

Abstract
Background
Apamin-sensitive small conductance calcium-activated K current (IKAS) is upregulated during ventricular pacing and masks short-term cardiac memory (CM).
Objective
To determine the role of IKAS in long-term CM.
Methods
CM was created with 3-5 weeks of ventricular pacing and defined by a flat or inverted T-wave off pacing. Epicardial optical mapping was performed in both paced and normal ventricles. Action potential duration (APD80) was determined during RA pacing. Ventricular stability was tested before and after IKAS blockade. Four paced hearts and 4 normal hearts were used for western blotting and histology.
Results
There were no significant differences in either the echocardiographic parameters or in fibrosis levels between groups. Apamin induced more APD80 prolongation in CM than in normal ventricles (9.6% [8.8%-10.5%] vs 3.1% [1.9%-4.3%], p<0.001). Apamin significantly lengthend the APD80 in the CM model at late activation sites, indicating significant IKAS upregulation at those sites. The CM model also had altered Ca2+ handling as the 50% Ca2+ transient duration and amplitude were increased at distal sites compared to a proximal site (near the pacing site). After apamin, the CM model had increased VF inducibility (paced vs control, 33/40 (82.5%) vs 7/20 (35%) P<0.001), and longer VF durations (124 vs 26 seconds, P<0.001).
Conclusions
Chronic ventricular pacing increases Ca2+ transients at late activation sites which activates IKAS to maintain repolarization reserve. IKAS blockade increases VF vulnerability in chronically paced rabbit ventricles.
Keywords: cardiac memory, electrophysiology, ventricular fibrillation, ion channels
Introduction
Unnecessary right ventricular (RV) pacing may cause cardiomyopathy in patients with preserved left ventricular systolic function1 and increased morbidity and mortality in patients with reduced ejection fraction with implantable cardioverter-defibrillators.2 The mechanisms of the detrimental effects of ventricular pacing remain incompletely understood. Ventricular pacing is known to produce both short and long term cardiac memory (CM) characterized by T-wave changes associated with altered activation sequence.3 Recent studies have shown that ventricular pacing is associated with cytosolic calcium (Cai) accumulation at late activation sites.4 The increased Cai can cause significant changes of cardiac repolarization, including increased sodium-calcium exchange current (INCX) that tend to prolong the action potential duration (APD) and promote afterdepolarizations.5 On the other hand, the increased Cai could also activate apamin sensitive small conductance Ca2+ activated K (SK) current (IKAS), which helps to reduce the APD and preserve the repolarization reserve.6 Because of the presence of these two opposing effects, ventricular pacing can cause transient APD prolongation at late activation sites leading to a positive APD-activation time (AT) relationship (short term CM) within 5-min of pacing.7 Continued pacing for an hour or more can normalize the APD-AT relationship due to IKAS upregulation in late activation site.6 Therefore, short term CM is transient and is well compensated by the upregulation of IKAS. However, the mechanism of long term CM remains unclear. We hypothesize that IKAS is upregulated at late activation sites during chronic ventricular pacing in rabbit ventricles but is of insufficient magnitude to suppress the manifestation of CM. Apamin, which blocks IKAS, aggravates APD prolongation at the late activation sites, reduces repolarization reserve and increases ventricular vulnerability to fibrillation. The purpose of the present study was to test these hypotheses.
Methods
The study protocol was approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine and the Methodist Research Institute, Indianapolis, Indiana, and conformed to the regulations for humane care and treatment of animals established by the NIH. A total of 29 female New Zealand white rabbits (3.5-5.0 kg) were used in this study. Among them, 20 were used for chronically ventricular pacing and 9 normal rabbit hearts that were not paced were used as controls. A detailed methods section is included in the Online Supplement.
Pacing-Induced long-term CM and Optical Mapping
Ventricular epicardial pacing of either the right ventricle (n=9) or the left ventricle (n=11) was used to induce long-term CM.8 Ventricles were paced at 270 beats per min (bpm) for 3-5 weeks. Each week, the T-wave from ECG Lead II was assessed. Chronic CM was defined by either a flat or inverted T-wave (Figure 1). Echocardiography was performed both before the pacemaker implantation and at follow up. Optical mapping was performed on both paced hearts and normal hearts of both the RV and LV (Online Supplement Figure 1) as previously described.8 Excised hearts were Langendorff perfused at 25 to 35 mL/min with Tyrode’s solution and stained with Rhod-2 AM (1.48 μmol/L) for Cai and RH237 (10 μmol/L) for Vm mapping. Contractility was blocked with blebbistatin (15 – 20 μmol/L). To prevent induction of short term CM the hearts remained in normal sinus rhythm except for specific programmed stimulation during optical recordings.6
Figure 1.
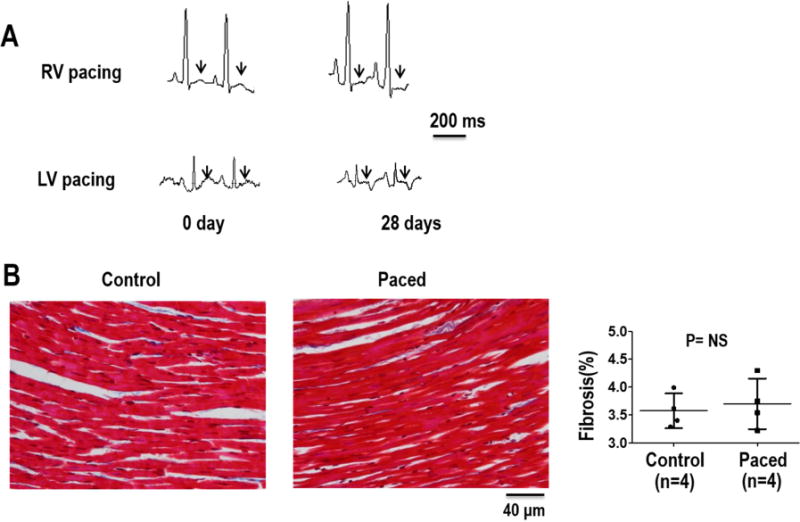
Evolution of CM over 28 days of ventricular pacing (VP). Depicted is lead II from a rabbit heart with RV pacing and with LV pacing (A) showing the T-wave at baseline, and a flat or inverted T-wave after pacing was discontinued on day 28. (B) Amount of fibrosis from Masson’s trichrome staining of ventricular tissue samples from structurally normal hearts (control) and chronically paced hearts. Summary data shows that there was no significant difference in fibrosis levels between the CM model and the control group.
Protocol I – Electrophysiological Effects of Cardiac Memory
The CM model was created in 7 hearts with RV pacing and 3 hearts with LV pacing to determine if LV pacing showed the same characteristics as RV paced hearts. Five normal hearts were used as controls. The ventricular APD80 was determined in each heart by pacing the right atrial appendage at 300 ms, and optical maps were acquired after at least 30 paced beats. To determine the role of IKAS, apamin (100 nmol/L) was then added to the perfusate and the protocol was repeated after 30 minutes. At that concentration, apamin is a highly specific IKAS blocker.9 After radiofrequency ablation of the atrioventricular (AV) node, pacing was performed from the RV apex. A S1/S2/S3 (short/long/short) pacing protocol (S1 30 beats with S1-S1 300 ms, a long S1-S2 of 1000 ms or 2000ms and a S2-S3 starting from 300 ms and gradually shortened to the ventricular effective refractory period (ERP)) was used to simulate the ECG characteristics that initiates early after-depolarizations (EADs) and torsades de pointes (TdP) ventricular tachycardia in the clinical setting.10 Finally, a dynamic ventricular pacing protocol was used to determine arrhythmia inducibility. If VF was not induced, burst pacing (PCL 50 ms, pacing duration 10 s) was used to induce VF. Any VF that occurred was allowed to continue for 180 s before a defibrillation attempt was made. After the return to sinus rhythm, the heart was allowed to rest for 1 min before continuing the pacing protocol. VF induction was attempted 4 times in each heart. Phase singularities (PS) were used to quantify VF characteristics.11
Protocol II – Effects of Cardiac Memory on Arrhythmia Inducibility
In protocol I, all arrhythmia inducibility data was acquired after apamin infusion. To determine the effects of CM on arrhythmia inducibility and the role of IKAS, 3 additional hearts with chronic LV pacing were used to acquire data on ventricular stability at baseline and after apamin infusion. In each heart, AV nodal ablation was performed, and to determine the inducibility of any EADs and VF, ventricular pacing was performed as described in protocol I. Any ventricular arrhythmias that resulted from the pacing were noted and then apamin (100 nmol/L) was added to the perfusate and the protocol was repeated after 30 min.
Western Blotting
Among the 3 different subtypes of SK channels, SK2 is known to be the most sensitive to apamin and contribute the most to IKAS.12 To determine the protein expression of SK2 channels within the myocardial tissue, Western blotting was performed on tissue acquired from sites distal and proximal to the chronic pacing site in 4 chronically paced hearts and from similar areas in 4 normal hearts.13
Histology
For histology, ventricular tissue from 4 normal hearts and 4 chronically paced hearts was embedded in paraffin, sectioned and stained using Masson’s trichrome stain. The collagen content was quantified by identifying and counting the number of blue-staining pixels as a percentage of the total tissue area using digital photomicrographs in Adobe Photoshop CS6 software.
Quantitative PCR
Quantitive polymerase chain reaction (PCR) was used to determine the expression of transcripts encoding kcnn2 (SK2) and kcnn3 (SK3) in tissue acquired from sites distal and proximal to the chronic pacing site in 3 chronically paced hearts.
Statistical Analysis
Data are presented as mean and 95% confidence interval (CI). Paired Student t tests were used to compare variables measured at baseline and after apamin infusion. An independent-sample t test was used to compare variables measured between groups. A one way ANOVA was used to compare SK2 protein expression between different groups, and a paired t-test was used to compared RNA levels between the proximal and distal sites. Categorical parameters between groups comparing VF vulnerability were compared by either a chi-square test or the Fisher exact test. A 2-sided P value of ≤0.05 was considered statistically significant.
Results
All rabbits survived the chronic pacing protocol. Long term CM was assessed by T-wave changes in the ECG (flat or inverted T-wave after pacing). As shown in Figure 1, after 28 days of ventricular pacing, the ECG shows a T-wave that changed to flat or inverted in both RV and LV paced hearts. There were no significant differences in echocardiographic based measurements of LV size and function between baseline and after chronic pacing. Ventricles demonstrated an LV end-diastolic dimension of 12.2 mm [95% CI, 11.3–13.3] vs 12.1 mm [95% CI,11.4–12.8], for 3-5 weeks pacing and baseline, respectively; P=0.649), end-systolic dimension (8.2 mm [95% CI, 7.3–9.1] vs 8.0 mm [95% CI, 7.4–8.7]; P=0.637), fractional shortening (32.9% [95% CI, 29.4–36.4] vs 32.4% [95% CI, 28.6–36.2]; P=0.81) and ejection fraction (69.3%[95% CI, 64.5–74.1] vs 69.6% [95% CI, 64.3–74.9] P=0.907). From histological analysis shown in Figure 1B, there was no difference in the amount of fibrosis between chronically paced hearts and normal hearts.
Effects of Apamin on the APD in CM
Figure 2A shows APD80 maps (measured during RA pacing at 300 ms PCL) before and after apamin infusion in normal ventricles, in RV paced ventricles and in LV paced ventricles studied in Protocol 1. Corresponding optical tracing of action potentials from a single representative pixel from the site marked with an “A” are also shown in the figure. At baseline, the APD80 distant from the pacing site had no or minimal prolongation while the APD80 closer to the pacing site was prolonged. After apamin, the APD80 was prolonged throughout the mapped region, indicating significant IKAS upregulation at sites remote from pacing (arrows) in both RV and LV paced ventricles. There was little or no APD80 prolongation in normal ventricles. Figures 2B–2D summarize the effects of chronic pacing on APD80 distribution at an atrial pacing cycle length (PCL) of 300 ms in all ventricles studied in Protocol I. As the summary data shows, apamin induced more APD80 prolongation in paced ventricles than in normal ventricles (paced vs Normal, 9.6% [8.8%-10.5%] vs 3.1% [1.9%-4.3%], p<0.001, Figure 2D), with no differences between RV and LV paced ventricles. APD heterogeneity has been shown to be an important factor that contributes to increased vulnerability to ventricular arrhythmias.14 Figure 2C shows that after apamin the standard deviation (SD) of APD80 significantly increased at 300 PCL in CM hearts, but not in normal hearts.
Figure 2. APD distribution before and after IKAS blockade by apamin.
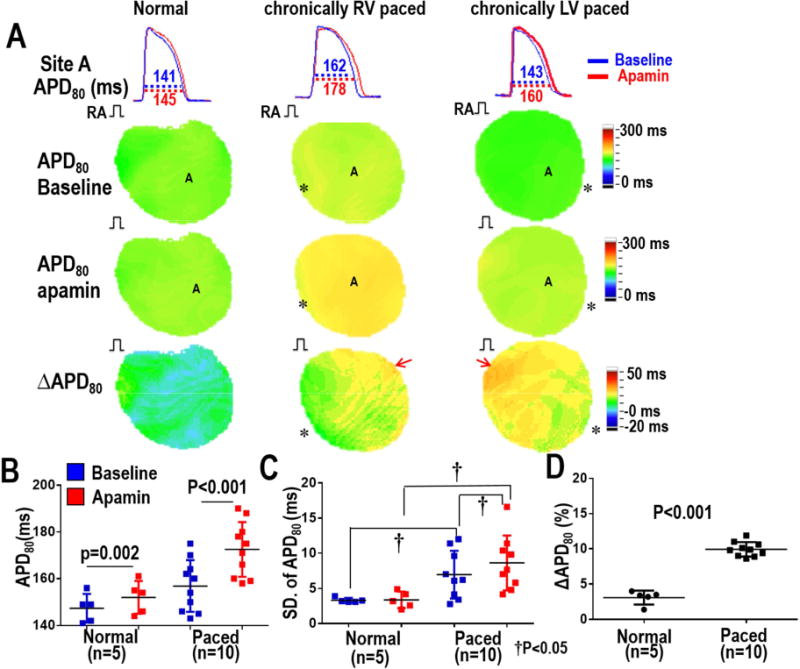
Optical Vm mapping was performed during RA pacing at a 300 ms PCL. A shows APD tracings (first row) from site A marked in the color maps below. Example APD80 maps are shown at baseline (second row), after apamin (100 nmol/L) infusion (third row) and the ΔAPD80 between baseline and apamin (4th row). Arrows point to a significant lengthening of the APD80 at the late activation site in both RV and LV paced ventricles. Asterisks indicate the sites of chronic pacing. B – Summary APD80 data showing that apamin prolonged the APD80 in normal and CM ventricles. C – Standard Deviation (SD) of APD80 is larger in CM ventricles compared to normal ventricles both before and after apamin infusion. Apamin significantly incresed the SD of APD80 in CM ventricles, but not in normal ventricles. D – APD80 was significantly prolonged after apamin infusion in CM ventricles compared to normal ventricles.
Characteristics of Ca2+ distribution in the CM model
We investigated the effects of the chronic pacing on the Ca2+ cycling characteristics by comparing the 50% Ca2+ transient duration (CaTD50). Sample Ca2+ signals from a normal heart and a paced heart are shown in figure 3. Overlapping the signals demonstrates that both RV and LV paced hearts have a longer CaTD50 than normal hearts. Summary data in panel C shows a significant increase in the CaTD50 in paced hearts compared to normal hearts. Ca2+ signals recorded with optical mapping also revealed a spatial gradient of the Ca2+ transients in the paced hearts. In both the RV and LV chronically paced hearts, Ca2+ fluorescence was increased at distal sites (site remote from the pacing site) compared to proximal sites (near the pacing site). However, this relationship did not exist in the normal ventricles. Sample normalized Ca2+ signals (F/F0) from both the proximal and distal locations are shown in Figure 4. For all chronically paced ventricles, the F/F0 measured was 1.070 (N=10, CI 1.052-1.088) near the chronic pacing site (early activation site), which was significantly lower than the F/F0 (1.103, N=10, CI 1.084-1.123, P<0.001) measured at the opposite site of the ventricle (late activation sites).
Figure 3. CaTD50 changes in normal and chronically paced hearts.
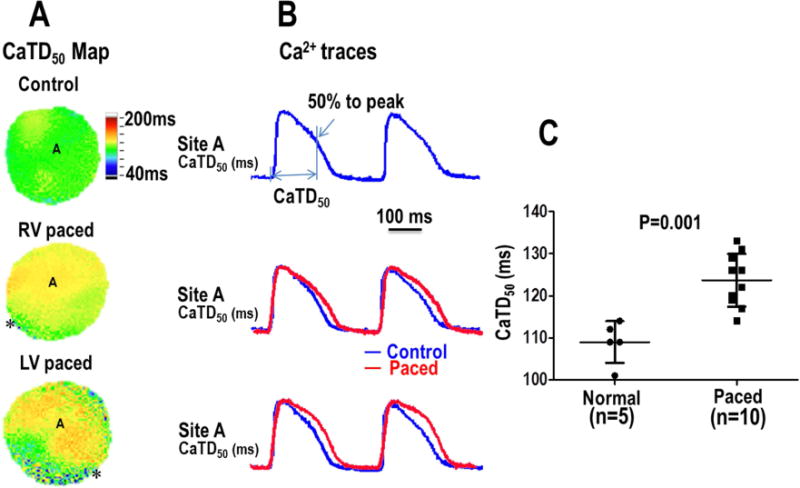
A – CaTD50 maps from optical mapping of rabbit hearts from both control and paced groups. Maps were recorded during atrial pacing (PCL of 300ms). Asterisks indicate the site of chronic ventricular pacing prior to explantation of the heart. B – Example Cai transient recordings show that the decay was prolonged in the CM model. Summary data is shown in Panel C.
Figure 4.
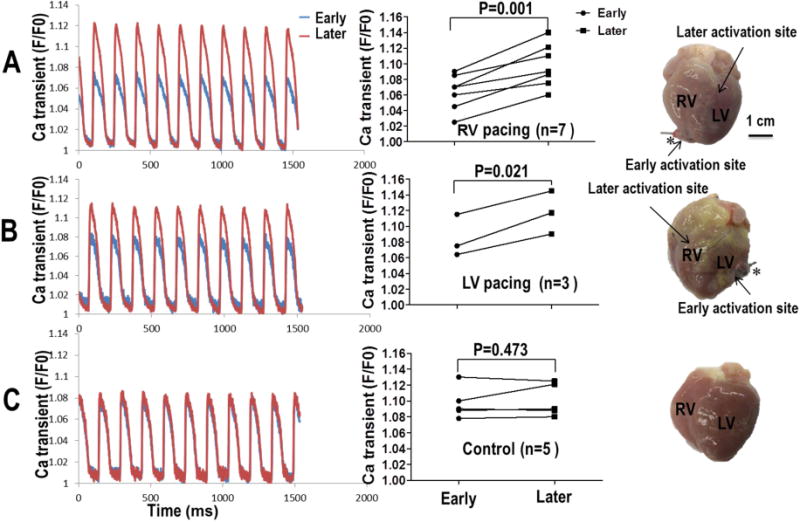
Effects of chronic pacing on Cai fluorescence transients in the CM model (A – RV pacing, B – LV pacing) as compared to control (C). All optical tracings were obtained during atrial pacing. Asterisks indicate the site of chronic ventricular pacing prior to explantation of the heart. Ca2+ fluorescence levels were increased at late activation sites compared to early activation sites near the location of chronic pacing in CM model (Panels A and B). However, the same was not observed in Panel C (normal control).
Chronic ventricular pacing is proarrhythmic
In protocol I, a dynamic ventricular pacing protocol was used to determine arrhythmia inducibility between normal and paced hearts. For all hearts studied, after apamin, paced ventricles developed APD alternans (Figure 5A) at a longer PCL than the control group (232ms [CI, 194–270] vs 180 [95% CI, 160–200 P=0.006] (Figure 5B). No spontaneous arrhythmias were observed during the study. In 10 paced hearts and 5 normal hearts, VF induction was attempted in each heart 4 times. VF vulnerability increased (paced vs control, 33/40(82.5%) vs 7/20(35%) P<0.001) (Figure 5C), with a longer duration in paced hearts compared to control hearts (124 vs 26 seconds, P<0.001) (Fig 5D).
Figure 5. Ventricular vulnerability to fibrillation after IKAS blockade in normal and chronically paced ventricles (Protocol I).
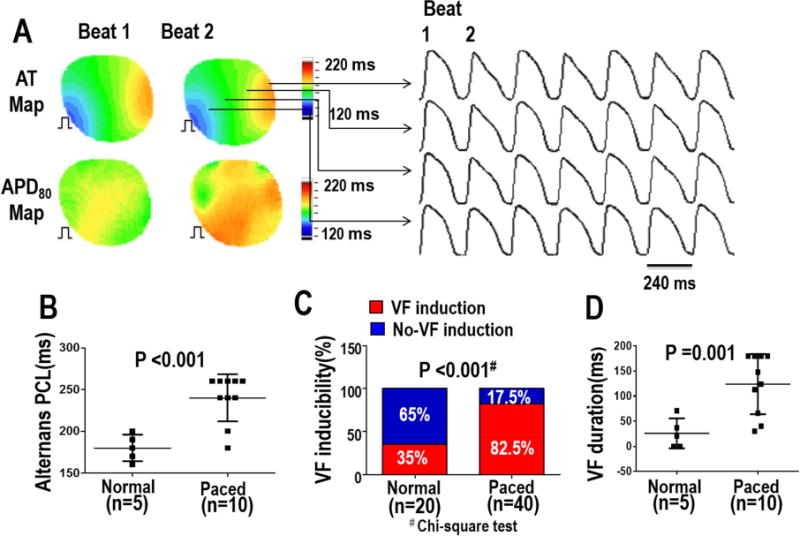
A - Representative traces of Vm alternans in the CM model after apamin infusion. Corresponding AT and APD maps from two consecutive ventricular activations are shown. B - Apamin prolonged the PCL threshold of 2:2 alternans. C - VF inducibility was tested 4 times in each heart. VF was able to be induced in all 10 paced ventricles, and in 3 of the 5 normal ventricles. D - After apamin infusion, VF duration was longer in chronic paced hearts compared to control hearts. The pacing sites during the mapping study were the same as that during chronic ventricular pacing
 . AT – activation time.
. AT – activation time.
In protocol II, 3 additional hearts were studied to investigate ventricular vulnerability to fibrillation at baseline and after apamin infusion. Figure 6A shows representative examples of optical mapping traces at baseline and after addition of apamin demonstrating a significant prolongation of the APD80 at a PCL of 1000 ms after apamin infusion. At baseline, only one of the three paced hearts developed a TdP arrhythmia. However, after apamin infusion, all of the three paced hearts developed a TdP arrhythmia during short-long-short pacing. An example of TdP induced with short-long-short pacing is shown in Figure 6B. In addition, the site of earliest activation of the TdP occurred at the sites farthest from the chronic pacing site (Figure 6C). In these 3 CM hearts, there were 5 VF episodes at baseline and 11 episodes after apamin infusion. Examples of phase maps during VF with marked phase singularities (black arrows) at baseline and after apamin infusion is shown in Figure 7A and 7B. Figure 7C summarizes the number of phase singularities in all hearts studied in Protocol II. There was a significant increase of phase singularities after apamin.
Figure 6. Optical mapping of pacing-induced Tdp in Protocol II (AV block).
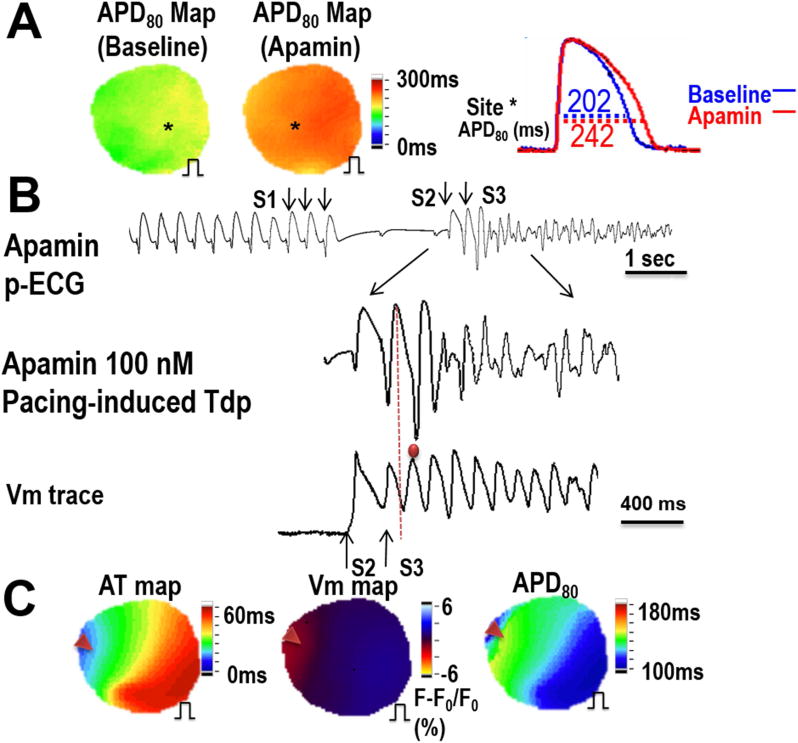
A – APD80 maps and example signals at baseline and after apamin infusion in the CM model. Shown is that the APD is significantly prolonged at a PCL of 1000 ms. B - EAD beat induced TdP during short-long-short pacing after apamin infusion (300-2000-220ms). C - The earliest activation site of an TdP occurred at a site distal from the chronic pacing site. Red arrows point to the origin of the TdP. An AT map is shown along with a fluorescence map which shows an area with light red color (Vm gradient change), which is the earliest activation site of the earliest TdP beat. An APD80 map shows that the earliest activation site correlated to the longest APD. The pacing during optical mapping was performed from the same site as during chronic LV pacing
 .
.
Figure 7.
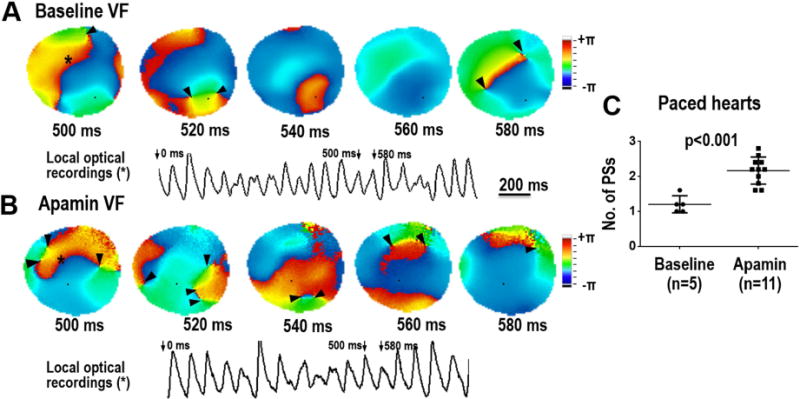
Effect of IKAS blockade on the wave breaks during VF in the CM model. Panels A and B show consecutive phase maps sampled at 20 ms during VF at baseline and after apamin infusion. Phase singularities (PS) are indicated by black arrowheads. A corresponding optical recording of VF from the site indicated by the asterisk is also shown. Note that the numbers of PS were significantly increased after apamin infusion. Summary data is shown in Panel C.
SK2 Protein and RNA Expression
Western blotting was performed in 4 normal hearts and 4 chronically paced hearts (2 RV pacing and 2 LV pacing). As the results in Figure 8 show, there was no significant difference in the SK2 protein expression between normal and chronically paced hearts. However, qPCR analysis showed that there was a significant increase in both SK2 (0.20 [95% CI, 0.08-0.31] vs. 0.27 [95% CI, 0.21-0.32], p=0.048) and SK3 (0.21 [95% CI, 0.13-0.28] vs. 0.24 [95% CI, 0.13-0.34], p=0.045) levels at the distal site compared to the proximal site.
Figure 8.
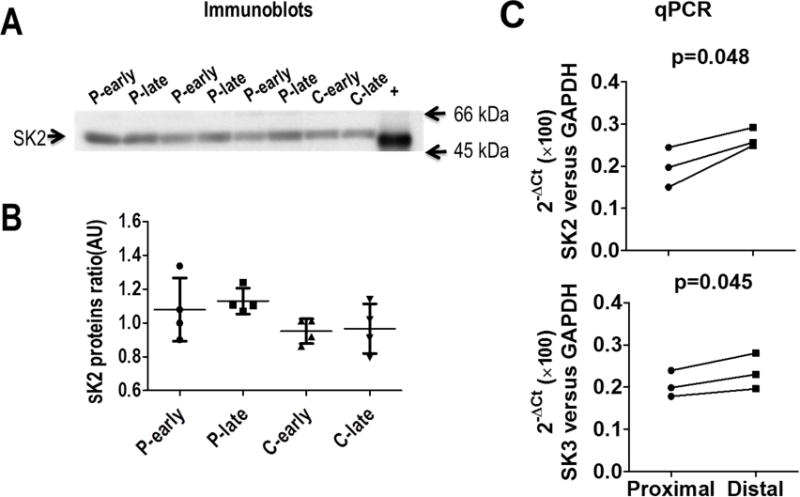
Protein and mRNA analyses. (A) Western blot analyses of the SK2 protein levels from chronically paced ventricles and in normal controls. (B) There was no significant difference in the SK2 protein levels in paced ventricles compared to normal ventricles or between proximal and distal sites. (C) RNA levels show a significant increase in SK2 and SK3 expression at the distal site compared to the proximal site.
Discussion
This study has shown that CM develops with persistent ventricular pacing, which results in a repolarization gradient across the epicardial surface during atrial pacing, with the longest APD80 near the chronic pacing site and the shortest APD80 at the distal sites within the mapping field of view. Infusion of IKAS inhibitor apamin significantly lengthened the APD80 with the greatest increase of APD80 at the locations distal from the chronic pacing site. In addition, an increase in the Ca2+ transients was also observed at these distal sites. Putting this information together, chronic pacing resulted in Cai accumulation and a robust activation of IKAS at late activation sites. Because of the increased IKAS, the late activation sites had a shorter APD than the early activation sites. This abnormal distribution of APD persisted when ventricular pacing was turned off, resulting in longer APD80 at the apical pacing sites than at the base. This abnormal distribution of APD80 results in abnormal patterns of T-waves during sinus or atrially paced rhythms (chronic CM).
IKAS blockade and ventricular vulnerability to fibrillation
In paced ventricles, inhibiting IKAS increased VF vulnerability and the incidence of TdP induced with EADs. These data support that IKAS plays a significant compensatory role in CM and protects against arrhythmia initiation. Several commonly used drugs, including amiodarone15 and ondansetron,16 are known to block IKAS. Whether or not IKAS inhibition increases arrhythmia risk for pacemaker patients deserves further investigation.
Mechanisms of cardiac memory
CM is characterized by a change in T-wave morphology resulting from either electrophysiological or structural remodeling induced by either ventricular pacing or myocardial stretch.17, 18 Varying degrees of CM has been shown after minutes to hours of pacing (producing “short-term” CM) and after 2 to 5 weeks of pacing (“long-term” CM). Two weeks of pacing is required to achieve a steady-state change in the T-wave. Molecular mechanisms of CM have been extensively reviewed recently.19,20 Short-term CM that is observed within minutes of ventricular pacing is thought to occur from modulation and modification of angiotensin II which has been shown to regulate Ito. Long-term CM, seen after longer periods of ventricular pacing and is longer lasting (days to weeks), includes alterations in AT1 receptors, stretch-activated receptors,21, 22 connexins23, 24 and in ion channels specifically Ito, Ikr, and ICa,L which results in an altered repolarization gradient. The mechanisms underlying this ion channel remodeling revolves around changes in gene transcription and protein synthesis.19,20 This study is the first to show that in addition to the currents mentioned above, IKAS may also play a role in long-term CM.
Clinical Implications
This study provided new insights into the mechanisms of CM. The importance of IKAS in human ventricular repolarization is supported by a previous study that showed apamin prolonged the APD in the failing human ventricular myocardium.25 In CM models, we have observed an increased APD heterogeneity, steepened maximal slope of APDR, increased PCL threshold for alternans and increased spatially discordant APD alternans after IKAS inhibition, factors which increase vulnerability to ventricular arrhythmias. These findings suggest drugs that block IKAS may be unsafe for patients with chronic ventricular pacing. A second clinical implication is that IKAS upregulation helps to shorten the APD80 and counteract the detrimental effects of Ca2+ accumulation at sites distal from pacing. Therefore, only a minority of patients develop pacing-induced cardiomyopathy after prolonged periods of RV pacing.1
Study Limitations
We only studied the acute effects of apamin in an ex vivo perfused heart model. We did not investigate the chronic administration of a SK channel blocker in an ambulatory cardiac memory model. In addition, we did not investigate SK1 or SK3 protein levels as a reliable antibody is not available for rabbit hearts.
Conclusions
Chronic ventricular pacing increases Cai at late activation sites which helps to activate IKAS to shorten the APD and maintain repolarization reserve. Inhibiting IKAS significantly lengthens the APD at late activation sites and increases VF vulnerability. IKAS blockade may contribute to the underlying mechanism of sudden cardiac death in patients with permanent ventricular pacing.
Supplementary Material
Acknowledgments
We thank Nicole Courtney, Jessica Warfel, Glen Schmeisser for their assistance.
Sources of Funding
This study was supported in part by National Institutes of Health grants P01 HL78931, R01 HL71140, R41 HL124741 and R42 DA043391, a Medtronic-Zipes Endowment, the Charles Fisch Cardiovascular Research Award endowed by Dr Suzanne B. Knoebel of the Krannert Institute of Cardiology, and the Indiana University Health-Indiana University School of Medicine Strategic Research Initiative.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- 1.Khurshid S, Epstein AE, Verdino RJ, Lin D, Goldberg LR, Marchlinski FE, Frankel DS. Incidence and predictors of right ventricular pacing-induced cardiomyopathy. Heart Rhythm. 2014 Sep;11:1619–1625. doi: 10.1016/j.hrthm.2014.05.040. [DOI] [PubMed] [Google Scholar]
- 2.Wilkoff BL, Cook JR, Epstein AE, Greene HL, Hallstrom AP, Hsia H, Kutalek SP, Sharma A, Dual C, Investigators VVIIDT Dual-chamber pacing or ventricular backup pacing in patients with an implantable defibrillator: the Dual Chamber and VVI Implantable Defibrillator (DAVID) Trial. JAMA. 2002 Dec 25;288:3115–3123. doi: 10.1001/jama.288.24.3115. [DOI] [PubMed] [Google Scholar]
- 3.Rosenbaum MB, Blanco HH, Elizari MV, Lazzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol. 1982 Aug;50:213–222. doi: 10.1016/0002-9149(82)90169-2. [DOI] [PubMed] [Google Scholar]
- 4.Jeyaraj D, Wan X, Ficker E, Stelzer JE, Deschenes I, Liu H, Wilson LD, Decker KF, Said TH, Jain MK, Rudy Y, Rosenbaum DS. Ionic bases for electrical remodeling of the canine cardiac ventricle. Am J Physiol Heart Circ Physiol. 2013 Aug 1;305:H410–419. doi: 10.1152/ajpheart.00213.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM, Chen-Izu Y. Sodium and calcium regulation in cardiac myocytes: from molecules to heart failure and arrhythmia. The Journal of physiology. 2015 Mar 15;593:1327–1329. doi: 10.1113/JP270133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan Y-H, Tsai W-C, Ko J-S, Yin D, Chang P-C, Rubart M, Weiss JN, Everett T, Lin S-F, Chen P-S. Small conductance calcium-activated potassium current is activated during hypokalemia and masks short term cardiac memory induced by ventricular pacing. Circulation. 2015;132:1377–1386. doi: 10.1161/CIRCULATIONAHA.114.015125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costard-Jackle A, Goetsch B, Antz M, Franz MR. Slow and long-lasting modulation of myocardial repolarization produced by ectopic activation in isolated rabbit hearts. Evidence for cardiac “memory”. Circulation. 1989 Nov;80:1412–1420. doi: 10.1161/01.cir.80.5.1412. [DOI] [PubMed] [Google Scholar]
- 8.Chua SK, Chang PC, Maruyama M, et al. Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res. 2011;108:971–979. doi: 10.1161/CIRCRESAHA.110.238386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu CC, Ai T, Weiss JN, Chen PS. Apamin does not inhibit human cardiac Na+ current, L-type Ca2+ current or other major K+ currents. PLoS One. 2014;9:e96691. doi: 10.1371/journal.pone.0096691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang PC, Turker I, Lopshire JC, Masroor S, Nguyen BL, Tao W, Rubart M, Chen PS, Chen Z, Ai T. Heterogeneous upregulation of apamin-sensitive potassium currents in failing human ventricles. J Am Heart Assoc. 2013 Feb;2:e004713. doi: 10.1161/JAHA.112.004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valderrabano M, Chen PS, Lin SF. Spatial Distribution of Phase Singularities in Ventricular Fibrillation. Circulation. 2003;108:354–359. doi: 10.1161/01.CIR.0000080322.67408.B4. [DOI] [PubMed] [Google Scholar]
- 12.Weatherall KL, Seutin V, Liegeois JF, Marrion NV. Crucial role of a shared extracellular loop in apamin sensitivity and maintenance of pore shape of small-conductance calcium-activated potassium (SK) channels. Proc Natl Acad Sci U S A. 2011 Nov 8;108:18494–18499. doi: 10.1073/pnas.1110724108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh YC, Chang PC, Hsueh CH, Lee YS, Shen C, Weiss JN, Chen Z, Ai T, Lin SF, Chen PS. Apamin-sensitive potassium current modulates action potential duration restitution and arrhythmogenesis of failing rabbit ventricles. Circ Arrhythm Electrophysiol. 2013 Apr;6:410–418. doi: 10.1161/CIRCEP.111.000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robert E, Aya AG, de la Coussaye JE, Peray P, Juan JM, Brugada J, Davy JM, Eledjam JJ. Dispersion-based reentry: mechanism of initiation of ventricular tachycardia in isolated rabbit hearts. Am J Physiol. 1999 Feb;276:H413–423. doi: 10.1152/ajpheart.1999.276.2.H413. [DOI] [PubMed] [Google Scholar]
- 15.Turker I, Yu C-C, Chang P, Chen Z, Sohma Y, Lin S-F, Chen P-S, Ai T. Amiodarone Inhibits Apamin-Sensitive Potassium Currents. PLoS One. 2013;8:e70450. doi: 10.1371/journal.pone.0070450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko J-S, Hassel J, Celestino-Soper P, et al. The role of small conductance Ca activated K current in the genesis of drug-induced long QT syndrome. Heart Rhythm. 2015;12:S489. (abstract) [Google Scholar]
- 17.Shvilkin A, Danilo P, Jr, Wang J, Burkhoff D, Anyukhovsky EP, Sosunov EA, Hara M, Rosen MR. Evolution and resolution of long-term cardiac memory. Circulation. 1998;97:1810–1817. doi: 10.1161/01.cir.97.18.1810. [DOI] [PubMed] [Google Scholar]
- 18.Patberg KW, Shvilkin A, Plotnikov AN, Chandra P, Josephson ME, Rosen MR. Cardiac memory: mechanisms and clinical implications. Heart Rhythm. 2005 Dec;2:1376–1382. doi: 10.1016/j.hrthm.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 19.Ozgen N, Rosen MR. Cardiac memory: a work in progress. Heart Rhythm. 2009 Apr;6:564–570. doi: 10.1016/j.hrthm.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Jeyaraj D, Ashwath M, Rosenbaum DS. Pathophysiology and clinical implications of cardiac memory. Pacing Clin Electrophysiol. 2010 Mar;33:346–352. doi: 10.1111/j.1540-8159.2009.02630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeyaraj D, Wilson LD, Zhong J, Flask C, Saffitz JE, Deschenes I, Yu X, Rosenbaum DS. Mechanoelectrical feedback as novel mechanism of cardiac electrical remodeling. Circulation. 2007 Jun 26;115:3145–3155. doi: 10.1161/CIRCULATIONAHA.107.688317. [DOI] [PubMed] [Google Scholar]
- 22.Kooshkabadi M, Whalen P, Yoo D, Langberg J. Stretch-activated receptors mediate cardiac memory. Pacing Clin Electrophysiol. 2009 Mar;32:330–335. doi: 10.1111/j.1540-8159.2008.02240.x. [DOI] [PubMed] [Google Scholar]
- 23.Spragg DD, Akar FG, Helm RH, Tunin RS, Tomaselli GF, Kass DA. Abnormal conduction and repolarization in late-activated myocardium of dyssynchronously contracting hearts. Cardiovasc Res. 2005 Jul 1;67:77–86. doi: 10.1016/j.cardiores.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Patel PM, Plotnikov A, Kanagaratnam P, Shvilkin A, Sheehan CT, Xiong W, Danilo P, Jr, Rosen MR, Peters NS. Altering ventricular activation remodels gap junction distribution in canine heart. J Cardiovasc Electrophysiol. 2001 May;12:570–577. doi: 10.1046/j.1540-8167.2001.00570.x. [DOI] [PubMed] [Google Scholar]
- 25.Yu CC, Corr C, Shen C, Shelton R, Yadava M, Rhea I, Straka S, Fishbein MC, Chen Z, Lin SF, Lopshire JC, Chen PS. Small conductance calcium-activated potassium current is important in transmural repolarization of failing human ventricles. Circ Arrhythm Electrophysiol. 2015 Apr 23;8:667–676. doi: 10.1161/CIRCEP.114.002296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.