin Elmer, CA, USA). Bioluminescent and RFP positive cells were grown in culture and characterized for stable luminescence in vitro and tumorigenic potential in vivo. Cells were maintained in a humidified environment of 5% CO2 at 37°C. KX-01 was provided by Athenex pharmaceuticals (Buffalo, NY). Tamoxifen pellets were purchased from Innovative Research of America (Sarasota, FL).
Tumor Xenograft Study
Female nude mice (4–6 weeks old; BALB/c nude) were purchased from Charles River and maintained in pathogen-free conditions. The use and care of animals in this study is approved by the Institutional Animal Care and Use Committee protocol #2941R2 from Tulane University New Orleans, LA. Xenograft procedures and KX-01 oral dosing was done as described in our previous studies (17,18). Briefly, we used the MDA-MB-231 xenograft model and tested two doses of KX-01 (1 and 5 mg/kg body weight, BID by oral gavage) for 30 days. 5 mg/kg KX-01 resulted in significant tumor growth inhibition associated with increased apoptosis and microtubule disruption. 1 mg/kg KX01 exhibited a more modest tumor growth inhibition but no significant apoptosis or microtubule disruption was detected in the tumors (18). The present study used KX-01 at 1 mg/kg b. wt., a dose that inhibits Src kinase activity. 5 × 106 MDA-MB-231 cells were injected bilaterally into the mammary fat pads of nude mice and tumors were allowed to grow to ~100 mm3. Mice were randomly divided into 4 treatment groups (N=5 mice, 7-10 tumors/group). Group 1 received pure distilled water by oral gavage BID which served as vehicle control, group 2 was treated with KX-01 (1mg/kg b.wt BID), group 3 mice were implanted with a tamoxifen pellet (5 mg, 60-day release) above the shoulder using a 10-gauge trochar, and group 4 mice were implanted with tamoxifen and treated with KX-01 (1mg/kg b.wt BID). All mice were sacrificed on day 40 due to large tumor size exceeding 1000 mm3 in the vehicle and tamoxifen alone groups. Tumor diameters were measured twice a week using digital calipers and tumor volume was calculated as 0.523 × LM2 (where L is large diameter and M is small diameter). At sacrifice, tumors were removed from the mice and either immediately snap frozen with liquid nitrogen and stored at −80°C, or fixed with 10% formalin solution for immunohistochemical staining.
Bioluminescent imaging (BLI)
A similar experiment as described for MDA-MB-231 xenografts was carried out with MDA-MB-468 xenografts using MDA-MB-468 luc+ cells. Approximately 5 × 106 MDA-MB-468 luc+ cells were injected into the mammary fat pads of nude mice to form primary tumors. Bioluminescent imaging was performed with a highly sensitive, cooled CCD camera mounted in a light-tight specimen box (IVIS XRMS; Perkin Elmer). Imaging and quantification of signals were controlled by the acquisition and analysis software Living Image (Perkin Elmer). For in vivo imaging, animals were given the substrate D-luciferin by intraperitoneal injection at 150 mg/kg in DPBS Dulbecco’s Phosphate Buffered Saline (Invitrogen) and anesthetized (1–3% isoflurane). Mice were then placed into the IVIS box containing a light-tight camera with continuous exposure to 1–2% isoflurane. Imaging times ranged from 1 sec to 3 min. depending on the tumor and the time point. Generally, one animal was imaged at a time. The low levels of light emitted from the bioluminescent tumors or cells were detected by the IVIS camera system, integrated, digitized, and displayed. Regions of interest from displayed images were identified around the tumor sites and were quantified as total photon counts or photons/sec using Living Image® software (Perkin Elmer). Background bioluminescence in vivo was in the region of 1 × 104 photon counts or 1–2 × 105 photons/s. Tissues were subsequently fixed in 10% formalin (Sigma, St. Louis, MO) and prepared for IHC evaluation.
Immunohistochemistry
Immunohistochemical (IHC) staining was performed on 10% neutral buffered formalin fixed paraffin-embedded tumor samples as described previously (17,18). Briefly, sections mounted on slides were deparaffinized in xylene, dehydrated in ethanol, rinsed in water and antigen retrieval was carried out with 0.01 M citrate buffer (pH 6.0) for 20 min in a steamer and then incubated with 3% hydrogen peroxide for 5 min. After washing with PBS, sections were blocked by incubation in 10% normal goat serum for 30 min, followed by overnight incubation with primary antibody. The source of the primary antibody and the dilutions used for IHC were as follows, ERα (1:100), Ki67 (prediluted) (NeoMarkers, Fremont, CA), PR (1:100; Thermo Scientific, Fremont, CA), vimentin (1:100; Vector labs, Burlingame, CA), E-cadherin (1:400) β-catenin (1:800), total Src (1:200) and phospho-Y416 Src (1:100) from Cell Signaling Technology Inc. After overnight incubation with primary antibody, slides were washed with PBS followed by 30 minutes incubation with biotinylated secondary antibody (Vector labs), rinsed in PBS and incubated with ABC reagent (Vector labs) for 30 min. The stain was visualized by incubation in 3, 3-diaminobenzidine (DAB) and counterstained with Harris hematoxylin. Internal negative control samples incubated with either non-specific rabbit IgG, or 10% goat serum instead of the primary antibody showed no specific staining. Slides were dehydrated and mounted with Permount (Fisher). Slides were visualized using a Nikon OPTIPHOT microscope and randomly selected bright field microscope images (magnification, × 200) were captured by Nikon Digital Sight High-Definition color camera (DS-Fi1) using NIS-Elements BR software.
Quantitative real-time RTPCR
Total RNA was extracted from MDA-MB-231, MDA-MB-468, and MCF-7 (positive control) tumors using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. 5 μg of total RNA was reverse transcribed to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen). In the real-time PCR step, PCR reactions were performed in triplicates with 1 μl cDNA per reaction and primers specific for ERα (Hs01046818_m1) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs99999905_ml) provided by Inventoried Gene Assay Products (Applied Biosystems, Foster City, CA) using the Fast Start 2× Taqman probe master (Roche Diagnostics, Mannheim, Germany) in a iQ5 Biorad thermocycler. Thermal cycling was initiated at 94°C for 4 min followed by 40 cycles of PCR (94°C, 15 s; 60°C, 30 s). GAPDH was used as an endogenous control and vehicle control was used as a calibrator. The relative changes of gene expression were calculated as: fold change in gene expression, 2−ΔΔCt = 2−[ΔCt (treated samples) − ΔCt (untreated control samples)], where ΔCt = Ct (ERα) − Ct (GAPDH) and Ct represents threshold cycle number. The real-time rtPCR was performed in triplicates and repeated at least two times.
Chromatin Immunoprecipitation (ChIP) assay for the ERα promoter
Tumor samples that were snap frozen in liquid nitrogen and stored at −80°C were used for ChIP assays. MDA-MB-231 tumors from the treatment groups 1) vehicle control (VC), 2) KX-01, 3) tamoxifen (TAM), and 4) TAM + KX-01 were used. MCF-7 tumors were used as a positive control for ERα expression. The ChIP assay was performed using the Magna ChIP G tissue kit according to the manufacturer’s protocol (Millipore) and our previous studies (23). Briefly, a 5 mm3 tumor tissue piece was obtained using a micro-dissection punch and the sample was dispersed in 1 ml Magna ChIP G tissue stabilization solution with protease inhibitors and then cross-linked using 1% formaldehyde treatment (prepared fresh; 270 μl of 37% formaldehyde [Sigma] to 10 ml of PBS). Glycine (125 mM) was used to quench the formaldehyde and block further cross linking. After centrifugation at 800 × g at 4°C for 5 min, the pellet was rinsed in PBS, suspended in 500 μl Magna ChIP G tissue lysis buffer, vortexed well and incubated on ice for 15 min. Cells were then centrifuged at 800 × g at 4°C for 5 min and the supernatant was removed. The cell pellet was re-suspended in 125 μl Magna ChIP dilution buffer in a 1.5 ml tube, and the samples were sonicated using the Bioruptor automatic sonicator (Diagenode, Denville, NJ) at 4°C for 12 cycles of 30 seconds “ON”/30 seconds “OFF” to shear chromatin and generate DNA fragments of 200-1000 base pairs. 5 μl (1%) of the content was removed and saved in 4° C as input. The sheared cross-linked chromatin was immunoprecipitated (IP) using ChIP-validated antibodies to acetyl-histone H3, acetyl-histone H3- Lys9 (H3K9), trimethyl-histone H3-Lys9 (Upstate Biotechnology), HDAC1 and RNA Pol II (Santa Cruz Biotechnology). Each IP reaction consisted of 125 μl of chromatin + 375 μl of dilution buffer with protease inhibitors + 20 μl of protein G magnetic beads + 5 μg of primary antibody. The IP reactions were incubated at 4° C overnight with rotation. IgG from the same species as the primary antibodies served as negative controls. Magnetic beads were separated using a magnetic separator (Biolabs) and the supernatant was discarded. The Protein G magnetic beads-antibody-chromatin complex was incubated with a series of wash buffers provided in the Magna ChIP G tissue Kit: one time each for 5 min each wash on a rotating platform followed by magnetic clearance and careful removal of the supernatant fractions: 500 μl low salt immune complex wash buffer, 500 μl high salt immune complex wash buffer, 500 μl LiCl immune complex wash buffer, 500 μl TE buffer. Following immunoprecipitation, protein-DNA cross-links were reversed by adding 100 μl Magna Chip elution buffer with proteinase K and incubated at 62° C for 2 h with shaking followed by incubation at 95 ° C for 10 min. Samples were allowed to cool to room temperature and the magnetic beads were separated and supernatant was transferred to a new tube and DNA was purified using spin columns according to the manufactures protocol. ChIP-purified DNA was amplified by standard PCR using primers for the ERα promoter (sense, 5’-GAACCGTCCGCAGCTCAAGATC-3’; antisense, 5’GTCTGACCGTAGACCTGCGCGTTG-3’) yielding a 150 bp fragment using the following reactions conditions: 2 μl of ChIP purified DNA or 1% total input DNA, 200 nmol/L of each primer, 1.5 mmol/L MgCl2, 200 μmol/L dNTP, 10X PCR gold buffer (Applied Biosystems), and 2 units of Hot start AmpliTaq Gold DNA polymerase (Applied Biosystems) in a total volume of 20 μl. The reaction was initiated at 94°C for 4 min followed by 30 cycles of PCR (94°C, 30 s; 56°C, 30 s; 72°C, 1 min), and extended at 72°C for 5 min. After amplification, PCR products were separated on a 1.5% agarose gel and visualized by ethidium bromide staining using a Gel Doc 2000 instrument (Bio-Rad, Hercules, CA). All ChIP assays were performed three times yielding similar results.
Methylation-specific PCR (MSP) analysis
Genomic DNA was isolated from MDA-MB-231 tumors treated with VC, TAM, KX-01 and TAM + KX-01, and from MCF-7 tumors using the QIAamp DNA mini kit DNeasy tissue kit (QIAGEN) according to manufacturer’s instructions. 500 ng of genomic DNA was bisulfite treated using the EZ DNA Methylation kit (Zymo Research) according to manufacturer’s directions. The bisulphite treatment converts unmethylated cytosine residues, but not methylated cytosines, to uracil (detected as thymine following PCR). 100 ng of bisulfite converted DNA was used as a template for methyl-specific PCR. ERα-positive MCF-7 tumor was used as an unmethylated (U) control for the ERα promoter; whereas vehicle treated ERα-negative MDA-MB-231 tumors were considered as a methylated control for the ERα promoter. The methylation status of bisulphite-modified DNA at the critical region of the ERα promoter CpG islands was characterized by Methyl-Specific PCR using the following primers (8,11,24):
ER unmethylated (U) Forward primer: 5’GGTGTATTTGGATAGTAGTAAGTTTGT 3’; Reverse primer: 5’CCATAAAAAAAAACCAATCTAACCA 3’;
ER methylated (M) Forward primer: 5’GTGTATTTGGATAGTAGTAAGTTCGTC 3’; Reverse primer: 5’CGTAAAAAAAACCGATCTAACCG 3’.
The PCR mixture contained 100 ng DNA, 200 nmol/L of each primer, 1.5 mmol/L MgCl2, 200 μmol/L dNTPs, 10X PCR gold buffer (Applied Biosystems), and 2 Units of Hot start AmpliTaq Gold DNA polymerase (Applied Biosystems) in a total volume of 20 μl. The reaction was initiated at 95°C for 5 min followed by 31 cycles of PCR (95°C, 30 s; 55°C, 30 s; 72°C, 30 s), and extended at 72°C for 5 min. PCR products were subjected to electrophoresis on 2% agarose gels and visualized by ethidium bromide staining. Pictures were taken using a Gel Doc 2000 instrument. Assays were performed three times yielding similar results.
Bisulfite sequencing analysis
Bisulfite modification was carried out using Zymo Research EZ Methylation kit (D5004). 200 - 500 ng of sample DNA was used for bisulfite modification followed by the PCR amplification. -533 to +120 from ATG site (−172 to +481 from transcriptional start site TSS) methylation sequencing was performed by EpigenDx, Hopkinton, MA to determine the site-specific methylation changes in the ERα promoter region.
Statistical analysis
Statistical significance was evaluated using the Student t tests (P < 0.05; 2-tailed) and one way ANOVA followed by Tukey multiple comparison test. Data were expressed as mean ± SD. P < 0.05 was considered statistically significant. The mean and SD were calculated using Microsoft Excel or Graph pad Prism.
RESULTS
Oral administration of KX-01 sensitized ERα-negative MDA-MB-231 and MDA-MB-468 breast tumor xenografts to tamoxifen
When MDA-MB-231 tumor volumes reached ~80-100 mm3, mice were treated with vehicle (ultrapure water), KX-01 at 1 mg/kg BID, tamoxifen (5mg pellet; 60 day release), or tamoxifen + KX-01 continuously for up to 40 days. KX-01 used at 1 mg/kg b.wt. resulted in some tumor growth inhibition beginning at day 18 (Fig. 1A), but the drug efficacy at 1 nM was less compared to our previous study that used 5 mg/kg KX-01 (18). Tumor growth inhibition by 1 mg/kg b.wt. KX-01 alone was correlated with inhibition of Src kinase (Supplementary Fig. S1) indicating that Src kinase inhibition likely contributed to KX-01 efficacy in MDA-MB-231 tumors. Mice implanted with tamoxifen pellet alone did not exhibit tumor growth inhibition compared to the vehicle control (Fig. 1A). On day 40, mice in the control and tamoxifen treatment groups had to be sacrificed due to high tumor burden. At day 40, KX-01 alone and tamoxifen + KX-01 reduced tumor volume by 59% and 70%, respectively, compared to vehicle. Tumor volume for the tamoxifen + KX-01 group was significantly reduced compared to the KX-01 alone group (Fig. 1A, B), and the difference in tumor volume between these treatments increased at day 48 (P< 0.01) and at day 60 (P< 0.001). The final tumor weights (day 60) for the tamoxifen + KX-01 group was 32% lower compared to the KX-01 alone group (Fig.1F).
Figure 1.
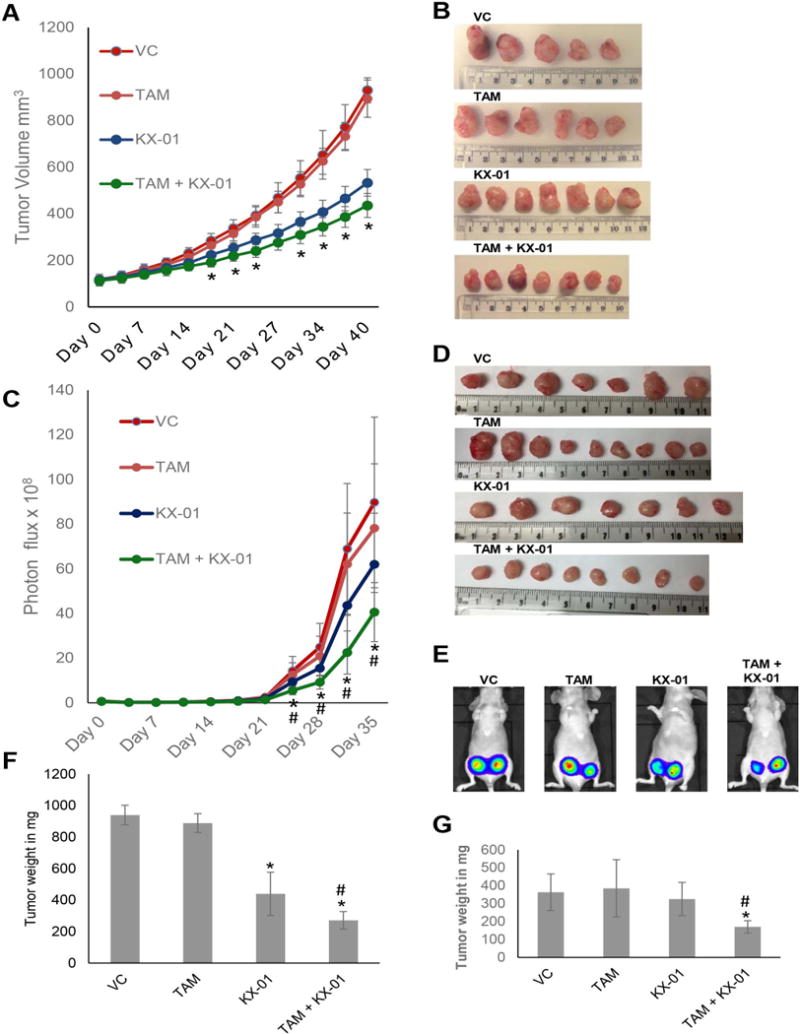
KX-01 treatment re-sensitized MDA-MB-231 and MDA-MB-468 breast tumor xenografts to tamoxifen. A) Athymic NUDE mice bearing MDA-MB-231 tumors (~100 mm3) were separated into four treatment groups randomly. Five animals/group (1-2 tumors/animal) were used for each treatment group for a total of n = 7-10 tumors/group: group 1 received VC, ultra-pure distilled water for each treatment administration; group 2 was implanted with 60 day release TAM pellet (5mg) was implanted subcutaneously near the neck; group 3 were treated with KX-01 1 mg/kg b.wt by oral gavage twice daily; group 4 were treated with TAM + KX-01. Tumors were measured twice a week by caliper and tumor volumes were calculated as described in the ‘Materials and Methods’. Data is represented as mean tumor volume in mm3 ± SD. *, P < 0.05 compared to KX-01 alone by student’s t-test. B) MDA-MB-231 tumors were excised and photographed. C) Identical experiment with MDA-MB-468-luc+ breast tumor measuring tumor volume in response to TAM and KX-01 treatment by Bioluminescent Imaging (BLI) using IVIS XRMS animal imager. * P<0.05 compared to VC, TAM alone and # P<0.05 compared to KX-01 alone. D) MDA-MB-468 tumors were excised and photographed. E) BLI of representative BALB/c mice treated with VC, TAM, KX-01 or TAM + KX-01 on day 35 imaged after D-luciferin injection using the IVIS XRMS small animal imager as described in materials and methods. After necropsy MDA-MB-231 (F) and MDA-MB-468 (G) tumors from all four treatment groups were removed from mice and weighed. The bar represents mean tumor weight (mg) ± SD. VC, vehicle control; TAM, tamoxifen.* P<0.05 compared to VC, TAM and # P<0.05 compared to KX-01.
In MDA-MB-468 tumors, tamoxifen alone (10 mg pellet; 60 day release) and KX-01 alone (1 mg/g b.wt. BID) had no effect on tumor volume compared to vehicle (Fig. 1C-E) demonstrating resistance of MDA-MB-468 tumors to both drugs. However co-treatment with tamoxifen + KX-01 reduced tumor volume 67% compared to vehicle (Fig.1C-E). The final tumor weights for the tamoxifen + KX-01 group was 43% lower compared to the KX-01 alone group (Fig. 1G).
KX-01 induced expression of ERα in MDA-MB-231 and MDA-MB-468 tumor xenografts
MDA-MB-231 and MDA-MB-468 tumor sections were examined for the effect of KX-01 on protein levels of ERα and the ERα target PR which is a marker for a functional ERα signaling pathway. IHC analysis revealed that ERα and PR expression was absent in tumors from the vehicle control and tamoxifen treated group in both MDA-MB-231 and MDA-MB-468 tumors, but KX-01 alone and KX-01 + TAM significantly increased ERα and PR expression (Fig. 2A-D). These results demonstrate that treatment with KX-01 in two TNBC xenograft tumors (MDA-MB-231 and MDA-MB-468) resulted in re-expression of ERα, a requirement for tumor sensitivity to tamoxifen. Proliferation marker Ki67 was significantly reduced in tumors treated with TAM + KX-01 compared to KX-01 alone or control treatment (Fig. 2A-D).
Figure 2.
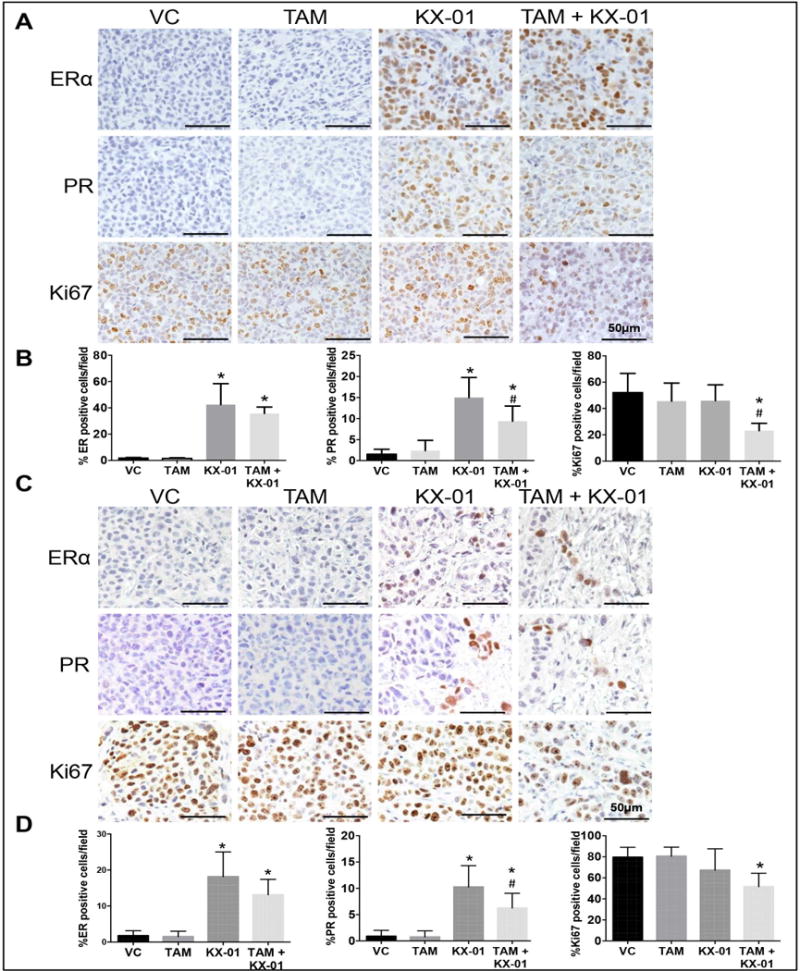
KX-01 induced ERα and PR re-expression in MDA-MB-231 and MDA-MB468 tumor xenografts. For the experiments described in Figure 1A, IHC for ERα, PR and Ki67 was performed in paraffin embedded MDA-MB-231 (A) and MDA-MB-468 (C) tumors sections. Bright field microscopic images (original magnification, × 100) were photographed. Quantitation of ERα, PR and Ki67 staining in MDA-MB-231 (B) and MDA-MB-468 (D) tumor sections. Nuclear staining positive cells (brown color stained cells) and total number of cells (blue color hematoxylin stained cells) were counted in three random microphotograph images from three tumor samples from VC, TAM, KX-01 and KX-01 + TAM group. The total cell number in each image was calculated by counting hematoxylin-positive cells using Image J particle count command, and DAB-positive cells (brown color nuclear staining) were also counted the same way after performing color deconvolution command and expressed as nuclear staining (%). The data are represented as the mean positive staining (%) with SD. *, P < 0.05 significantly different compared to VC and #, P < 0.05 significantly different compared to KX-01 was determined using one-way ANOVA and Tukey Post-hoc test. VC, vehicle control, TAM, tamoxifen.
To further evaluate the KX-01 effect on re-expression of ERα, we assessed two TNBC patient derived xenograft (PDX) tumors. PDX tumors propagated in mice were excised and cultured in medium ex vivo with vehicle or KX-01 (25, 50 nmol/L) for 72 hrs. KX-01 (25 and 50 nmol/L) increased ERα mRNA 2.7 and 3.4 fold, respectively (Supplementary Fig. S2).
To further assess the restoration of ERα signaling by KX-01, MDA-MB-231 tumors were assessed for expression of additional ERα target proteins, c-myc, cyclin D1 and pS2. KX-01 induced expression of cyclin D1 and pS2 protein. Co-treatment of KX-01 + tamoxifen resulted in suppression of c-myc, cyclin D1 and pS2 protein levels (Supplementary Fig. S3). These results indicate that KX-01 could restore ERα target proteins in MDA-MB-231 tumors and that co-treatment with tamoxifen could suppress the KX-01 induced expression.
It is possible that KX-01 sensitized tumor cells to off-target effects of tamoxifen to induce apoptosis (25). To address this possibility, the level of apoptosis was measured in tumors from all treatment groups in MDA-MB-231 tumors (Supplementary Fig. S4). Tamoxifen alone did not induce apoptosis. KX01 induced a very modest level the apoptosis, and there was no additional apoptosis in the KX01 + tamoxifen treatment group. To further address off-target effects of tamoxifen, and a requirement for ERα re-expression to sensitize tumors to tamoxifen, we used another TNBC xenograft model, MDA-MB-157, that does not express significant ERα protein in response to KX-01 treatment (Supplementary Fig. S5A). Tamoxifen alone did not induce apoptosis, KX-01 alone induced a modest level of apoptosis, and KX-01 + tamoxifen induced the same level of apoptosis as KX-01 alone in MDA-MB-157 tumors (Supplementary Fig. S6). Tamoxifen did not result in tumor growth inhibition in the presence and absence of KX-01 (Supplementary Fig. S5B). These data indicate that when KX-01 does not result in significant ERα protein expression in these TNBC tumors, the tumors were not sensitized to tamoxifen growth inhibition. The data with MDA-MB-157 tumors further indicate that KX-01 targets mechanisms other than ERα re-expression that contribute to the anti-tumor efficacy.
ERα re-expression and sensitivity to tamoxifen was reversible upon KX-01 withdrawal
To determine the reversibility of ERα re-expression and tamoxifen sensitivity by KX-01 in MDA-MB-231 tumors, tumor bearing animals were treated with KX-01 for 14 days, KX-01 treatment was withdrawn, and then the animals were then divided and treated with either tamoxifen or vehicle for an additional 16 days. There was no significant difference in tumor volume between the tamoxifen and vehicle treatment groups at day 30 (Supplementary Fig. S7A). The tumors in both treatment groups did not exhibit significant ERα expression (Supplementary Fig. S7B) as compared to continuous KX-01 treatment (Fig. 2A, C). MCF-7 tumor sections were used as a positive control that demonstrated a robust ERα expression (Supplementary Fig. S7C).
KX-01 treatment increased epithelial markers and reduced mesenchymal markers inMDA-MB-231 and MDA-MB-468 tumors
ERα expression is a marker for a well-differentiated breast tumor with epithelial-like phenotype. Since MDA-MB-231 tumors exhibit a mesenchymal phenotype, the re-expression of ERα by KX-01 suggested that the tumors may have undergone a mesenchymal to epithelial transition (MET). Both MDA-MB-231 and MDA-MB-468 tumors express the mesenchymal marker vimentin and exhibit β-catenin staining in the cytoplasm and nucleus. These tumors are negative for the epithelial marker E-cadherin, a cell-cell adhesion protein that is increased by Src inhibition (26). KX-01 treatment increased E-cadherin expression in tumor cell membranes and markedly reduced vimentin expression (Fig. 3A, B). Nuclear β-catenin contributes to breast tumorigenesis by regulating genes that are involved in proliferation, invasion, and EMT (27). When β-catenin is expressed in the cell membrane with E-cadherin, signaling-competent nuclear β-catenin levels diminished and cell proliferation and invasion were suppressed (28). β-catenin as located predominantly in the nucleus of untreated MDA-MB-231 and MDA-MB-468 tumors. KX-01 treatment resulted in marked reduction in nuclear β-catenin and redistribution to the cell membrane (Fig. 3A-D). Re-expression of ERα protein and the epithelial marker E-cadherin by KX-01 in MDA-MB-231 was further demonstrated by Western blot (Supplementary Fig. S8). Taken together, these data demonstrate that KX-01 induced epithelial markers and suppressed mesenchymal markers in two TNBC xenograft tumors.
Figure 3.
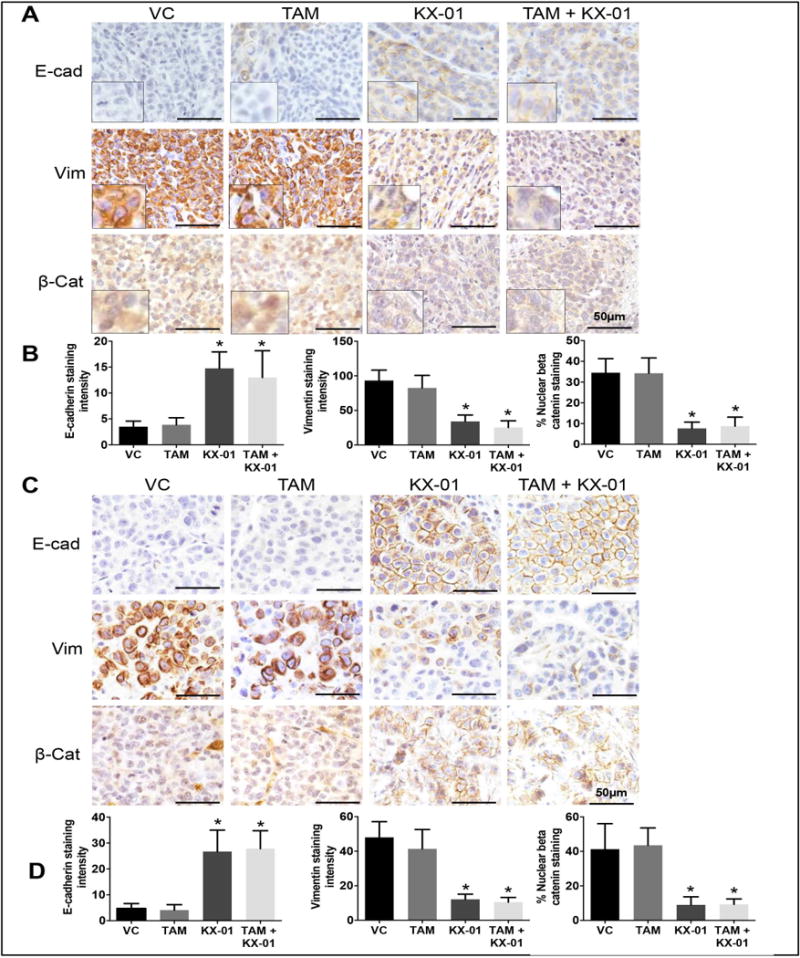
KX-01 increased epithelial markers and reduced mesenchymal markers in both MDA-MB-231 and MDA-MB-468 tumor xenografts. Paraffin tumor sections from MDA-MB231 (A), MDA-MB-468 (B) tumors were analyzed by IHC using antibodies against E-cadherin, vimentin and β-catenin. Bright field microscopic images for IHC staining with antibodies to E-cadherin, vimentin and β-catenin (original magnification, × 200) were photographed and representative photomicrographs are presented. The data are represented as the mean staining intensity for E-cadherin and vimentin, and % nuclear β-catenin staining with SD. *, P < 0.05 significantly different compared to VC by student’s t-test. VC, vehicle control, TAM, tamoxifen.
KX-01 induced histone modifications in the ERα promoter region of MDA-MB-231 tumors
We sought to investigate the mechanisms involved in ERα re-expression mediated by treatment with KX-01. KX-01 resulted in 3-4 fold increase in ERα mRNA in MDA-MB-231 tumors (Fig. 4A). Previous studies have reported that histone acetylation and methylation in the ERα promoter regulate expression in MDA-MB-231 cells in vitro (24,29–31). Histone modification patterns in MDA-MB-231 tumors were analyzed by Chromatin Immunoprecipitation (ChIP) assays using antibodies to both transcriptionally active (acetyl-H3, acetyl-H3Lys9) and inactive (trimethyl-H3Lys9) markers of chromatin (24). KX-01 and KX-01 + tamoxifen resulted in enrichment of the active histone acetylation chromatin markers, acetyl-H3 and acetyl-H3 Lys9 (Fig. 4B). The inactive, trimethyl-H3Lys9 mark was not changed in any of the treatment groups compared to vehicle (Fig. 4B). Remarkably, KX-01 treatment resulted in HDAC1 dissociation from the ERα promoter, and a concomitant recruitment of RNA polymerase II (Fig. 4B). KX-01 did not alter histone deacetylase 1 (HDAC1) levels or activity in tumors (Supplementary Fig. S9). Collectively, these data demonstrate that KX-01 induced alterations in histone acetylation that were consistent with a recruitment of RNA polymerase II to the ERα promoter and transcriptional increase in ERα mRNA (Fig.4A).
Figure 4.
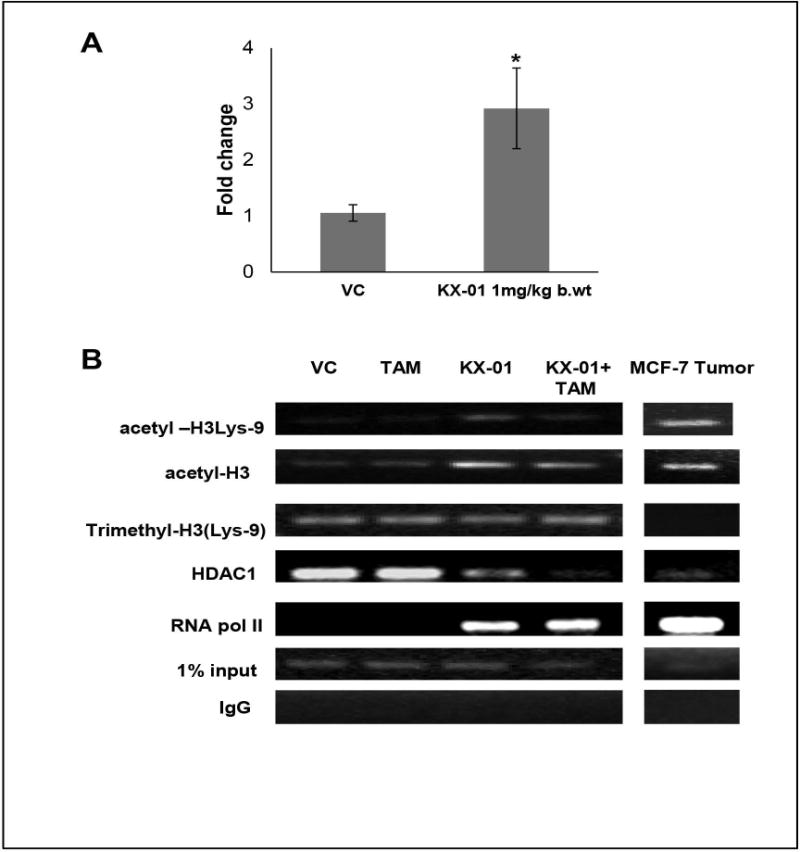
KX-01 induced ERα mRNA expression by alteration of histone acetylation marks in the ERα promoter in MDA-MB-231 tumors. A) Expression of ERα mRNA was measured by real-time PCR in MDA-MB-231 tumors treated with VC, KX-01 (1 mg/kg b.wt.) and data was expressed as fold change with SD. *, P < 0.05 significantly different compared to VC by student’s t-test. B) Histone modification patterns were analyzed by the chromatin immunoprecipitation (ChIP) assay. Cross-linked chromatin prepared from ERα negative MDA-MB-231 tumor xenografts and ERα positive MCF-7 were immunoprecipitated with antibodies to HDAC1, RNA Pol II, and antibodies to chromatin markers acetyl-H3Lys9, acetyl-H3, trimethyl-H3Lys9. Rabbit IgG was used as a negative control. The immunoprecipitates were subjected to PCR analysis using primer pairs directed against the ERα promoter CpG islands (see Materials and Methods). ERα-positive MCF-7 cells served as a control for histone acetylation marks present in an actively transcribed ERα promoter. 5 μl (1%) aliquots of chromatin taken before immunoprecipitation of total chromatin (500 μl) were used as input controls. Chromatin aliquots eluted from immunoprecipitations with non-specific IgG antibody were used as negative controls. Gel photographs presented are representative of experiments that were repeated three or more times. VC, vehicle control; TAM, tamoxifen.
KX-01 did not alter DNA methylation status of the ERα promoter in MDA-MB-231 tumors
The ERα promoter in human ERα negative breast cancer cell lines is highly methylated at CpG islands (29). More than 25% of ERα-negative breast cancer cells exhibit aberrant methylation in the ERα promoter suggesting that DNA methylation plays a critical role in regulating ERα expression (11,32). The methylation status of the ERα promoter region covering +375 to +495 CpG islands was examined in MDA-MB-231 tumors using Methylation Specific PCR (MSP) analysis. As a control, the ERα promoter region in ERα positive MCF-7 breast cancer cells was predominantly unmethylated (Fig. 5B, lanes 9-10). In contrast, the ERα promoter in vehicle treated MDA-MB-231 tumors was partially hypermethylated (Fig. 5B, lanes 1-2). Treatment with KX-01 or tamoxifen alone or in combination did not significantly alter the methylation status of the ERα promoter in MDA-MB-231 tumors (Fig. 5B, lanes 3-7). These data indicated that re-expression of ERα in MDA-MB-231 tumors by KX-01 was not the result of alteration in the methylation status of the ERα CpG islands. To elucidate the effects of methylation on the ERα promoter region, we examined the methylation status of the ERα promoter region from −66 to −356 covering most of the CpG dinucleotides. Bisulfite-sequencing was used to examine ERα methylation patterns MDA-MB-231 tumors. ERα-positive MCF-7 breast cancer cells served as control. The ERα promoter region of MCF-7 cells maintained an unmethylated status, whereas the ERα promoter of MDA-MB-231 tumors was hyper-methylated on CpG islands (~80%) (Fig. 5C). There was no significant change in the methylation status of the ERα promoter in MDA-MB-231 tumors from animals treated with vehicle (84.8 ± 14.7%), tamoxifen (78± 10%), KX-01 (75± 12%), and KX-01 + tamoxifen (81.4± 15.1%) (Fig. 5C), indicating that alterations in DNA methylation does not contribute to ERα re-expression by KX-01. These results indicated that KX-01-induced changes in histone modifications of the ERα promoter was of greater importance for ERα re-expression that were changes in DNA methylation in TNBC.
Figure 5.

Effect of KX-01 on ERα promoter methylation status in MDA-MB-231 tumors. A) A schematic overview of CpG island (denoted by red triangles) of the ERα promoter region is shown. ATG, start codon; TSS transcription start site. B) The methylation pattern of the ERα promoter was analyzed using a MSP (Methylation Specific PCR) with previously reported primer set ER5 (4) in all four treatment groups (VC, TAM, KX-01, TAM + KX-01). ERα positive MCF-7 tumors were used as a control for the unmethylated ERα promoter and H20 used as negative no template control. A representative gel photograph is presented and the experiment was repeated three times. C) The DNA methylation status of the ERα promoter in tumors from VC, TAM, KX-01, TAM + KX-01 treatment mice were detected by sodium bisulfite methylation sequencing. MCF-7 and no template were used as controls. U, Unmethylated; M, methylated; VC, vehicle control; TAM, tamoxifen.
DISCUSSION
Surgery, chemotherapy and radiation are mainstays for therapeutic management of TNBC. Targeted therapy is limited in TNBC due to the paucity of druggable targets such as ERα and HER2/neu. ERα is silenced in TNBC through epigenetic changes including DNA methylation and altered histone acetylation and hyper activation of kinases (24,29,33). In the present study, clinical Src/pretubulin inhibitor KX-01 resulted in a robust re-expression of ERα in TNBC tumor models that coincided with activating epigenetic marks in the ERα promoter. Tumors treated with low dose KX-01 became sensitized to the endocrine therapy agent tamoxifen and also exhibited a decrease in mesenchymal markers and an increase in epithelial markers. This study describes a novel application of a clinical peptidomimetic Src kinase inhibitor, KX-01, for TNBC resulting in ERα re-expression that occurs through epigenetic changes in the tumor. ERα re-expression and mesenchymal to epithelial reprogramming of TNBC tumors by KX-01, may sensitize tumors to tamoxifen or other endocrine therapy agents and limit metastatic spread of TNBC.
Restoration of ERα expression in TNBC patients is an appealing treatment strategy that could sensitize tumors to endocrine therapy and avoid or reduce the levels of cytotoxic chemotherapy needed for disease management. In contrast to irreversible genetic mutations, epigenetic changes such as occur in the ERα gene are potentially reversible (34) making these changes amenable to pharmacological interventions (35). Currently there are no agents that achieve re-expression of ERα in the clinic although certain HDAC inhibitors, demethylating agents, epigallocatechin-3-gallate (a major polyphenol in green tea) and arsenic trioxide have been shown to re-express ERα in experimental models (36,37). Src expression and activity is inversely correlated with ERα levels in human primary breast cancers (15,38,39) suggesting that Src kinase inhibition may be a strategy for re-expression of ERα and restoration of sensitivity to endocrine therapies.
The present study identified a previously unknown preclinical application of KX-01 at low doses that results in the re-expression of ERα in TNBC tumors. KX-01 is a novel peptidomimetic compound with two identified MOA’s (18,19); inhibition of Src kinase that was evident at both low dose (1 mg/kg BID; Supplementary Fig. S1) and high dose (5 mg/kg BID) KX-01 in MDA-MB-231 tumors (18), and; microtubule disruption evident only at higher doses (≥5 mg/kg BID) (18) [Supplementary Fig S10]. At these higher doses of ≥5 mg/kg BID, KX-01 did not result in re-expression of ERα in MDA-MB-231 tumors (data not shown). The re-expression of ERα only at the lower KX-01 dose suggests that the Src inhibition MOA of KX-01, but not microtubule disruption, was contributing to the ERα re-expression. These dose-dependent changes in the MOA and action of KX-01 are reminiscent of other drugs such as cyclophosphamide which exhibits an immunosuppressive and tumoricidal effect at high dose, but an immuo-stimulatory effect at low dose (40).
Histone acetylation/deacetylation is the most prominent posttranslational modification of histones and a crucial determinant of gene expression (41). Histone deacetylation of core histones generates an overall positive charge on lysine residues that when reacted with negatively charged DNA, results in a more compact nucleosome that limits transcription due to the physical inability of RNA polymerase to access the DNA (42). Histone acetylation results in an open chromatin structure leading to active gene transcription. Previous studies showed that deacetylated histones were associated with the inactive ERα promoter in MDA-MB-231 cells, whereas acetylated histones were associated with the active ERα promoter in MCF-7 breast cancer cells (30). The present study demonstrated that KX-01 significantly altered histone acetylation of the ERα promoter of MDA-MB-231 tumors without altering DNA methylation. Continuous treatment with KX-01 was necessary to maintain ERα expression in the tumor as removal of the drug resulted in tumors that no longer expressed ERα. This reversible ERα re-expression by KX-01 that occurred concomitant with histone acetylation changes is consistent with the findings that dynamic and reversible chromatin modifications regulate gene expression. Previous studies demonstrated that silenced ERα could be re-expressed solely by changes in histone acetylation without alteration of the DNA methylation pattern. Treatment with the HDAC inhibitor LBH589 or epigallocatechin-3-gallate restored ERα mRNA and protein expression in MDA-MB-231 cells without demethylation of the CpG islands within the ERα promoter (8,43). Taken together, these studies support the hypothesis that changes in histone acetylation alone can restore the expression of the silenced ERα gene without altering the DNA methylation state at the ERα promoter, and provide a mechanistic explanation for the transcriptional activation of the silenced ER gene by KX-01.
There are several potential mechanisms by which inhibition of Src by KX-01 may alter histone modifications and impact chromatin structure in the ERα promoter. Yu et. al. reported that Src phosphorylated the HDAC Inhibitor of Growth 1 (ING1), resulting in nuclear to cytoplasmic localization and decrease in protein stability (44) . C-terminal Src kinase (Csk)-binding protein (Cbp)/PAG1 expression was repressed via Src-mediated alterations in histone H4 acetylation and trimethylation of histone H3/lysine 27 in the Cbp promoter and associated changes in HDAC activity (45). v-Src transformed NIH-3T3 cells exhibited elevated HDAC1 leading to repression of the Src-suppressed C kinase substrate (SSeCKS) and altered histone marks in the promoter (46) . Src was also shown to phosphorylate and increase the activity of HDAC3 (47). Src may activate a transcriptional repressor to associate with chromatin and/or alter its subcellular localization. Src phosphorylated Transcription Factor II-I (TFII-I) and enhanced its transcriptional repressor function that was associated with recruitment of HDAC1 and was sufficient to suppress transcription of SSeCKS/Gravin/Akap12 (48). Inhibition of Src prevented gene silencing mediated by Krüppel-like factor 16 (KLF16), a transcription factor with domains that regulate acetylases and HDAC’s (49). Our data indicated that HDAC1 activity is not impacted by KX-01 but rather, HDAC association with the ERα promoter was lost and presumably a co-repressor complex was dissociated. Loss of HDAC1 from the ERα promoter would alter histone acetylation in the ERα promoter. Further experimentation will define the precise molecular mechanisms for KX-01 de-repression of the ERα promoter in TNBC.
Inhibition of Src kinase by KX-01, separate from effects on ERα re-expression, contributed to anti-tumor drug efficacy in MDA-MB-231, MDA-MB-157, but not in MDA-MB-468 tumors (Fig. 1; Supplementary Fig. S5). Notably, KX-01 did not induce ERα re-expression in MDA-MB-157 tumors although anti-tumor efficacy was still evident (Supplementary Fig. S5). The MDA-MB-157 tumors were resistant to tamoxifen treatment as also occurred in MDA-MB-231 tumors when KX-01 was withdrawn and there was no ERα re-expression (Supplementary Fig. S7). It was reported that MDA-MB-468 cells were resistant to the Src kinase inhibitor dasatinib in vitro (13) and we found that MDA-MB-468 tumors were also resistant to low dose KX-01 (Fig. 1C). Taken together, these data indicate that KX-01 efficacy at low dose is tumor specific and is mediated by two mechanisms; Src kinase inhibition and/or re-expression of ERα that sensitizes tumors to tamoxifen. It is likely that the re-expression of ERa by KX-01 is linked to the Src kinase inhibition by the agent.
Tamoxifen sensitivity in the MDA-MB-231 and MDA-MB-468 tumors occurred only after KX-01 treatment suggesting that ERα re-expression is needed to restore tamoxifen sensitivity to TNBC tumors. As PR is a marker for a functional ERα signaling pathway, the re-expression of PR is also a candidate biomarker for a tumor that would be responsive to endocrine therapy. MDA-MB-231 and MDA-MB-468 tumors are histologically different. MDA-MB-231 cells were derived from an adenocarcinoma, the cells are highly invasive expressing mesenchymal markers and are representative of late-stage breast cancer (50). MDA-MB-468 cells were derived from a patient with a histologically different tumor (ductal carcinoma) and exhibit less mesenchymal features than MDA-MB-231 cells (51). The sensitization to endocrine therapy by KX-01 of these two TNBC models that have different features may serve as a paradigm for future studies of tumor response to KX-01 for endocrine therapy sensitivity.
Inhibition of Src kinase has been shown to prevent ERα protein degradation in breast cancer (15). In addition to increasing ERα mRNA in triple negative tumors, it is possible that KX01 may also inhibit ongoing degradation of ERα protein. We measured ERα mRNA in three TNBC cell lines with and without KX01 treatment. Compared to MCF-7 cells that express robust levels of ERα mRNA, all TNBC cell lines had very low (MDA-MB-468, MDA-MB-157) to barely detectable (MDA-MB-231) ERα mRNA that was markedly increased by KX01 treatment (Supplementary Fig. S11). In MDA-MB-157 tumors, KX01 markedly increased ERα mRNA without a significant increase in ERα protein (Supplementary Fig. S5A) suggesting that KX-01 treatment has a greater effect on inducing ERα mRNA than in inhibiting ERα protein turnover in these tumors. Although the major contribution of KX01 to ERα re-expression in MDA-MB-231 and MDA-MB-468 tumors is likely increased ERα mRNA, we cannot exclude that some of the ERα protein re-expression observed may be due to KX-01 inhibiting ERα protein turnover.
In addition to re-expression of ERα/PR by KX-01, MDA-MB-231 and MDA-MB-468 tumors also exhibited an increase in the epithelial marker E-cadherin expression, and a concomitant decrease in mesenchymal markers nuclear β-catenin and vimentin. Epithelial to mesenchymal transition (EMT) has been recognized as a critical feature of embryogenesis, organogenesis and has been shown to play a critical role in cancer progression and metastasis (52). Human breast cancers exhibit a strong direct correlation between ERα and E-cadherin expression and studies have shown that ERα signaling can regulate E-cadherin expression and EMT (53,54). Additionally, Src inhibition has been shown to inhibit EMT and reduce metastasis in many cancers including breast cancer (26,55).
Although the effect on primary TNBC growth inhibition by KX-01 + tamoxifen was modest, the paradigm of ERα re-expression by a clinical agent, KX-01, provides opportunity to test additional endocrine therapy agents and/or combination with other non-endocrine therapies that are effective in ER positive breast cancer. Since TNBC is frequently metastatic, another potential clinical benefit of KX-01 is the ability to induce MET that could limit metastatic spread. In this regard, a number of differentiation therapies induce MET and limit breast cancer metastasis (56–60). It is possible that patient tumors exhibiting silenced ERα that is due predominantly to chromatin remodeling (deacetylated histones) and that also exhibit elevated Src kinase may be candidates for KX-01 therapy to re-express ERα and induce MET in the tumors.
Supplementary Material
Acknowledgments
This study was supported, in part by DK068432 to BGR, LA CaTS Center grant 1 U54 GM104940 to BCB, and CA125806 to MEB. This work has been supported in part by the Bio-specimen Core Laboratory of the Louisiana Cancer Research Consortium. We thank Mr. Tripp Frasch for assistance with preparing figures.
References
- 1.Byar DP, Sears ME, McGuire WL. Relationship between estrogen receptor values and clinical data in predicting the response to endocrine therapy for patients with advanced breast cancer. European journal of cancer. 1979;15(3):299–310. doi: 10.1016/0014-2964(79)90041-0. [DOI] [PubMed] [Google Scholar]
- 2.Giacinti L, Claudio PP, Lopez M, Giordano A. Epigenetic information and estrogen receptor alpha expression in breast cancer. The oncologist. 2006;11(1):1–8. doi: 10.1634/theoncologist.11-1-1. [DOI] [PubMed] [Google Scholar]
- 3.McGuire WL. Hormone receptors: their role in predicting prognosis and response to endocrine therapy. Seminars in oncology. 1978;5(4):428–33. [PubMed] [Google Scholar]
- 4.Lapidus RG, Nass SJ, Davidson NE. The loss of estrogen and progesterone receptor gene expression in human breast cancer. J Mammary Gland Biol Neoplasia. 1998;3(1):85–94. doi: 10.1023/a:1018778403001. [DOI] [PubMed] [Google Scholar]
- 5.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(15 Pt 1):4429–34. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 6.Ovcaricek T, Frkovic SG, Matos E, Mozina B, Borstnar S. Triple negative breast cancer - prognostic factors and survival. Radiology and oncology. 2011;45(1):46–52. doi: 10.2478/v10019-010-0054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast cancer research and treatment. 2003;81(3):177–86. doi: 10.1023/A:1026146524737. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Q, Atadja P, Davidson NE. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer biology & therapy. 2007;6(1):64–9. doi: 10.4161/cbt.6.1.3549. [DOI] [PubMed] [Google Scholar]
- 9.Fan J, Yin WJ, Lu JS, Wang L, Wu J, Wu FY, et al. ER alpha negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. Journal of cancer research and clinical oncology. 2008;134(8):883–90. doi: 10.1007/s00432-008-0354-x. [DOI] [PubMed] [Google Scholar]
- 10.Raha P, Thomas S, Munster PN. Epigenetic modulation: a novel therapeutic target for overcoming hormonal therapy resistance. Epigenomics. 2011;3(4):451–70. doi: 10.2217/epi.11.72. [DOI] [PubMed] [Google Scholar]
- 11.Lapidus RG, Nass SJ, Butash KA, Parl FF, Weitzman SA, Graff JG, et al. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer research. 1998;58(12):2515–9. [PubMed] [Google Scholar]
- 12.Yang X, Ferguson AT, Nass SJ, Phillips DL, Butash KA, Wang SM, et al. Transcriptional activation of estrogen receptor alpha in human breast cancer cells by histone deacetylase inhibition. Cancer research. 2000;60(24):6890–4. [PubMed] [Google Scholar]
- 13.Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/"triple-negative" breast cancer cell lines growing in vitro. Breast cancer research and treatment. 2007;105(3):319–26. doi: 10.1007/s10549-006-9463-x. [DOI] [PubMed] [Google Scholar]
- 14.Tryfonopoulos D, Walsh S, Collins DM, Flanagan L, Quinn C, Corkery B, et al. Src: a potential target for the treatment of triple-negative breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology/ESMO. 2011;22(10):2234–40. doi: 10.1093/annonc/mdq757. [DOI] [PubMed] [Google Scholar]
- 15.Chu I, Arnaout A, Loiseau S, Sun J, Seth A, McMahon C, et al. Src promotes estrogen-dependent estrogen receptor alpha proteolysis in human breast cancer. J Clin Invest. 2007;117(8):2205–15. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fallah-Tafti A, Foroumadi A, Tiwari R, Shirazi AN, Hangauer DG, Bu Y, et al. Thiazolyl N-benzyl-substituted acetamide derivatives: synthesis, Src kinase inhibitory and anticancer activities. European journal of medicinal chemistry. 2011;46(10):4853–8. doi: 10.1016/j.ejmech.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 17.Anbalagan M, Carrier L, Glodowski S, Hangauer D, Shan B, Rowan BG. KX-01, a novel Src kinase inhibitor directed toward the peptide substrate site, synergizes with tamoxifen in estrogen receptor alpha positive breast cancer. Breast Cancer Res Treat. 2012;132(2):391–409. doi: 10.1007/s10549-011-1513-3. [DOI] [PubMed] [Google Scholar]
- 18.Anbalagan M, Ali A, Jones RK, Marsden CG, Sheng M, Carrier L, et al. Peptidomimetic Src/pretubulin inhibitor KX-01 alone and in combination with paclitaxel suppresses growth, metastasis in human ER/PR/HER2-negative tumor xenografts. Molecular cancer therapeutics. 2012;11(9):1936–47. doi: 10.1158/1535-7163.MCT-12-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hangauer D, Smolinski M, Bu Y, Teegarden P, Qu J, Kazim L, Hegab T, Quinn J, Gao L, Gelman I. Discovery of KX2-391: A Phase II Src Signaling Inhibitor with a Second MOA. Whistler, British Columbia: 2010. Apr 22, [Google Scholar]
- 20.Antonarakis ES, Heath EI, Posadas EM, Yu EY, Harrison MR, Bruce JY, et al. A phase 2 study of KX2-391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer chemotherapy and pharmacology. 2013;71(4):883–92. doi: 10.1007/s00280-013-2079-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowan BG, Narayanan R, Weigel NL. Analysis of receptor phosphorylation. Methods Enzymol. 2003;364:173–202. doi: 10.1016/s0076-6879(03)64011-5. [DOI] [PubMed] [Google Scholar]
- 22.ClinicalTrial.gov [homepage on the Internet]. Identifier: NCT01764087. January 2013.
- 23.Duplessis TT, Williams CC, Hill SM, Rowan BG. Phosphorylation of Estrogen Receptor alpha at serine 118 directs recruitment of promoter complexes and gene-specific transcription. Endocrinology. 2011;152(6):2517–26. doi: 10.1210/en.2010-1281-en.2010-1281[pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma D, Blum J, Yang X, Beaulieu N, Macleod AR, Davidson NE. Release of methyl CpG binding proteins and histone deacetylase 1 from the Estrogen receptor alpha (ER) promoter upon reactivation in ER-negative human breast cancer cells. Molecular endocrinology. 2005;19(7):1740–51. doi: 10.1210/me.2004-0011. [DOI] [PubMed] [Google Scholar]
- 25.Liu CY, Hung MH, Wang DS, Chu PY, Su JC, Teng TH, et al. Tamoxifen induces apoptosis through cancerous inhibitor of protein phosphatase 2A-dependent phospho-Akt inactivation in estrogen receptor-negative human breast cancer cells. Breast Cancer Res. 2014;16(5):431. doi: 10.1186/s13058-014-0431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nam JS, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8(7):2430–6. [PubMed] [Google Scholar]
- 27.Hatsell S, Rowlands T, Hiremath M, Cowin P. Beta-catenin and Tcfs in mammary development and cancer. Journal of mammary gland biology and neoplasia. 2003;8(2):145–58. doi: 10.1023/a:1025944723047. [DOI] [PubMed] [Google Scholar]
- 28.Wong AS, Gumbiner BM. Adhesion-independent mechanism for suppression of tumor cell invasion by E-cadherin. The Journal of cell biology. 2003;161(6):1191–203. doi: 10.1083/jcb.200212033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer research. 1994;54(10):2552–5. [PubMed] [Google Scholar]
- 30.Sabnis GJ, Goloubeva O, Chumsri S, Nguyen N, Sukumar S, Brodie AM. Functional activation of the estrogen receptor-alpha and aromatase by the HDAC inhibitor entinostat sensitizes ER-negative tumors to letrozole. Cancer research. 2011;71(5):1893–903. doi: 10.1158/0008-5472.CAN-10-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Meeran SM, Patel SN, Chen H, Hardy TM, Tollefsbol TO. Epigenetic reactivation of estrogen receptor-alpha (ERalpha) by genistein enhances hormonal therapy sensitivity in ERalpha-negative breast cancer. Molecular cancer. 2013;12:9. doi: 10.1186/1476-4598-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer research. 2001;61(19):7025–9. [PubMed] [Google Scholar]
- 33.Bayliss J, Hilger A, Vishnu P, Diehl K, El-Ashry D. Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(23):7029–36. doi: 10.1158/1078-0432.CCR-07-0587. [DOI] [PubMed] [Google Scholar]
- 34.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nature reviews Drug discovery. 2006;5(1):37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 35.Santos-Rosa H, Caldas C. Chromatin modifier enzymes, the histone code and cancer. European journal of cancer. 2005;41(16):2381–402. doi: 10.1016/j.ejca.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 36.Belguise K, Guo S, Sonenshein GE. Activation of FOXO3a by the green tea polyphenol epigallocatechin-3-gallate induces estrogen receptor alpha expression reversing invasive phenotype of breast cancer cells. Cancer research. 2007;67(12):5763–70. doi: 10.1158/0008-5472.CAN-06-4327. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W, Wang L, Fan Q, Wu X, Wang F, Wang R, et al. Arsenic trioxide re-sensitizes ERalpha-negative breast cancer cells to endocrine therapy by restoring ERalpha expression in vitro and in vivo. Oncology reports. 2011;26(3):621–8. doi: 10.3892/or.2011.1352. [DOI] [PubMed] [Google Scholar]
- 38.Elsberger B, Tan BA, Mitchell TJ, Brown SB, Mallon EA, Tovey SM, et al. Is expression or activation of Src kinase associated with cancer-specific survival in ER-, PR- and HER2-negative breast cancer patients? The American journal of pathology. 2009;175(4):1389–97. doi: 10.2353/ajpath.2009.090273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anbalagan M, Moroz K, Ali A, Carrier L, Glodowski S, Rowan BG. Subcellular Localization of Total and Activated Src Kinase in African American and Caucasian Breast Cancer. Plos One. 2012;7(3) doi: 10.1371/journal.pone.0033017. doi ARTN e33017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nature reviews Immunology. 2008;8(1):59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 41.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Brinkman JA, El-Ashry D. ER re-expression and re-sensitization to endocrine therapies in ER-negative breast cancers. Journal of mammary gland biology and neoplasia. 2009;14(1):67–78. doi: 10.1007/s10911-009-9113-0. [DOI] [PubMed] [Google Scholar]
- 43.Skliris GP, Nugent ZJ, Rowan BG, Penner CR, Watson PH, Murphy LC. A phosphorylation code for oestrogen receptor-alpha predicts clinical outcome to endocrine therapy in breast cancer. Endocr Relat Cancer. 2010;17(3):589–97. doi: 10.1677/ERC-10-0030ERC-10-0030[pii]. [DOI] [PubMed] [Google Scholar]
- 44.Yu L, Thakur S, Leong-Quong RY, Suzuki K, Pang A, Bjorge JD, et al. Src regulates the activity of the ING1 tumor suppressor. PLoS One. 2013;8(4):e60943. doi: 10.1371/journal.pone.0060943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki K, Oneyama C, Kimura H, Tajima S, Okada M. Down-regulation of the tumor suppressor C-terminal Src kinase (Csk)-binding protein (Cbp)/PAG1 is mediated by epigenetic histone modifications via the mitogen-activated protein kinase (MAPK)/phosphatidylinositol 3-kinase (PI3K) pathway. J Biol Chem. 2011;286(18):15698–706. doi: 10.1074/jbc.M110.195362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bu Y, Gelman IH. v-Src-mediated down-regulation of SSeCKS metastasis suppressor gene promoter by the recruitment of HDAC1 into a USF1-Sp1-Sp3 complex. J Biol Chem. 2007;282(37):26725–39. doi: 10.1074/jbc.M702885200. [DOI] [PubMed] [Google Scholar]
- 47.Longworth MS, Laimins LA. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene. 2006;25(32):4495–500. doi: 10.1038/sj.onc.1209473. [DOI] [PubMed] [Google Scholar]
- 48.Bu Y, Gao L, Gelman IH. Role for transcription factor TFII-I in the suppression of SSeCKS/Gravin/Akap12 transcription by Src. Int J Cancer. 2011;128(8):1836–42. doi: 10.1002/ijc.25524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daftary GS, Lomberk GA, Buttar NS, Allen TW, Grzenda A, Zhang J, et al. Detailed structural-functional analysis of the Kruppel-like factor 16 (KLF16) transcription factor reveals novel mechanisms for silencing Sp/KLF sites involved in metabolism and endocrinology. J Biol Chem. 2012;287(10):7010–25. doi: 10.1074/jbc.M111.266007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cailleau R, Olive M, Cruciger QV. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In vitro. 1978;14(11):911–5. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 51.Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. Journal of the National Cancer Institute. 1974;53(3):661–74. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009;119(6):1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.da Silva BB, dos Santos AR, Pires CG, Correa-Lima MA, Pereira-Filho JD, dos Santos LG, et al. E-cadherin expression in estrogen receptor-positive and negative breast carcinomas of postmenopausal women. European journal of gynaecological oncology. 2010;31(1):90–3. [PubMed] [Google Scholar]
- 54.Ye Y, Xiao Y, Wang W, Yearsley K, Gao JX, Shetuni B, et al. ERalpha signaling through slug regulates E-cadherin and EMT. Oncogene. 2010;29(10):1451–62. doi: 10.1038/onc.2009.433. [DOI] [PubMed] [Google Scholar]
- 55.Liu X, Feng R. Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta biochimica et biophysica Sinica. 2010;42(7):496–501. doi: 10.1093/abbs/gmq043. [DOI] [PubMed] [Google Scholar]
- 56.Li X, Li P, Liu C, Ren Y, Tang X, Wang K, et al. Sinomenine hydrochloride inhibits breast cancer metastasis by attenuating inflammation-related epithelial-mesenchymal transition and cancer stemness. Oncotarget. 2017 doi: 10.18632/oncotarget.14593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dasgupta A, Sawant MA, Kavishwar G, Lavhale M, Sitasawad S. AECHL-1 targets breast cancer progression via inhibition of metastasis, prevention of EMT and suppression of Cancer Stem Cell characteristics. Sci Rep. 2016;6:38045. doi: 10.1038/srep38045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pattabiraman DR, Bierie B, Kober KI, Thiru P, Krall JA, Zill C, et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science. 2016;351(6277):aad3680. doi: 10.1126/science.aad3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alam F, Al-Hilal TA, Park J, Choi JU, Mahmud F, Jeong JH, et al. Multi-stage inhibition in breast cancer metastasis by orally active triple conjugate, LHTD4 (low molecular weight heparin-taurocholate-tetrameric deoxycholate) Biomaterials. 2016;86:56–67. doi: 10.1016/j.biomaterials.2016.01.058. [DOI] [PubMed] [Google Scholar]
- 60.Qin G, Xu F, Qin T, Zheng Q, Shi D, Xia W, et al. Palbociclib inhibits epithelial-mesenchymal transition and metastasis in breast cancer via c-Jun/COX-2 signaling pathway. Oncotarget. 2015;6(39):41794–808. doi: 10.18632/oncotarget.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.