

Abstract
JC virus (JCV) is a human polyomavirus and the etiologic agent of the demyelinating disease progressive multifocal leukoencephalopathy (PML). PML is observed in patients with underlying immunocompromising conditions, suggesting that neuro-immune interactions between peripheral immune cells and neuro-glia play an important role in controlling viral reactivation in the brain. There is little known about the immunobiology of JCV reactivation in glial cells and the role of immune, glial, and viral players in this regulation. We have previously showed that agnoprotein, a small JCV regulatory protein, is released from infected cells and internalized by neighboring bystander cells. Here we have investigated the possible role of extracellular and intracellular agnoprotein in the neuroimmune response to JC virus. Our findings suggest that glial cells exposed to agnoprotein secrete significantly less GM-CSF, which is mediated by agnoprotein induced suppression of GM-CSF transcription. Likewise, monocytes treated with agnoprotein showed altered differentiation and maturation. In addition, monocytes and microglial cells exposed to agnoprotein showed a significant reduction in their phagocytic activities. Moreover, when an in vitro blood-brain barrier model was used, agnoprotein treatment resulted in decreased monocyte migration through the endothelial cell layer in response to activated astrocytes. All together, these results have revealed a novel immunomodulatory function of agnoprotein during JCV infection within the CNS and open a new avenue of research to better understand the mechanisms associated with JCV reactivation in patients who are at risk of developing PML.
Keywords: JC virus, PML, agnoprotein, GM-CSF, neuroinflammation, immunomodulation, migration, differentiation, monocytes, blood-brain barrier
INTRODUCTION
JC virus is a human polyomavirus which has been found to infect between 70–90 percent of the human population (Holman et al, 1991; San-Andreas et al, 2003). Generally, following initial infection, JCV remains latent within the body at various proposed sites; such as the brain, kidneys, tonsillar stromal cells, and hematopoietic progenitor cells (Ferenczy et al, 2012; Monaco et al, 1996; Monaco et al, 1998). In rare cases, JCV can reactivate following the onset of immunosuppression, which results in progressive multifocal leukoencephalopathy (PML) development. PML is characterized by lytic infection of oligodendrocytes, resulting in multifocal lesions throughout the brain and the secondary destruction of neurons (Houff et al, 1988). Primarily, PML is mainly defined in HIV/AIDS patients, however, it is also found rarely in patients on therapeutic immunomodulatory therapies, such as monoclonal antibody therapies to treat autoimmune diseases, such as rituximab for rheumatoid arthritis or natalizumab for multiple sclerosis (Major, 2010). The general consensus among all risk factors of PML development is either a reduced T-cell population, suppression of T-cell function or an increase in B-cell proliferation, however, the exact mechanism of JCV reactivation remains to be elucidated. While the majority of the human population has been infected with JC virus and produce circulating antibodies, these antibodies do not prevent PML. While evidence has suggested that cell-mediated immunity (i.e., T cell mediated CTL response) is the main mechanism controlling JC virus infection (Du Pasquier and Koralnik., 2003; Koralnik, 2002; Jelcic et al., 2016), some drugs that are associated with the development of PML deplete or block B cells, suggesting that B cells may also play a role in controlling JC virus infection. During PML development, there is a lack of widespread inflammation within the brain, which appears to contradict what would be expected following viral reactivation in glial cells (Richardson, 1961; von Einsiedel et al., 1993; Koralnik, 2002; Cinque et al, 2009; Ferenczy et al, 2012; SantaCruz et al., 2016). There is little known about the immunobiology of JCV reactivation in glial cells and the role of immune, glial, and viral players in this regulation. Given the fact that the natural occurrence of PML is strongly associated with immunosuppression and limited inflammation in the brain, the functional and molecular interaction between JCV regulatory proteins and neuroimmune signaling is likely to play a major role in reactivation of JCV and the progression of the lytic viral life cycle leading to the development of PML.
The JCV genome is a double-stranded circular DNA genome which contains a bidirectional non-coding control region (NCCR), which separates the early and late coding regions (Frisque et al, 1984; Sariyer et al, 2010). Prior to DNA replication, the early coding region of JCV is transcribed, resulting in expression of the JCV regulatory T-antigen proteins, including large T-antigen, small t-antigen, and the T-prime splice variants, which are expressed following the alternative splicing of the early viral transcript (Trowbridge et al, 1995). After early coding region transcription, both DNA replication of the genome as well as transcription of the late coding region of JCV occur simultaneously. The late coding region of JCV encodes the viral capsid proteins, VP1, VP2, and VP3, as wells as the small regulatory agnoprotein. The function of JCV T-antigens primarily involves promoting viral gene replication through various functions within the cell (Bollag et al, 2000; Tavis et al, 1994; White et al, 2006). From the late region, the viral capsid proteins are essential for functional infection, and they function in viral entry into permissive host cells through interactions with both sialic acid-containing receptors and serotonin receptor on the cell membrane (Gee et al, 2004; Elphick et al., 2004).
The late region of JCV also encodes a small regulatory protein denoted agnoprotein. During JCV infection of permissive cells, agnoprotein is found primarily in the cytoplasm, with perinuclear localization (Okada et al, 2001, Del Valle et al, 2002). These results are confirmed within the PML brain, with immunohistochemical staining showing similar cytoplasmic and perinuclear localization of agnoprotein within lesion sites (Okada et al, 2002). In terms of function, much remains to be elucidated for agnoprotein. Currently, there are limited studies assessing agnoprotein function. One study suggests that agnoprotein functions as a viroporin, a viral protein which serves to increase plasma membrane permeability, since agnoprotein deletion mutants are defective in virus release and viral propagation (Suzuki et al, 2010). This hypothesis is bolstered by studies showing that JCV agnoprotein mutants release viral particles that are defective in viral DNA (Sariyer et al, 2011). Likewise, the mature virions of JCV agnoprotein mutants are irregularly shaped and have decreased VP1 capsid formation due to dysregulated interactions between agnoprotein and VP1 (Suzuki et al, 2012). Aside from its role in virion formation, during JCV infection, agnoprotein interacts with JCV T-antigen to enhance T-antigen’s DNA binding activity to the viral origin of replication, resulting in significantly increased viral replication (Saribas et al, 2012). Furthermore, we have showed that agnoprotein is released by infected glial cells during viral propagation, and present in the extracellular matrix (Otlu et al, 2014). The release and detection of agnoprotein in the extracellular matrix have suggested a novel role of agnoprotein in the molecular pathogenesis of JCV infection in the brain.
One possible role for the secreted agnoprotein could be an extracellular immunomodulatory function to facilitate the invasion of the virus within the affected tissue. Here, we investigated the role of agnoprotein in modulating the host-immune response to JC virus infection in the CNS. Our results suggest that agnoprotein is released by cells and internalized by non-infected glial cells. Following agnoprotein internalization, GM-CSF release from glial cells was reduced due to agnoprotein mediated suppression of GM-CSF transcription. In addition, our data have also suggested that extracellular agnoprotein impairs monocyte/macrophage differentiation, inhibits their phagocytic activity, and impairs their migration through an in vitro blood-brain barrier composed of human primary endothelial and astrocyte cultures. These results have revealed a novel immunomodulatory function of agnoprotein and may suggest a possible mechanism of limited inflammation associated with PML lesions.
RESULTS
Agnoprotein is released and taken up by glial cells
During PML progression, oligodendrocytes become productively infected by JCV, culminating with the lysis of infected cells and widespread tissue damage within the CNS. However, as limited inflammation is seen during PML progression, regardless of the mechanism of immunosuppression, we have hypothesized that agnoprotein may serve to play an immunomodulatory role since agnoprotein is released by infected cells. To assess the role of agnoprotein, an oligodendrocyte cell line, TC620, was transiently transfected with an expression vector encoding agnoprotein, pcgT7-agnoprotein. At 48-hours post-transfection, whole cell lysates and growth media were collected to assess agnoprotein expression and release. Growth media was processed for immunoprecipitation to determine the presence of agnoprotein released by the cells. After immunoprecipitation, growth media samples and whole cell lysates were analyzed by Western blotting for detection of agnoprotein. As shown in Figure 1A, agnoprotein was present within the cell media following cellular expression of the protein. To ensure that extracellular agnoprotein was not the result of cell lysis, we assessed cellular viability following transfection using an MTT assay and found no significant changes in cellular viability following transfection (data not shown). To quantify released agnoprotein, an ELISA was completed using collected growth media from transfected cells, which showed that released agnoprotein concentration ranged between 45–55 ng/mL over multiple replicates (data not shown). To assess if extracellular agnoprotein is internalized by glial cells, primary human fetal astrocytes (PHFA) and primary human fetal microglia (PHFM) were treated with agnoprotein containing growth media (CM-agno) obtained from agnoprotein transfected cells. Primary cells were treated for 48 hours after which whole cell lysates were collected and analyzed for agnoprotein uptake via Western blotting. Both astrocytes and microglia showed agnoprotein uptake following conditioned growth media treatment (Fig.1B). To further assess extracellular agnoprotein uptake, primary human fetal astrocytes and microglia were treated with conditioned growth media from pcgT7-alone (CM-control) or pcgT7-agnoprotein (CM-agno) transfected TC620 cells. At 48 hours, cells were fixed and immunocytochemistry was completed to assess agnoprotein uptake. Both astrocytes and microglia showed robust agnoprotein uptake with perinuclear and cytoplasmic agnoprotein localization (Fig.1C).
Figure 1.
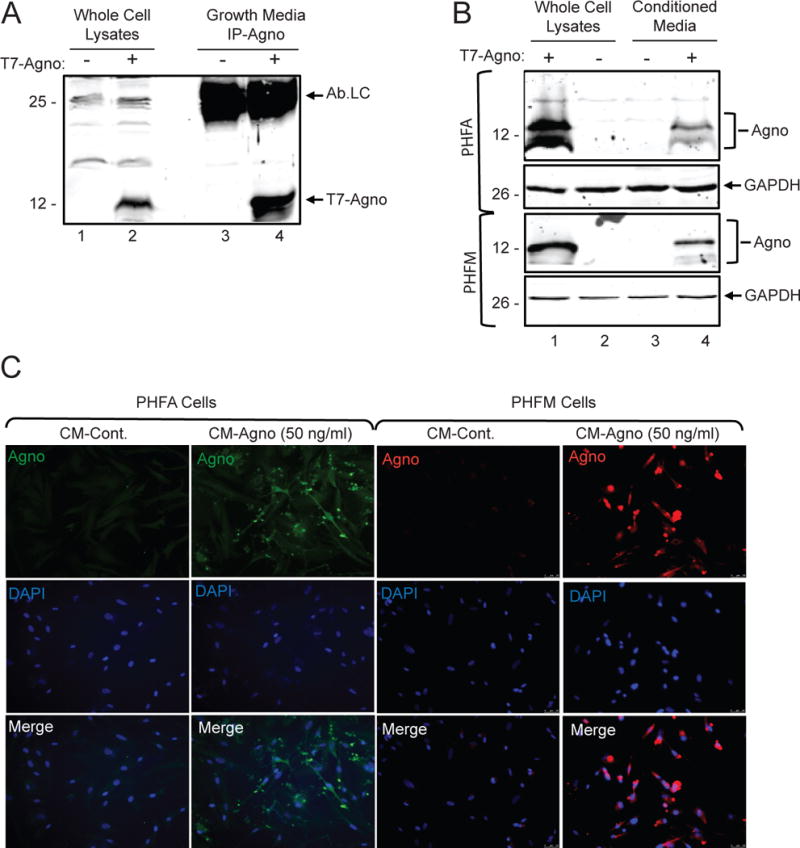
Agnoprotein is released and taken up by glial cells. A. TC620 cells were transfected with pcgT7-agno and whole cell extracts and growth media was collected at 48 hours. Growth media was subjected to immunoprecipitation for the detection of agnoprotein. In lane 1 and 2, whole cell protein extracts from untransfected and transfected cells were loaded as negative and positive controls of agnoprotein expression, respectively. Ab-Lc points the light chain of agnoprotein antibody. B. Whole cell extracts of primary glial cells show uptake of extracellular agnoprotein following conditioned media treatment. Primary human fetal astrocytes (PHFA) or primary human fetal microglia (PHFM) were either transfected with pcgT7-agno or treated with CM-control or CM-agno obtained from Tc620 cells as shown in panel A. At 48 hours post-transfection and treatments, cell lysates were prepared and analyzed via Western blotting for agnoprotein and GAPDH detection. C. Immunocytochemistry analysis of primary glial cells shows uptake of extracellular agnoprotein. PHFA and PHFM cells were treated with CM-control or CM-agno. At 48 hours, cells were fixed and processed for immunocytochemistry for the visualization of agnoprotein.
GM-CSF release is suppressed in astrocytes exposed to extracellular agnoprotein
As agnoprotein is released and internalized, we hypothesized that it may serve to modulate the immune response to the virus, potentially through dysregulating the cytokine profile of glial cells. To assess if agnoprotein altered the cytokine profile, primary human fetal astrocytes were treated with conditioned media from vector-transfected (CM-control) or agnoprotein-transfected (CM-agno) TC620 cells. At 24-hours, media was aspirated and fresh media was added. At 48-hours, media was collected for cytokine/chemokine profile analysis using a RayBioTech Human Cytokine C3 Array to assess the media concentration of 42 various cytokines. When comparing the released cytokine profiles of the cells, the release of GM-CSF was found to be significantly reduced following CM-agno treatment in contrast to CM-control treatment (Figure 2A). The intensities from the cytokine array membrane were quantified to determine relative growth media concentration of GM-CSF, and CM-agno treatment resulted in a forty percent decrease of released GM-CSF, suggesting that this phenomenon was specific to GM-CSF and not a widespread decrease in cytokine release by treated cells (Figure 2B). To ensure that changes in cytokine release were not due to the toxicity associated with treatments, primary human fetal astrocytes were either untreated or treated with CM-control or CM-agno, and after 48-hours were processed for an MTT assay and showed no significant alteration in cellular viabilities (Figure 2C).
Figure 2.
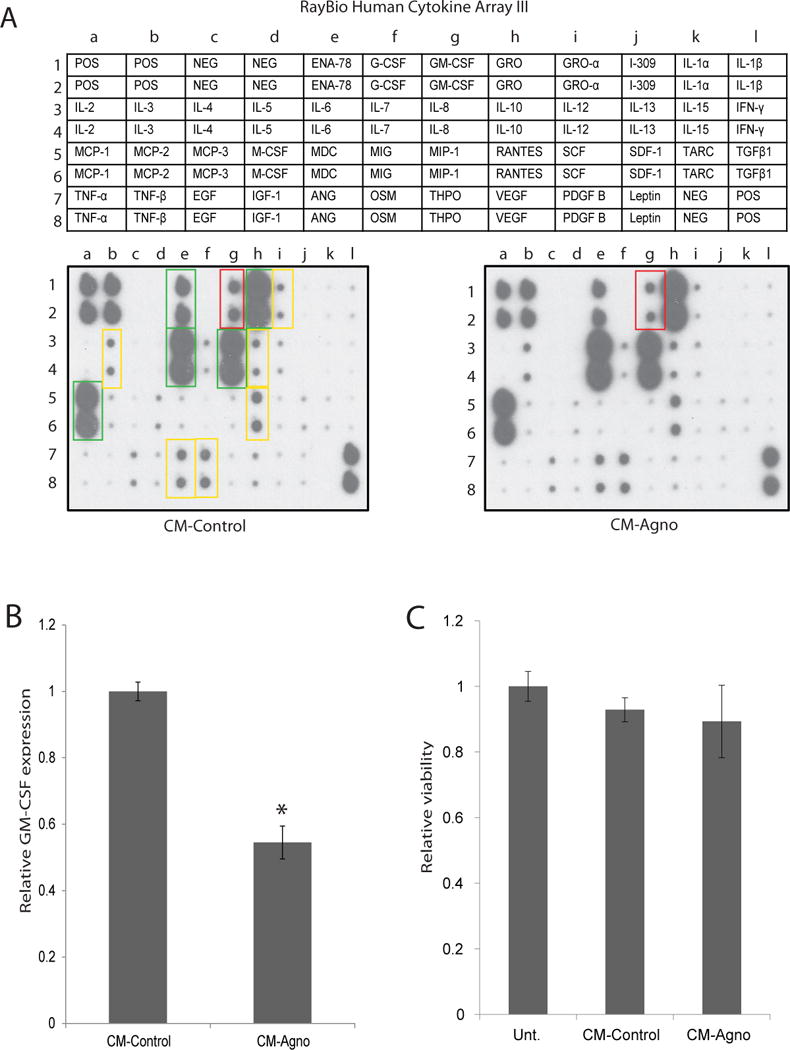
GM-CSF release by astrocytes exposed to CM-agno is suppressed. A. RayBioTech Human Cytokine Array C3 analysis of conditioned media from PHFA cells. PHFA cells were treated with CM-control or CM-agno obtained from Tc620 cells transfected with either control plasmid (CM-control) or pcGT7-agno plasmid (CM-agno) for 24 hours, after which media was changed and cells were exposed to fresh media. At 24 hours, media was collected and cytokine expression was determined using the cytokine array kit. Cytokine intensity was normalized to positive control signal intensities. Green boxes signify highly secreted cytokines, yellow boxes signify moderately secreted cytokines, and red box signify GM-CSF with a significant change in secretion following agnoprotein treatment by PHFA cells. B. Quantification of relative GM-CSF expression in the media following CM-control or CM-agno treatment. Intensities were normalized to positive control intensities from three tested membranes. C. MTT assay for the assessment of PHFA viability following treatment with CM-control or CM-agno for 24 hours. Viabilities were normalized to untreated PHFA cellular viability.
Agnoprotein inhibits GM-CSF transcription in glial cells
Subsequently, the mechanism resulting in decreased GM-CSF release was investigated. GM-CSF transcriptional activity was assessed through a luciferase assay. TC620 cells were transiently transfected with a luciferase reporter construct consisting of the −627 to +28 base pair promoter region of GM-CSF. Cells were transfected with either the luciferase construct alone, the luciferase construct and pcgT7-alone, or the luciferase construct and pcgT7-agnoprotein to determine the impact of agnoprotein expression on GM-CSF expression. At 48-hours, cells were harvested and processed for luciferase reporter assay. Expression of the luciferase reporter alone determined basal GM-CSF transcriptional activity, which was significantly suppressed by expression of agnoprotein, but not by control transfection (Figure 3A). As cytokine arrays were completed using conditioned growth media treatment as opposed to agnoprotein transfection, the impact of extracellular agnoprotein on GM-CSF transcriptional activity was also assessed. TC620 cells were transfected with the GM-CSF luciferase reporter construct and treated with CM-control or CM-agno. At 48 hours, cells were harvested and processed for luciferase assay. Treatment of cells with CM-control resulted in no change in GM-CSF transcriptional activity, whereas treatment with CM-agno resulted in significant suppression of GM-CSF transcriptional function (Figure 3B). These data suggest that agnoprotein expression or it’s uptake by cells is sufficient to suppress GM-CSF transcription.
Figure 3.
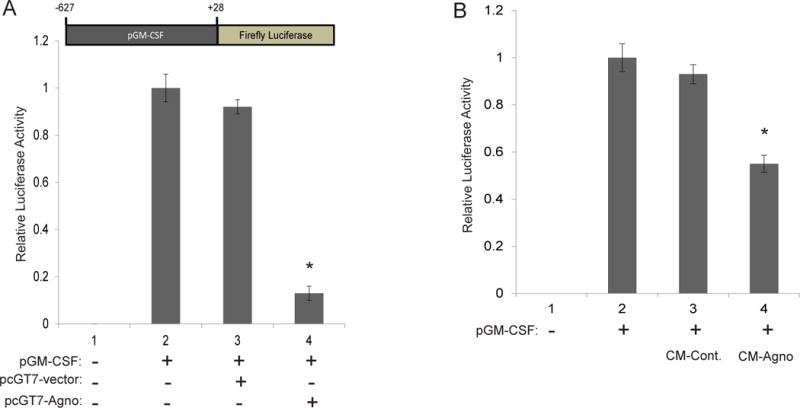
Agnoprotein suppresses GM-CSF transcription. A. Luciferase assay showing the effects of transfected agnoprotein on GM-CSF transcription. TC620 cells were co-transfected with pLuc.GM-CSF and the listed expression plasmids. At 48 hours post-transfections, cellular lysates were extracted and processed for luciferase assay analysis. Luminescence was determined and normalized to protein concentrations. Standard deviations were calculated from three independent experiments. Schematic of the luciferase reporter plasmid is presented in the upper panel of the graph. B. Luciferase assay showing the effects of agnoprotein treatments on GM-CSF transcription. TC620 cells were transfected with pLuc.GM-CSF and were either untreated or treated with CM-control or CM-agno. At 48 hours post-transfections, cellular lysates were extracted and processed for luciferase assay analysis. Luminescence was determined and normalized to protein concentrations for each sample. Standard deviations were calculated from three independent experiments.
GM-CSF inhibits JC virus early and late gene transcription
As GM-CSF transcription was suppressed by agnoprotein, the possibility arose that GM-CSF may regulate JCV transcription. To assess the impact of GM-CSF on JCV transcription, TC620 cells were transfected with a JCV bi-directional dual reporter construct, which allows for renilla reporter activity to be assessed for JCV-early transcription and luciferase reporter activity to be assessed for JCV-late transcription (Figure 4A). Expression of the dual reporter alone demonstrated basal levels of JCV early and late gene transcription in our system, which was used for normalization following treatment conditions. Following treatment with GM-CSF, JCV early transcription was suppressed in a dose-dependent manner (Figure 4B). For JCV early gene transcription, expression of T-antigen did not impact transcriptional activity and did not rescue GM-CSF mediated transcriptional suppression. When evaluating JCV late gene transcription, GM-CSF treatment significantly suppressed viral transcription in a dose-dependent manner, with 10 μg/mL treatment suppressing transcription to more than 50% of basal levels (Figure 4C). When cells were co-transfected with T-antigen, JCV late gene transcription was significantly induced. This induction was suppressed to basal levels following treatment with GM-CSF. These data suggest that GM-CSF suppresses JCV early and late gene transcription.
Figure 4.
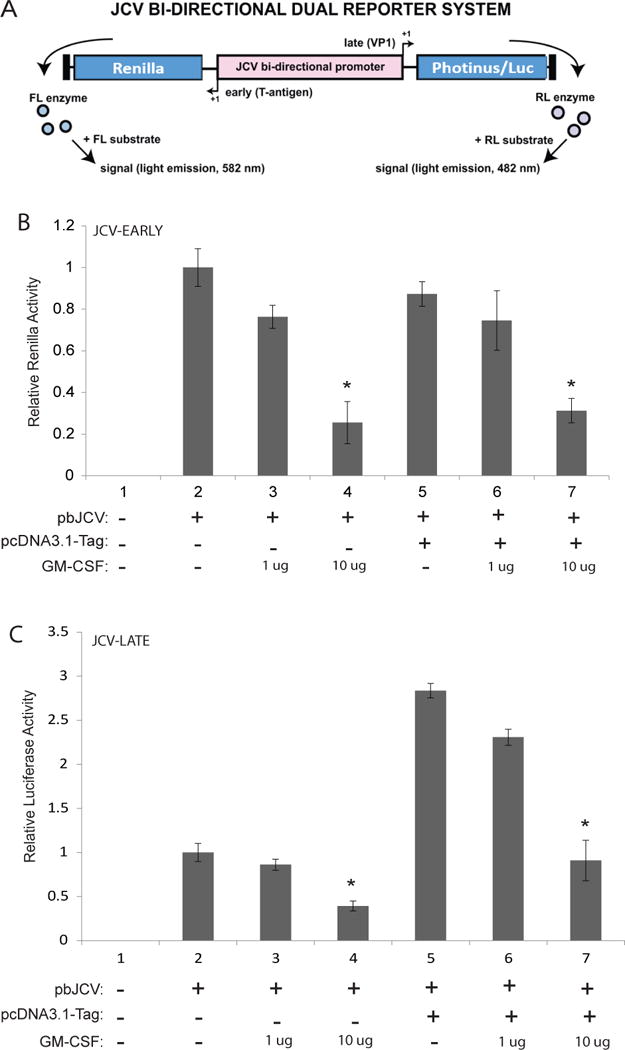
GM-CSF suppresses JCV-early and -late gene transcription. A. Schematic representation of the JCV bi-directional dual reporter system used to analyze JCV gene transcription. Expression of renilla protein is driven by JCV-early promoter whereas expression of luciferase protein is driven by JCV-late promoter activity. B. Renilla assay showing the effect of GM-CSF and T-antigen on JCV early promoter region transcriptional activity. TC620 cells were co-transfected with the JCV bi-directional dual reporter plasmid (pbJCV) and T-antigen as listed. Following transfection, cells were treated with GM-CSF at 1 and 10 ug/ml concentrations. At 48 hours post-transfections, cell lysates were harvested and processed for luciferase-renilla dual luciferase assay. Luminescence for JCV early-region transcriptional activity was determined by renilla luminescence and normalized to protein concentrations. Experiments were complicated in triplicate and standard deviations were calculated from the triplicates. C. Luciferase assay showing the effect of GM-CSF and T-antigen on JCV late promoter region transcriptional activity. TC620 cells were co-transfected with the JCV bi-directional dual reporter plasmid and T-antigen as listed. Following transfections, cells were treated with GM-CSF at 1 and 10 ug/ml concentrations. At 48 hours post-transfections, cell lysates were harvested and processed for luciferase-renilla dual luciferase assay. Luminescence for JCV late-region transcriptional activity was determined by luciferase luminescence and normalized to protein concentrations. Experiments were complicated in triplicate and standard deviations were calculated from the triplicates.
Agnoprotein treatment impairs U937 differentiation and maturation into macrophages
One function of GM-CSF during the inflammatory response is to induce the differentiation and maturation of monocytes into macrophages. As agnoprotein suppresses GM-CSF release and transcription in vitro, the ability of agnoprotein to directly impact monocytes was further assessed. To assess the impact of agnoprotein, an MBP-based bacterial expression system was utilized, allowing production and purification of recombinant agnoprotein. To ensure immunogenicity, samples were analyzed via Western blot, showing the expression of recombinant agnoprotein following factor Xa cleavage and purification (Figure 5A). To assess monocyte differentiation and maturation, the U937, a human monocytic cell line was utilized. For differentiation, U937 cells were induced for differentiation with PMA treatment. At 48 hours, cells were analyzed for differentiation, which was characterized by cellular attachment, as monocytes are non-adherent cells, while differentiated they express surface proteins allowing for cellular adhesion. U937 cells which were treated with PMA showed significantly increased cellular attachment as opposed to U937 cells with no PMA treatment. To assess the function of agnoprotein, U937 cells were treated with both conditioned media containing agnoprotein and recombinant agnoprotein. CM-agno treatment resulted in a significant reduction of cellular attachment as opposed to CM-control treatment, which had similar attachment to PMA-only U937 cells. Similarly, recombinant agnoprotein treatment resulted in a significant reduction of U937 cellular attachment as opposed to MBP-control treated cells (Figure 5B). These trends were also found during quantification over multiple experiments, with agnoprotein presence resulting in a significant decrease of attached cells (Figure 5C). To ensure that changes in U937 cellular attachments were not due to the toxicity associated with treatments, U937 cells were treated with both conditioned media containing agnoprotein and recombinant agnoprotein. After 48-hours, cells were processed for an MTT assay showing no significant alteration in cellular viability (Figure 5D). As agnoprotein was found to decrease U937 attachment, the ability of agnoprotein to dysregulate monocyte/macrophage surface marker expression was assessed. Under homeostatic conditions, CD16 marker expression is low on monocytes and high on macrophages; CD68 marker expression is low on monocytes and high on macrophages; and CD71 marker expression is higher on macrophages than monocytes. U937 cells were induced to differentiate with PMA and simultaneously treated with either CM-control or CM-agno. At 48 hours, cells were fixed with a 50% acetone-50% methanol solution and processed for immunocytochemistry. Following agnoprotein treatment, CD16 surface marker expression was significantly reduced on U937-macrophage like cells as opposed to CM-control treated cells (Figure 6A). Similarly, CD68 surface marker expression was reduced following agnoprotein treatment, with CM-agno treated cells having less CD68 fluorescence intensity (Figure 6B). However, the expression of CD71 was increased on U937 cells following agnoprotein treatment in contrast to control growth media treatment (Figure 6C). These data suggest that agnoprotein treatment is sufficient to suppress monocyte differentiation into macrophages and that agnoprotein can dysregulate the surface marker expression of macrophage-like cells.
Figure 5.
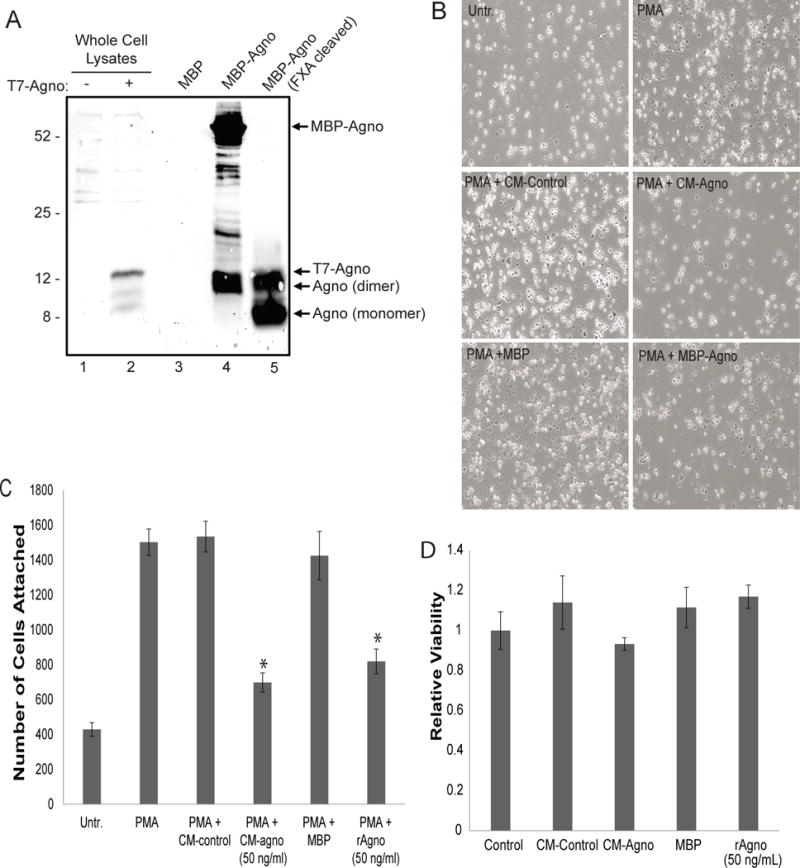
Agnoprotein inhibits U937 differentiation. A. Agnoprotein was expresses in E. coli as an MBP-agnoprotein complex and was purified by Factor Xa cleavage and analyzed by Western blotting. In lane 1 and 2, whole cell lysates from pcgT7-agnoprotein transfected TC620 cells served as negative and positive controls, respectively. Lane 3 contains MBP from the E. coli MBP-vector alone control. Lane 4 contains MBP-agnoprotein prior to factor Xa cleavage. Lane 5 shows cleaved and purified agnoprotein in both monomer and dimer forms. B. U937 cells were seeded in a 6-well tissue culture dish and induced for differentiation with PMA for two hrs. Simultaneously, the cells were untreated or treated with CM-control, CM-agno, MBP, or recombinant agnoprotein. Uninduced U937 served as a control condition. At 48 hours, adherent cells were imaged using phase contrast microscopy. Images are representative sections. C. U937 attachment following treatments at 48 hours was counted and shown as bar graph. D. MTT assay for the assessment of U937 viability following treatment with CM-control, CM-agno, and recombinant agnoprotein for 24 hours. Viabilities were normalized to untreated U937 cellular viability.
Figure 6.
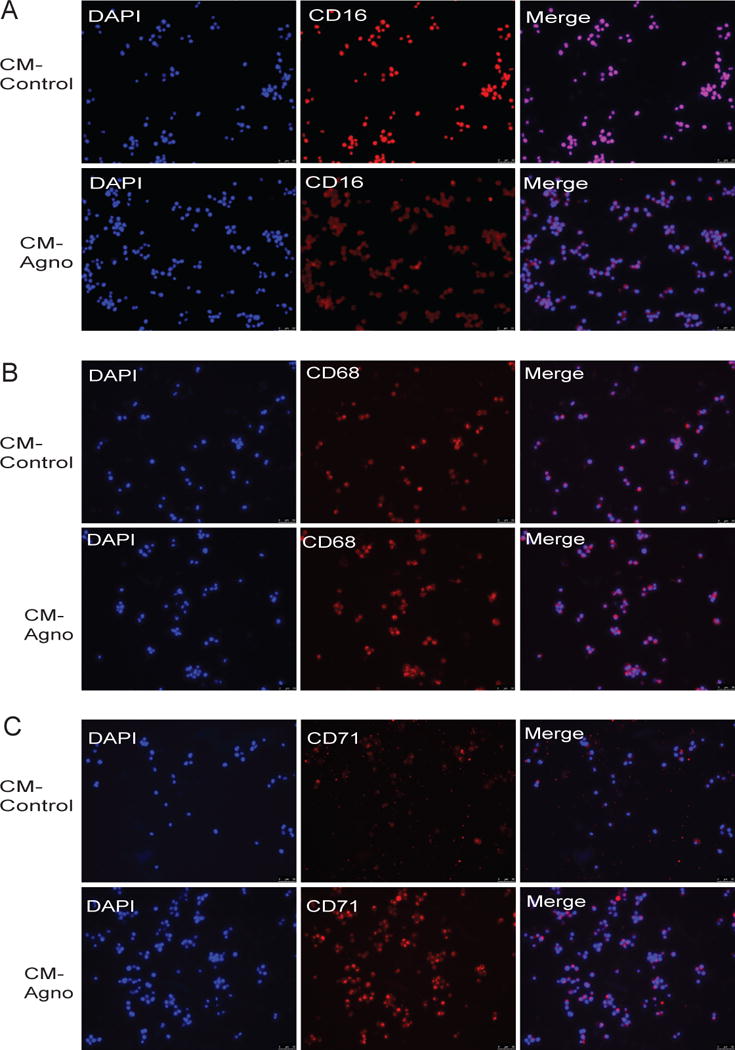
Agnoprotein dysregulates U937 surface marker expression during differentiation. U937 cells were seeded in 2-well tissue culture chamber slides and induced for differentiation with PMA. Simultaneously, cells were treated with CM-control (upper panel) or CM-agnoprotein (lower panel). At 72 hours, cells were fixed and processed for immunocytochemistry for CD16 (A), CD68 (B), or CD71 (C).
Phagocytic activity of macrophages and microglia is suppressed by extracellular agnoprotein
As agnoprotein was found to significantly impact monocyte differentiation and surface marker expression, the impact on monocyte function was further assessed. One major function of monocytes and macrophages is phagocytosis to clear cellular debris, foreign bodies, or apoptotic cells. To assess if agnoprotein affected phagocytosis, U937 monocytes, U937 macrophages, human primary macrophages, and primary human microglial cells were treated with either CM-agno or recombinant agnoprotein for 24 hours, after which they were exposed to fluorescently labeled E. coli, and allowed to phagocytose for 2 hours. After the 2-hour period, extracellular E. coli fluorescence was quenched with trypan blue and fluorescence readings were completed, allowing for quantification of the intensity of phagocytosed particles. For U937 monocytes, both PMA induction and agnoprotein treatment occurred simultaneously, while for U937-derived macrophages and human primary macrophages, monocyte cells were pretreated with PMA to induce their differentiation prior to agnoprotein treatment. For U937 monocytes, treatment of cells with either CM-agno or recombinant agnoprotein resulted in a significant reduction of phagocytic activity, with agnoprotein presence resulting in a ~50% decrease in the amount of phagocytosed E. coli particles (Figure 7A). A similar trend was seen for macrophages; however, the suppression was not as strong but significant, with agnoprotein treatment of both U937 macrophages and human primary macrophages resulting in a roughly ~40% suppression of phagocytic activity (Figure 7B, 7C). However, in vivo, the prominent macrophage-like cells that are present in the CNS, and thus the focal point of PML infection, are microglial cells, therefore, the effect of agnoprotein on microglial phagocytosis was assessed. Similarly, primary human fetal microglia possessed reduced phagocytic ability following treatment with agnoprotein (Figure 7D). These data taken together suggest that agnoprotein significantly suppresses the phagocytic ability of myeloid-derived cells in vitro.
Figure 7.
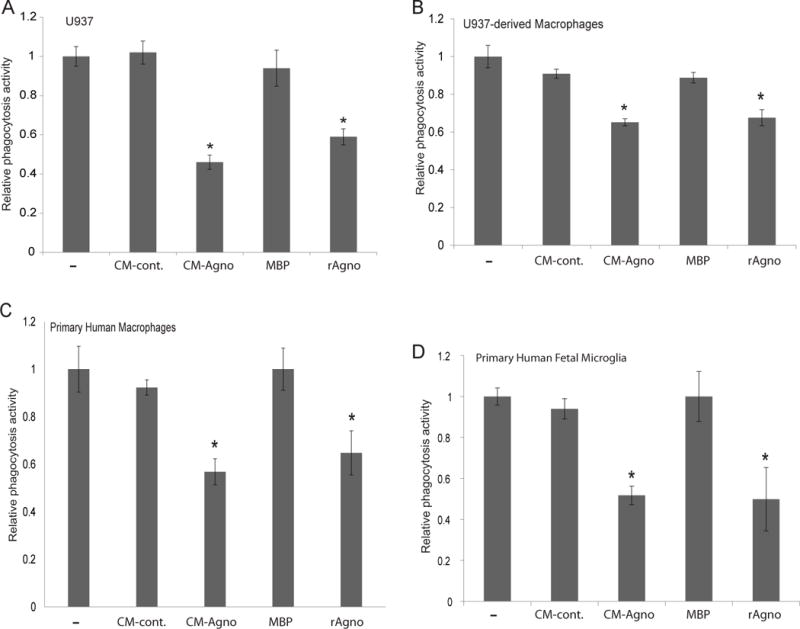
Agnoprotein inhibits phagocytic ability of myeloid-derived cells. U937 (A), U937-derived macrophages (B), human primary macrophages (C), and primary human fetal microglia (D) were plated in 96-well plates and induced with PMA and treated with CM-control, CM-agno, MBP, or recombinant agnoprotein. At 48-hours, cells were incubated for two hours with 200 μL of fluorescently labeled E. coli particles. Subsequently, the E. coli particles were removed and the fluorescence of extracellular particles was quenched with trypan blue treatment. Fluorescence values were read and corrected for background fluorescence. Values were normalized to PMA-induced conditions. Experiments were completed in triplicate and standard deviations were calculated from the triplicates.
Impact of Agnoprotein on Monocyte Migration in an in vitro BBB model
As agnoprotein impacted monocyte/macrophage surface marker expression and phagocytic function, it follows that agnoprotein may possess other immunomodulatory functions. To continue assessing the impact of agnoprotein on modulating the immune system during JCV infection, a blood-brain barrier model was used to determine if decrease leukocytic infiltration to sites of lytic infection may be partially due to the agnoprotein presence. To emulate the blood-brain barrier, primary human fetal astrocytes were plated in a 6-well tissue culture dishes, on top of which a 3.0 μm trans-well membrane insert was placed, on which primary human fetal brain endothelial cells were cultured (Figure 8A). To mimic the PML brain in which astrocytes display a reactive phenotype, the primary human fetal astrocytes were activated for 24-hours with10 μg/mL E. coli 026:B6 lipopolysaccharides (LPS). At 24-hours, LPS-positive media was aspirated from the model and one million primary human monocytes were placed into the top chamber above the insert, and migration was assessed at 3, 6, and 24-hours. Monocyte migration was significantly increased following LPS activation of monocytes at all the time points (data not shown). To assess if agnoprotein inhibited monocyte migration, astrocytes were treated with increasing concentrations of recombinant agnoprotein in combination with LPS activation. Without LPS activation, there was minimal migration of monocytes across the model BBB, with around 100,000 cells migrating after 24-hours. This migration could be explained by either background leakage through the 3.0 μm pore or by low level astrocytic activation by the FBS or other soluble factors present in the media. LPS induction resulted in high levels of monocyte migration, with slightly less than 800,000 monocytes migrating after 24-hours. This monocyte migration was reduced following treatment with concentrations of 50, 100, and 200 ng/mL of recombinant agnoprotein. Treatment with the highest concentration of agnoprotein suppressed monocyte migration by more than 50% over 24 hours, with only 300,000 monocytes migrating following 200 ng/mL agnoprotein treatment (Figure 8B). This experiment was replicated using the U937 monocyte cell line in place of primary human monocytes and resulted in similar trends, with agnoprotein treatment suppressing U937 migration in a dose-dependent manner (data not shown).
Figure 8.
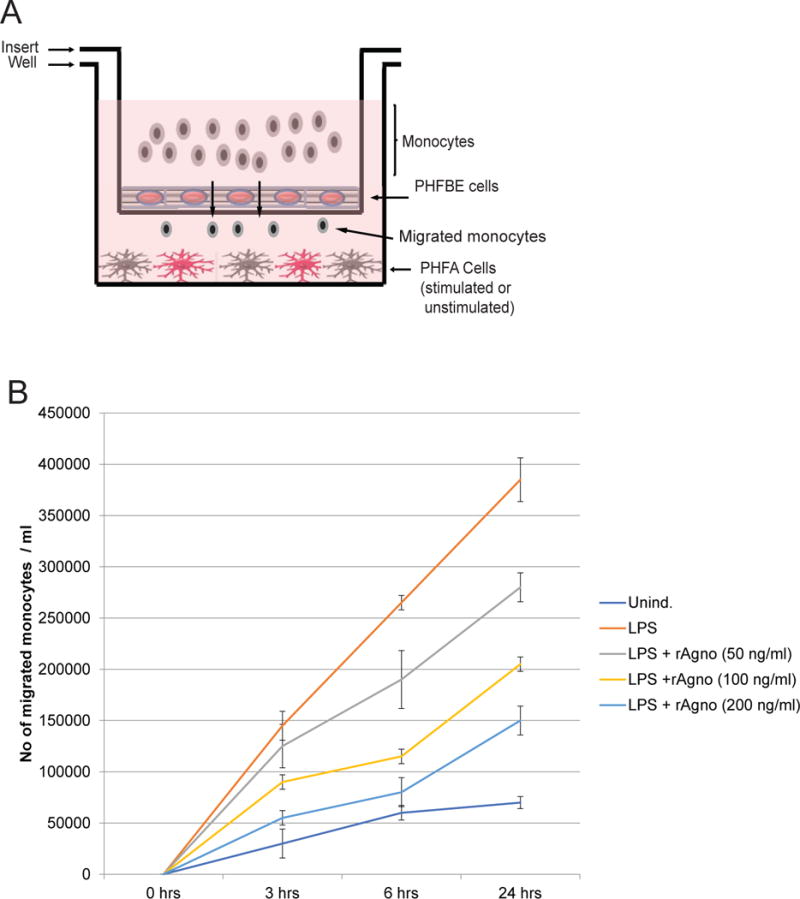
Agnoprotein decreases monocyte migration across a blood-brain barrier model. A. Schematic representation of the blood-brain barrier model used for the study. A corning 3.0 μm trans-well was inserted into a 6-well tissue culture dish. The 6-well tissue culture dish was seeded with PHFA that were stimulated with LPS or left unstimulated. The trans-well insert was seeded with primary human fetal brain endothelial (PHFBE) cells to form a confluent surface mimicking the blood-brain barrier. Primary human monocytes were seeded into the top chamber of the insert and allowed to migrate across the BBB-like membrane into the bottom portion of the system. Media from the bottom portion of the system was assessed for the presence of monocytes using cell-counting. B. PHFA cells were treated with recombinant agnoprotein at concentrations of 50, 100, and 200ng/mL. At 24 hours, cells were retreated with recombinant agnoprotein and simultaneously activated with LPS treatment. At 24 hours, LPS-containing media was removed and cells were retreated with agnoprotein in combination with the addition of one million human primary monocytes. Migrated cells in the bottom well were counted at 3, 6, and 24 hours after their addition into the top chamber. Experiment was completed in triplicate and standard deviations were calculated from the triplicates.
DISCUSSION
The immune system evolved to deal with a wide array of pathogens through two primary mechanisms, cellular immunity and humoral immunity. When discussing JC virus infection, cellular immunity appears to be the driving force behind immune control of the virus (Koralnik 2002). Generally, PML development is found in patients with conditions depleting either cellular immunity specifically, generally through conditions resulting in decreased T-cell populations or T-cell function, or conditions that suppress both arms of the immune system. One of the most prominent diseases which PML is associated with is AIDS, and AIDS patients are the cohort with the highest risk for developing PML (Berger et al, 2001). However, while suppression of the cellular arm of the immune system increases the risk for PML development, there is no significant correlation between suppression of the humoral immune system and PML development (Weber et al, 2001). Since cellular immunity is hypothesized to be the mechanism of JCV suppression, we investigated the potential of viral agnoprotein to negatively regulate aspects of the cellular immune responses within the CNS. In terms of agnoprotein functions, one potential function may be to modulate neural inflammation. While JCV infection and progression into the PML generally results in minimal inflammation, there also exists inflammatory JCV infections. JC virus encephalopathy is a gray matter disease caused by JCV infection of cortical pyramidal neurons, resulting in an inflammatory encephalopathy (Dang et al, 2012). The JCV strain which results in encephalopathy contains a 143 bp deletion in the agnoprotein gene, resulting in the expression of a truncated peptide (Dang et al, 2012). If agnoprotein serves to mediate inflammation, then the loss of agnoprotein in this variant may explain the resulting encephalitis compared to other JCV strains.
When looking at other viruses, there are many viral proteins which modulate the immune response to the virus, either to suppress immune cell function, to mask the virus, or other various functions. One prominent example is the CNS-targeting rabies virus. Like JCV, rabies virus is highly neurotropic and infects the CNS, resulting in a fatal infection. After initial rabies infection, the virus enters muscle tissue and initiates low levels of viral replication, however, muscle tissue is not a productive cellular site for the virus. Eventually, the virus infects muscle-associated motor endplates via binding to acetylcholine receptors at the neuromuscular junction, allowing for entry in the peripheral nervous system (Gluska et al, 2014). From the peripheral nervous system, the virus replicates and viral particles travel to the spine and into the CNS, where it replicates primarily in motor neurons (Hemachudha et al, 2013). In the CNS, the virus replicates and evades the immune response through the production and release of rabies virus phosphoprotein. Rabies phosphoprotein is a protein that is released from infected cells and serves to suppress the transcription of antiviral genes in infected and uninfected cells (Scott et al, 2016; Srithayakumar et al, 2014).
Like rabies virus, JCV infection of the CNS results in the production and release of a viral agnoprotein. Here, we investigated the ability of agnoprotein to act in a manner similarly to rabies virus phosphoprotein, in which we assessed the ability of agnoprotein to modulate the immune response to the virus. Our results demonstrate that agnoprotein is released from cells following expression, and that the protein is internalized by non-infected glial cells. Following this uptake, glial cells present with a dysregulated cytokine profile, marked by the decrease of GM-CSF release. We found that GM-CSF significantly suppresses JCV early and gene transcription in glial cells, but the expression of agnoprotein is sufficient to suppress GM-CSF transcription, effectively preventing the anti-viral effects of GM-CSF on JCV. We hypothesize that suppression of GM-CSF is important within the viral infection cycle, as it helps the virus evade immune surveillance by significantly reducing the infiltration of peripheral monocytes to sites of astrocytic activation (Figure 9). Our results also show that agnoprotein suppresses the function of myeloid-derived cells, primarily through suppressing phagocytosis. As phagocytosis is an important part of cellular immunity, this function of agnoprotein may be very important to the viral life cycle. Generally, during JCV infection, there is a reduction in immune function, with most comorbidities resulting in decreased T-cell populations. However, monocytes/macrophages and microglia remains functional in many of these patients. Of note, extracellular agnoprotein had no significant alteration in release of MCP-1 shown to be activated in the brain of patients with HIV-encephalitis and dementia (Cinque et al., 1998; Conant et al, 1998). Whether agnoprotein has any impact on MCP-1 activation in glial cells and MCP-1-mediated pro-inflammatory signaling in a PML-AIDS setting remains to be elucidated.
Figure 9.
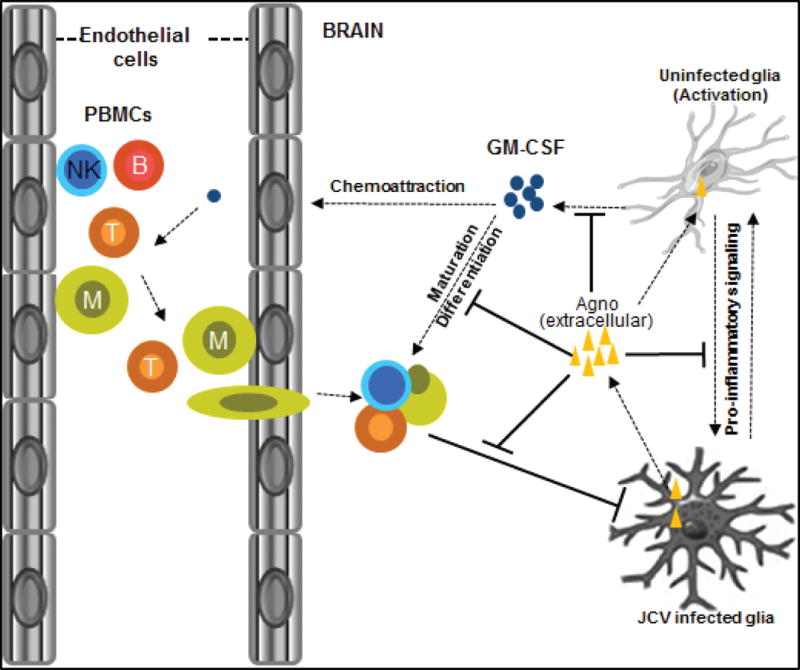
Schematic presentation of the role of agnoprotein in neuroimmune response to JC virus. Infected cells set the stage of pro-inflammatory signaling to the bystander uninfected cells to initiate neuroinflammation induced by GM-CSF activation. Upon activation, GM-CSF can serve as a chemo-attractant for the migration of lymphocytes through blood-brain barrier into the area of infection and contribute to their maturation and differentiation within the brain. Agnoprotein is expressed and released into the extracellular space by cells infected with JC virus and taken up by uninfected bystander glial cells. Pro-inflammatory signaling between infected and uninfected cells leading to the GM-CSF activation and neuroinflammation is disrupted by extracellular and intracellular agnoprotein leading to the development of PML lesions.
Agnoprotein is a highly basic phosphoprotein that possesses a major alpha-helix domain between amino acid residues 23–39 and a minor alpha-helix domain between amino acid residues 6–13 (Coric 2017; Saribas 2016). Following agnoprotein expression, the protein forms highly stable dimers and oligomer complexes which are dependent on amino acid residues 17–42, a region highly rich in leucine, isoleucine, and phenylalanine (Saribas 2011). Our data confirms to this, with western blot analysis showing a double-banding pattern for agnoprotein, showing both the presence of monomer and dimer forms of recombinant agnoprotein. However, it still remains to be elucidated whether both monomer and dimer agnoprotein structures play an immunomodulatory roles. JCV agnoprotein mutants lacking amino acid residues 17–42, the domain essential for dimer formation, show significantly decreased JCV replication, however, it is not clear if this impact is due to suppression of agnoprotein dimer formation or due to another mechanism (Saribas 2011).
Neuroinflammation can be induced by a myriad of insults, including as the consequence of infectious agents, such as measles, herpes, and human immunodeficiency virus, via autoimmune diseases, or through traumatic brain injury (Gendelman 2002). When discussing neuroinflammation in the context of infectious agents, there is a fine balance between too minimal of a response, in which the agent is not cleared, and too strong of a response, in which there is extensive neural tissue damage, resulting in neural cell destruction with subsequent neurodegenerative symptoms. An example of extensive damage as the result of an immune response is HIV-Associated Neurocognitive Disorder (HAND). During primary HIV infection, HIV enters the brain parenchyma through the migration of infected lymphocytes or monocytes across the blood-brain barrier in combination with transependymal migration, the migration of CSF containing free viral particles (Booss et al, 1987; Eggers et al, 2017). After neural entry, HIV encephalitis may occur, which is characterized by widespread infiltration of lymphocytes, macrophages, and multinucleated giant cells, fusions of macrophages, into the CNS to infected sites (Carrol et al, 2017; Eggers et al, 2017). This infiltration results in the induction of inflammation in the brain, primarily due to the release of pro-inflammatory cytokines by infiltrating cells, resulting in widespread neuroinflammation and tissue damage. While HIV results in increased neuroinflammation in the brain to clear the virus, JCV generally exhibits minimal neuroinflammation. Interestingly, JCV and HIV are highly comorbid, with PML occurring in a significant portion of AIDS patients (Cinque et al, 2009). Generally, PML lesions are devoid of lymphoplasmocytic infiltrates. However, some inflammatory forms of PML have been described in HIV-seropositive patients, especially after reconstitution of the immune system, resulting in PML-IRIS, or PML-immune reconstitution inflammatory syndrome (Du Pasquier et al, 2003). Since inflammatory PML is rare, even in the presence of comorbid conditions such as AIDS, it may suggest that JCV possesses a viral factor that regulates inflammation. Our data suggest that JCV agnoprotein may serve to modulate neuroinflammation leading to progressive enhancement of PML lesions. Since JCV reactivation and development of PML is mostly seen in HIV patients, the molecular interaction between JCV and HIV in the concept of modulating neuroimmune regulations remains to be elucidated.
In summary, our results demonstrate a novel immunomodulatory function of agnoprotein during JCV infection within the CNS and open a new avenue of research to better understand the mechanisms associated with JCV reactivation in patients who are at risk of developing PML.
MATERIALS AND METHODS
Ethics Statement
All samples were obtained and utilized in accordance with Temple University Human Subjects Protections and the approval of the Institutional Review Board.
Cell lines and culture
The human oligodendroglioma cell line, TC620, were obtained from American Type Culture Collection (ATCC) and were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and penicillin/streptomycin (100 μg/ml). TC620 cells were maintained at 37°C in a humidified environment with 5% CO2. The human monocytic cell line, U937, were obtained from American Type Culture Collection (ATCC) and were grown in RPMI 1640 media containing 10% heat-inactivated fetal bovine serum and penicillin/streptomycin (100 μg/mL). Primary human fetal astrocytes (PHFA) were obtained from the fetal brain of a donor provided by the Comprehensive NeuroAIDS Core (CNAC) facility at the Lewis Katz School of Medicine of Temple University and cultured in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F-12) containing 10% heat-inactivated fetal bovine serum (FBS), penicillin/streptomycin (100 µg/ml), GlutaMax (100 µg/mL) and insulin (100 µg/mL). Cultured PHFA were maintained at 37° C in a humidified atmosphere with 5% CO2. Primary human fetal microglia (PHFM) were obtained from the fetal brain of a donor provided by the CNAC facility at the Lewis Katz School of Medicine of Temple University and cultured in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F-12) containing 15% heat-inactivated fetal bovine serum (FBS), gentamicin (50 μg/mL), L-glutamine (100 μg/mL), fungizone (50 μg/mL), inulin (100 μg/mL), D-biotin (10 ng/mL) and NI supplement (Sigma #N6530). Cultured PHFM were maintained at 37° C in a humidified atmosphere with 5% CO2. Primary human fetal brain endothelial cells (PHFE) were obtained from the fetal brain of a donor provided by the CNAC facility at the Lewis Katz School of Medicine of Temple University and cultured in Clonetics Endothelial Cell Basal Medium-2 (EBM-2) supplemented with FBS, hydrocortisone, human fibroblast growth factor, vascular endothelial growth factor, R3-IGF, recombinant insulin-like growth factor, ascorbic acid, human epidermal growth factor, GA-1000, and heparin. Cultured PHFE were maintained at 37° C in a humidified atmosphere with 5% CO2. Conditioned growth media was collected from TC620 cells which were transfected with either pcgT7-control vector (CM-control) or pcgT7-agnoprotein vector (CM-agno). Briefly, TC620 were plated in 100 mm dishes and transfected with 10 μg of vector. At 48 hours, media was collected and centrifuged to remove cellular debris. Following centrifugation, immunoprecipitation and ELISA were completed using conditioned growth media to assess agnoprotein presence and concentration.
Plasmid Constructs
The pcgT7-agnoprotein expression vector construct was described previously (Otlu, O. et al 2014). The GM-CSF luciferase reporter construct, pGM-CSF, was kindly provided by Peter Cockerill (College of Medical and Dental Science, Institute of Biomedical Research, University of Birmingham, UK) and was described previously (Cockerill et al, 1999). The pcDNA3.1-T-antigen expression vector was described previously (Craigie et al 2016). The JCV bi-directional dual reporter system was created by cloning JCV NCCR region (4987-246, NC_001699.1) into the pSF-Renilla-Photinus vector (Sigma-Aldrich, OGS595) at NotI and EcoRI sites.
Western Blot Analysis
Cells were trypsinized and collected, after which whole cell protein extracts were washed with PBS and lysed with TNN lysis buffer (150 mM NaCl, 40 mM tris pH 7.4, 1% NP-40, 1 mM DTT, 1 mM EDTA) containing protease inhibitors. Purified protein extracts were denatured at 95° C for 10 minutes and resolved through sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After resolution, the gel was transferred to a 0.2-micron nitrocellulose membrane (Whatman, Germany) for 30 minutes at 250 mÅ at 4° C in transfer buffer (25 mM Tris pH 7.4, 200 mM glycine, 20% methanol). After transfer, membranes were blocked for one hour at room temperature with 10% non-fat dry milk in 1× phosphate-buffered saline containing 1% Tween-20 (PBST). After blocking, membranes were washed and incubated with primary antibodies overnight at 4°C. After primary antibody incubation, membranes were washed three times in PBST and incubated with secondary antibody at a 1:5000 dilution at room temperature for one hour. Following secondary antibody incubation, membranes were visualized with the Odyssey CLx Imaging System (LI-COR). Primary antibodies used were anti-agnoprotein (ab7903) and anti-GAPDH (Cell Signaling Technology). Secondary antibodies used were IRDye 680 RD goat anti-rabbit (LI-COR) and IRDye 800CW goat anti-mouse (LI-COR).
Luciferase Reporter Assay
TC620 cells were plated in 6-well tissue culture dishes and were transiently transfected with pLuc.GMCSF reporter plasmids in the presence or absence of the agnoprotein expression plasmid or pcgT7-alone as a control. At 48-hours post-transfection, cells were trypsinized and lysed using Promega reporter lysis buffer. After cell lysis, luciferase activity of samples was assessed using luciferase assay reagent (LAR II) provided by Promega. Luciferase activities were assessed using a Zylux Corporation Femtomaster FB12 luminometer and corrected for protein concentration. Values were then normalized to basal transcriptional levels, allowing for determination of fold change in transcriptional activity following agnoprotein expression.
TC620 cells were plated in 6-well tissue culture treated dishes and were transiently transfected with the pb.JCV bi-directional dual reporter system in the presence or absence of pcDNA3.1-T-antigen or pcDNA3.1-alone as control. Cells were then untreated or treated with 1 ug/mL or 10 ug/mL doses of GM-CSF over the 48-hour period. At 48-hours post-transfection, cells were trypsinized for collection and lysed using Promega reporter lysis buffer. After cell lysis, luciferase activities were assessed using luciferase assay reagent (LAR) provided by Promega. After luciferase activities were collected, Stop-and-Glow buffer was added to quench luciferase activity while inducing Renilla activity. Luciferase and Renilla activities were collected using Zylux Corporation Femtomaster FB12 luminometer and corrected for protein concentration. Values were then normalized to basal transcriptional levels and assessed for fold changes following GM-CSF treatment.
MBP protein work
The production and purification of MBP-agnoprotein were published previously (Saribas et al, 2011). Briefly, the pMAL-c5x-JCV-Agno-FL plasmid was transformed into DH5α E. coli cells. Cells were grown overnight in 100 mL LB media supplemented with ampicillin (100 μg/mL). Cultures were then diluted 1:10 in fresh LB media supplemented with ampicillin (100 μg/mL) and glucose (2g/L) and grown at 37° C until the OD600 = 0.5. At this point, MBP protein production was induced with the addition of 0.5 mM IPTG and bacteria was grown at 37° C for 3 hours. Bacterial cells were then harvested by centrifugation at 10,000 RPM using a Thermo Scientific F12-6X500 LEX rotor and the resulting pellet was re-suspended in 10 mL amylose column buffer consisting of 20 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA. Re-suspended bacteria were incubated on ice for 30 minutes in the presence of lysozyme and protease inhibitor cocktail, after which lysis was completed via sonication. The lysates were centrifuged at 15,000 RPM for 30 minutes using a Sorvall HB-6 rotor, after which clear lysates were obtained. Clear lysates were then incubated with 800 μL of amylose fast flow resin beads (New England Biolabs) overnight at 4° C, allowing for optimal binding of MBP to the resin beads. Following overnight incubation, beads were spun down and supernatant was removed, after which beads were resuspended in amylose column buffer. Proteins were eluted from beads utilizing 10 mM maltose buffer as the elution solution, after which lysates were spun and the supernatant was collected in which the MBP-agnoprotein complex was present. Agnoprotein was then cleaved from the MBP complex using the protease Factor Xa (New England Biolabs, #P8010S), which cleaves the fusion protein at the Ile-(Glu-Asp)-Gly-Arg cleavage site. For cleavage, 50 μg of MBP-agnoprotein fusion protein complex was treated with 1 μL of Factor Xa in 2 mM CaCl2 buffer overnight at 23°C. Following cleavage, remaining MBP was removed using incubation with a 1:1 volume of amylose fast flow resin beads and incubation overnight at 4° C. Agnoprotein presence was assessed using Western blotting with anti-agnoprotein antibody.
U937 Monocyte differentiation assay
U937 cells were plated in a 6-well tissue culture treated at a density of 1×106 cells per well. Cells were induced for differentiation through a two hour pulse of 100 ng/ml phorbol myristate acetate (PMA). PMA was removed and cells were treated with CM-control, CM-agno, MBP, or recombinant agnoprotein. CM-treatments were 50% conditioned media, 50% fresh media, resulting in final agnoprotein concentration of 25 ng/mL media. Recombinant agnoprotein and MBP treatments were completed at a final concentration of 25 ng/mL of protein. At 24, 48, and 72 hours, media and unattached cells were removed from culture and treatments were repeated. At each time point, five phase contrast images were completed of various regions in each to assess cellular attachment as a model of differentiation into macrophage like cells. Attached cells from each image were counted and compiled into a graph to quantify attached cells within each condition.
Phagocytosis assay
U937 cells, human primary macrophages, or microglia were cultured in RPMI 1640 for 3 days prior to phagocytosis assay. The Vybrant phagocytosis assay kit (Thermo Fisher #V6694) was used to assess phagocytic ability. Briefly, cells were treated for 48 hours prior to phagocytosis assay completion, with either CM-control, CM-agno (25 ng/mL), MBP (25 ng/mL) or recombinant agnoprotein (25 ng/mL). At 48 hours, media was removed and 100 μL of fluorescein-labeled E. coli K-12 bioparticles were added, after which cells were incubated for 2 hours at 37° C. After 2 hours, E. coli particles were aspirated and cells were incubated for 1 minute at room temperature with trypan blue to quench extracellular fluorescence from non-phagocytosed E. coli particles. Particle uptake was assessed with a Fisher Scientific Multiskan FC microplate reading using 480 nm excitation and 520 nm emission wavelengths. The change in phagocytosis was calculated using PMA-induced cells as the control to which treated cells were normalized to.
Cytokine array
Primary human fetal astrocytes were plated in 60 mm tissue culture treated dishes and were treated with CM-control or CM-agno for 24 hours. At 24 hours, conditioned media was removed and replaced with fresh DMEM:F12 media, in which cells were incubated for 24 hours. At 24 hours, media was collected and centrifuged to remove cellular debris and processed with the RayBiotech C-series human cytokine antibody array C3. Briefly, cytokine array membranes were blocked for 30 minutes at room temperature using blocking buffer provided by the manufacturer. After blocking, conditioned growth media for each condition was added into each well and incubated for 4 hours at room temperature. After growth media incubation, membranes were washed with provided wash buffers. Following washing steps, detection antibody cocktail was added into each well and incubated for 2 hours at room temperature. Membranes were washed with provided wash buffers following antibody incubation. Membranes were then incubated with HRP-streptavidin solution for 2 hours at room temperature, after which membranes were again washed. Signal detection was completed using provided detection buffers and membranes were analyzed through x-ray film development with a Kodak X-OMAT 2000A processor.
Immunoprecipitation
TC620 cells were seeded in 100 mm tissue culture treated dishes and grown until confluent. After confluence, cells were transiently transfected with pcgT7-alone or pcgT7-agnoprotein expression plasmids. At 48-hours, conditioned growth media was collected and centrifuged to remove cellular debris. Cleared growth media was diluted 1:1 with TNN lysis buffer (150 mM NaCl, 40 mM tris pH 7.4, 1% NP-40, 1 mM DTT, 1 mM EDTA) and preincubated with Protein G-Sepharose 4B conjugate (Life Technologies) to decrease background binding. Simultaneously, recombinant Protein G-Sepharose 4B conjugate was preincubated with anti-agnoprotein antibody. Diluted media was centrifuged and the bead-free media fraction was collected. The anti-agnoprotein incubated beads were added to the diluted media and rotated overnight at 4° C. After immunoprecipitation, media was removed and beads were washed with PBS (3×) and re-suspended in distilled water. Re-suspended beads were analyzed by Western blotting as described.
Immunocytochemistry
Primary human fetal astrocytes (PHFA) or primary human fetal microglia (PHFM) were cultured in 2-well tissue culture treated chamber slides (500,000 cells per well). Cells were treated with 1 mL of CM-control or CM-agnoprotein in combination with 1 mL of fresh PHFA or PHFM media at 0 and 24-hour time points. At 48 hours, cells were fixed with ice cold acetone-methanol (50:50) for 1 minute, washed five times with PBS, and blocked with 10% BSA in PBS for 1 hour at room temperature. After blocking, cells were incubated with anti-agnoprotein antibody, diluted at 1:300 in 5% BSA in PBS solution overnight at 4° C with gentle rocking. Cells were washed three times with PBS and incubated with secondary antibody solution, either 1:500 FITC-rabbit or 1:500 rhodamine-rabbit in 5% BSA in PBS solution for 2 hours at room temperature. Wells were washed five times with PBS and mounted with Vectashield mounting medium containing DAPI (Vector Laboratories). Glass coverslips were added prior to visualization with a Leica Fluorescent Microscope (Leica Biosystems Inc.). U937 cells were cultured in 2-well tissue culture treated chamber slides (500,000 cells per well). Cells were induced for differentiation through exposure to 100 ng/mL phorbol myristate acetate treatments. Simultaneously, cells were treated with 1 mL of CM-control or CM-agnoprotein in combination with 1 mL of fresh PHFA or PHFM media at 0-, 24-, and 48-hour time points. At 72 hours, cells were fixed with ice cold acetone-methanol (50:50) for 1 minute, washed five times with PBS, and blocked with 10% BSA in PBS for 1 hour at room temperature. After blocking, cells were incubated with anti-CD16, anti-CD68, or anti-CD71 antibody, diluted at 1:300 in 5% BSA in PBS solution overnight at 4° C with gentle rocking. Cells were washed three times with PBS and incubated with secondary antibody solution, 1:500 rhodamine-mouse in 5% BSA in PBS solution for 2 hours at room temperature. Wells were washed five times with PBS and mounted with Vectashield mounting medium containing DAPI (Vector Laboratories). Glass coverslips were added prior to visualization with a Leica Fluorescent Microscope (Leica Biosystems Inc.).
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Assay
PHFA or TC620 cells were plated in six-well plates at 1×106 cells per well and treated with 2 mL of CM-control or CM-agnoprotein in combination with 2 mL of fresh PHFA or PHFM media at 0 and 24-hour time points. At 48-hours, cells were incubated with 1 mL of MTT working solution (DMEM and 0.5 mg/mL MTT substrate) for 2 hours at 37° C. Metabolically produced formazan was solubilized with 1 mL acidic isopropanol (4 mM HCl in 100% isopropanol), resulting in a purple color which was measured via spectrophotometer at a wavelength of 570 nm with a background subtraction at 650 nm.
In Vitro Blood Brain Barrier Model
Primary human fetal astrocytes were seeded in a 6-well tissue culture dish at a concentration of 250,000 cells per well and grown until confluence at 37° C in a humidified environment with 5% CO2. After reaching confluence, primary human endothelia cells were seeded in a Corning Transwell permeable support containing a 3.0 μm polyester membrane at a concentration of 150,000 cells per membrane and placed into wells containing previously seeded astrocytes. The cells were grown for 24 hours at 37° C in a humified environment containing 5% CO2. At 24 hours, astrocytes were activated with LPS at concentrations of 1 or 10 μg/mL for 24 hours. After 24 hours, LPS-containing media was removed and one million primary human monocytes were added into the top chamber of the blood-brain barrier and allowed to migrate across the barrier in response to astrocyte activation. Monocyte migration was assessed through phase contrast imaging and cell counting using a hemocytometer at 3, 6, and 24 hours after monocyte addition. To assess the impact of agnoprotein on suppressing monocyte migration, primary human fetal astrocytes were seeded in a 6-well tissue culture dish at a concentration of 250,000 cells per well and grown until confluence at 37° C in a humidified environment with 5% CO2. After reaching confluence, primary human endothelial cells were seeded in a Corning Transwell permeable support containing a 3.0 μm polyester membrane at a concentration of 150,000 cells per membrane and placed into wells containing previously seeded astrocytes. After endothelial cell seeding, recombinant agnoprotein was added to the astrocyte culture at concentrations of 50, 100, or 200 ng/mL for 24 hours. At 24 hours, another treatment of agnoprotein occurred simultaneously with astrocyte activation using 10 μg/mL of LPS for 24 hours. After 24 hours, LPS containing media was removed and 1 million primary monocytes were added to the top chamber of the model in combination with another astrocyte treatment of agnoprotein. Monocyte migration was assessed through phase contrast imaging and cell counting using a hemocytometer at 3, 6, and 24 hours after monocyte addition.
Acknowledgments
We would like to thank Dr. Jennifer Gordon and Rahsan Sariyer for their assistance in generation and cloning a dual reporter construct for JCV NCCR region. We would like to thank Dr. Kamel Khalili for his support and insightful comments. We would like to also thank Dr. Martyn White for the critical reading of the manuscript. This study utilized services offered by core facilities of the Comprehensive NeuroAIDS Center (CNAC NIMH Grant Number P30MH092177) at Temple University Lewis Katz School of Medicine, Department of Neuroscience. This work was made possible by grants awarded by NIH to IKS (AI101192)
Footnotes
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: IKS and MC. Performed the experiments: MC and SC. Analyzed the data: MC and IKS. Contributed reagents/materials/analysis tools: IKS. Wrote the paper: MC and IKS.
References
- 1.Berger JR, Chauhan A, Galey D, Nath A. Epidemiological evidence and molecular basis of interactions between HIV and JC virus. J Neurovirol. 2001;7:329–338. doi: 10.1080/13550280152537193. [DOI] [PubMed] [Google Scholar]
- 2.Bollag B, Prins C, Snyder EL, Frisque RJ. Purified JC virus T and T’ proteins differentially interact with the retinoblastoma family of tumor suppressor proteins. Virology. 2000;274:165–178. doi: 10.1006/viro.2000.0451. [DOI] [PubMed] [Google Scholar]
- 3.Booss J, Harris SA. Neurology of AIDS virus infection: a clinical classification. Yale J Biol Med. 1987;60(6):537–543. [PMC free article] [PubMed] [Google Scholar]
- 4.Carroll A, Brew B. HIV-associated neurocognitive disorders: recent advances in pathogenesis, biomarkers, and treatment. [version 1; referees: 4 approved] F1000 Research. 2017;6(312) doi: 10.12688/f1000research.10651.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cinque P, Vago L, Mengozzi M, Torri V, Ceresa D, Vicenzi E, Transidico P, Vagani A, Sozzani S, Mantovani A, Lazzarin A, Poli G. Elevated cerebrospinal fluid levels of monocyte chemotactic protein-1 correlate with HIV-1 encephalitis and local viral replication. AIDS. 1998;12:1327–1332. doi: 10.1097/00002030-199811000-00014. [DOI] [PubMed] [Google Scholar]
- 6.Cinque P, Koralnik IJ, Gerevini S, Miro JM, Price RW. Progressive multifocal leukoencephalopathy in HIV-1 infection. Lancet Infect Dis. 2009;9(10):625–636. doi: 10.1016/S1473-3099(09)70226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cockerill PN, Bert AG, Roberts D, Vadas MA. The human granulocyte-macrophage colony-stimulating factor gene is autonomously regulated in vivo by an inducible tissue-specific enhancer. Immunology. 1999;96:15097–15102. doi: 10.1073/pnas.96.26.15097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA JID - 7505876. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coric P, Saribas AS, Abou-Gharbia M, Childers W, Condra JH, White MK, Safak M, Bouaziz S. Nuclear magnetic resonance structure of the human polyoma JC virus agnoprotein. J Cell Biochem. 2017 doi: 10.1002/jcb.25977. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craigie M, Regan P, Otalora YL, Sariyer IK. Molecular interplay between T-Antigen and splicing factor, arginine/serine-rich 1 (SRSF1) controls JC virus gene expression in glial cells. Virol J. 2015 Nov 24;12:196. doi: 10.1186/s12985-015-0426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dang X, Wüthrich C, Gordon J, Sawa H, Koranik IJ. JC virus encephalopathy is associated with a novel agnoprotein-deletion JCV variant. PLoS One. 2012;7(4):e35793. doi: 10.1371/journal.pone.0035793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Valle L, Gordon J, Enam S, Delbue S, Croul S, Abraham S, Radkahrishnan S, Assimakopoulou M, Katsetos CD, Khalili K. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J Natl Cancer Inst. 2002;94:267–273. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- 13.Du Pasquier RA, Koralink IJ. Inflammatory reaction in progressive multifocal leukoencephalopathy: harmful or beneficial. J Neurovirol. 2003;9:25–31. doi: 10.1080/13550280390195315. [DOI] [PubMed] [Google Scholar]
- 14.Eggers C, Arendt G, Hahn K, Husstedt IW, Mashchke M, Neuen-Jacob E, Obermann M, Rosenkranz T, Schielke E, Straube E. HIV-1-associated neurocognitive disorder: epidemiology, pathogenesis, diagnosis, and treatment. J Neurol. 2017 doi: 10.1007/s00415-017-8503-2. (epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004 Nov 19;306(5700):1380–3. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- 16.Ferenczy MW, Marshall LJ, Nelson CDS, Atwood WJ, Nath A, Khalili K, Major EO. Molecular biology, epidemiology, and pathogenesis of progressive multiple leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microb Rev. 2012;25:471–505. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51(2):458–469. doi: 10.1128/jvi.51.2.458-469.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gee GV, Tsomaia N, Mierke DF, Atwood WJ. Modeling a sialic acid binding pocket in the external loops of JC virus VP1. J Biol Chem. 2004;279:49172–49176. doi: 10.1074/jbc.M409326200. [DOI] [PubMed] [Google Scholar]
- 19.Gendelman H. Neural immunity: Friend or foe? Journal of Neurovirology. 2002;8(6):474–479. doi: 10.1080/13550280290168631. [DOI] [PubMed] [Google Scholar]
- 20.Gluska S, Zahavi EE, Chein M, Gradus T, Bauer A, Finke S, Perlson E. Rabies virus hijacks and accelerates the p75NTR retrograde axonal transport machinery. PLoS Pathogens. 2014;10(8):e1004348. doi: 10.1371/journal.ppat.1004348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemachudha T, Ugolini G, Wacharapluesadee S, Sungkarat W, Shuangshoti S, Laothamatas J. Human rabies: neuropathogenesis, diagnosis, and management. Lancet Neurology. 2013;12:498–513. doi: 10.1016/S1474-4422(13)70038-3. [DOI] [PubMed] [Google Scholar]
- 22.Holman RC, Janssen RS, Buehler JW, Zelasky MT, Hooper WC. Epidemiology of progressive multifocal leukoencephalopathy in the United States: analysis of national morality and AIDS surveillance data. Neurol. 1991;41:1733–1736. doi: 10.1212/wnl.41.11.1733. [DOI] [PubMed] [Google Scholar]
- 23.Houff SA, Major EO, Katz DA, Kufta CV, Sever JL, Pittaluga S, Roberts JR, Gitt J, Saini N, Lux W. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N Engl J Med. 1988;318:301–305. doi: 10.1056/NEJM198802043180507. [DOI] [PubMed] [Google Scholar]
- 24.Koralnik IJ. Overview of the cellular immunity against JC virus in progressive multifocal leukoencephalopathy. J Neurovirol. 2002;8:59–65. doi: 10.1080/13550280290167894. [DOI] [PubMed] [Google Scholar]
- 25.Jelcic I, Jelcic I, Kempf C, Largey F, Planas R, Schippling S, Budka H, Sospedra M, Martin R. Mechanisms of immune escape in central nervous system infection with neurotropic JC virus variant. Ann Neurol. 2016;79(3):404–18. doi: 10.1002/ana.24574. [DOI] [PubMed] [Google Scholar]
- 26.Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- 27.Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol. 1996;70:7004–7012. doi: 10.1128/jvi.70.10.7004-7012.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monaco MC, Shin J, Major EO. JC virus infection in cells from lymphoid tissue. Dev Biol Stand. 1998;94:115–122. [PubMed] [Google Scholar]
- 29.Okada Y, Endo S, Takahashi H, Sawa H, Umemura T, Nagashima K. Distribution and function of JCV agnoprotein. Journal Neurovirology. 2001;7:302–306. doi: 10.1080/13550280152537148. [DOI] [PubMed] [Google Scholar]
- 30.Okada Y, Sawa H, Endo S, Orba Y, Umemura T, Nishihara H, Stan AC, Tanaka S, Takahashi H, Nagashima K. Expression of JC virus agnoprotein in progressive multifocal leukoencephalopathy brain. Acta Neuropathol. 2002;104:130–136. doi: 10.1007/s00401-002-0526-8. [DOI] [PubMed] [Google Scholar]
- 31.Otlu O, De-Simone FI, Otalora YL, Khalili K, Sariyer IK. The agnoprotein of polyomavirus JC is released by infected cells: evidence for its cellular uptake by uninfected neighboring cells. Virology. 2014;0:88–95. doi: 10.1016/j.virol.2014.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richardson EP., Jr Progressive Multifocal Leukoencephalopathy. N Engl J Med. 1961;265:815–823. doi: 10.1056/NEJM196110262651701. [DOI] [PubMed] [Google Scholar]
- 33.San-Andreas FJ, Rubio R, Castilla J, Pulido F, Palao G, de Pedro I, Costa J, del Palacia A. Incidence of acquired immunodeficiency syndrome-associated opportunistic diseases and the effect of treatment on a cohort of 1115 patients infected with human immunodeficiency virus. Clin Infect Dis. 2003;36:1177–1185. doi: 10.1086/374358. [DOI] [PubMed] [Google Scholar]
- 34.SantaCruz KS, Roy G, Spigel J, Bearer EL. Neuropathology of JC virus infection in progressive multifocal leukoencephalopathy in remission. World J Virol. 2016;12(5(1)):31–7. doi: 10.5501/wjv.v5.i1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saribas AS, Arachea BT, White MK, Viola RE, Safak M. Human polyomavirus JC small regulatory agnoprotein forms highly stable dimers and oligomers: implications for their roles in agnoprotein function. Virology. 2011;420:51–65. doi: 10.1016/j.virol.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saribas AS, White MK, Safak M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: evidence for its involvement in viral DNA replication. Virology. 2012;433:12–26. doi: 10.1016/j.virol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saribas AS, Coric P, Hamazaspyan A, Davis W, Axman R, White MK, Abou-Gharbia M, Childers W, Condra JH, Bouaziz S, Safak M. Emerging from the unknown: structural and functional features of agnoprotein of polyomaviruses. J Cell Physiol. 2016;231(10):2115–2127. doi: 10.1002/jcp.25329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sariyer IK, Khalili K. Regulation of human neurotropic JC virus replication by alternative splicing factor SF2/ASF in glial cells. PLoS ONE. 2010;6(1):e14630. doi: 10.1371/journal.pone.0014630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sariyer IK, Saribas AS, White MK, Safak M. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J. 2011;8:255. doi: 10.1186/1743-422X-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott TP, Nel LH. Subversion of the immune response by rabies virus. Viruses. 2016;8(3) doi: 10.3390/v8080231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Srithayakumar V, Sribalachandran H, Rosatte R, Nadin-Davis SA, Kyle CJ. Innate immune responses in raccoons after raccoon rabies virus infection. J Gen Virol. 2014;95:16–25. doi: 10.1099/vir.0.053942-0. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, Nagashima K, Hall WW, Sawa H. The Human Polyoma JC Virus Agnoprotein Acts as a Viroporin. PLoS Pathogens. 2010;6(3):e1000801. doi: 10.1371/journal.ppat.1000801. http://doi.org/10.1371/journal.ppat.1000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki T, Semba S, Sunden Y, Orba Y, Kobayashi S, Nagashima K, Kimura T, Hasegawa H, Sawa H. Role of JC virus agnoprotein in virion formation. Microbiol Immunol. 2012;56:639–646. doi: 10.1111/j.1348-0421.2012.00484.x. [DOI] [PubMed] [Google Scholar]
- 44.Tavis JE, Trowbridge PW, Frisque RJ. Converting the JCV T antigen Rb binding domain to that of SV40 does not alter JCV’s limited transforming activity but does eliminate viral viability. Virology. 1994;199:384–392. doi: 10.1006/viro.1994.1136. [DOI] [PubMed] [Google Scholar]
- 45.Trowbridge PW, Frisque RJ. Identification of three new JC virus proteins generated by altnerative splicing of the early viral mRNA. Neurovirol. 1995;1:195–206. doi: 10.3109/13550289509113966. [DOI] [PubMed] [Google Scholar]
- 46.von Einsiedel RW, Fife TD, Aksamit AJ, Cornford ME, Secor DL, Tomiyasu U, Itabashi HH, Vinters HV. Progressive multifocal leukoencephalopathy in AIDS: a clinicopathologic study and review of the literature. J Neurol. 1993;240(7):391–406. doi: 10.1007/BF00867351. [DOI] [PubMed] [Google Scholar]
- 47.Weber F, Goldmann C, Krämer M, Kaup FJ, Pickhardt M, Young P, Petry H, Weber T, Lüke W. Cellular and humoral immune response in progressive multifocal leukoencephalopathy. Ann Neurol. 2001;49:636–642. [PubMed] [Google Scholar]
- 48.White MK, Khalili K. Interaction of retinoblastoma protein family members with large T-antigen of primate polyomaviruses. Oncogene. 2006;25:5286–5293. doi: 10.1038/sj.onc.1209618. [DOI] [PubMed] [Google Scholar]