

Abstract
After several decades of widespread use for mapping elemental ions and small molecular fragments in surface science, secondary ion mass spectrometry (SIMS) has emerged as a powerful analytical tool for molecular imaging in biology. SIMS has primarily been used as a qualitative technique; although the distribution of a single analyte can be accurately determined, it is difficult to map the absolute quantity of a compound, or even to compare the relative abundance of one molecular species to that of another. We describe a method for quantitative SIMS imaging of small molecules in agar-based microbial communities. The microbes are cultivated on a thin film of agar, dried under nitrogen, and imaged directly with SIMS. Using optical microscopy we show that the area of the agar is reduced by 26% ± 2% standard deviation during dehydration but the overall biofilm morphology and analyte distribution are largely retained. We detail a quantitative imaging methodology in which the ion intensity of each analyte is (1) normalized to an external quadratic regression curve, (2) corrected for isomeric interference, and (3) filtered for sample-specific noise and lower and upper limits of quantitation. The end result is a two-dimensional surface density image for each analyte. The sample preparation and quantitation methods are validated by quantitatively imaging four alkyl-quinolone and alkyl-quinoline N-oxide signaling molecules (including Pseudomonas quinolone signal) in Pseudomonas aeruginosa colony biofilms. We show that the relative surface densities of the target biomolecules are substantially different from values inferred through direct intensity comparison and that the developed methodologies can be used to quantitatively compare as many ions as there are available standards.
Keywords: Mass Spectrometry Imaging, Secondary Ion Mass Spectrometry Imaging, Quantitative MSI, Quinolones, PQS, HQNO, C9-PQS, NQNO, Biofilms, Pseudomonas aeruginosa
Graphical Abstract
Simultaneous improvements to primary ion sources and mass spectrometers have facilitated the emergence of secondary ion mass spectrometry (SIMS) as a viable tool for small-molecule imaging in biology.1 The introduction and wide-scale incorporation of polyatomic and cluster ion sources has improved ionization efficiencies for intact molecular ions and extended the upper mass detection limit above m/z 1000.2,3 Academic work4–6 inspired instrument manufacturers to incorporate tandem MS (MS2)-capable mass analyzers into commercial SIMS instruments,7,8 an essential development that enables the differentiation of isomers, which are pervasive in biological systems. Among other examples, biomolecular SIMS imaging has been applied to examine single cells,9–11 neurons,6,12,13 bacterial biofilms,14–20 and brain tissue from songbirds,7,21 rodents,22,23 and Drosophila.24–26
As evidenced by the 2016 announcement of the National Microbiome Initiative,27 the need for tools and workflows that reveal microbial chemistry is increasingly urgent.28 Characterizing the role of small molecules in bacterial life is essential for not only understanding the microorganisms themselves, but also the ecosystems that they inhabit and influence. In a process termed quorum sensing, collaborative communities of bacteria produce and sense small molecules, (e.g., acyl homoserine lactones, cyclic peptides, and quinolones) to communicate with one another and coordinate collective behavior. 29 Beyond signaling, bacterial communities utilize small molecules to cohesively modify their local environment, enhance nutrient uptake, colonize new territory, and defend themselves against competing organisms. Because of the simple sample preparation requirements, and an innate capability for two- and in some cases three-dimensional small molecule imaging, SIMS is wonderfully positioned to facilitate the exploration of microbial chemistry.
As discussed in a recent Account,30 the large variations in both chemical composition and surface architecture make microbial communities exceptionally challenging to study with mass spectrometry imaging (MSI). Further complexity arises from the cultivation requirements; biofilms and other microbial communities are often grown on semi-solid agar, which must be dehydrated prior to introduction to the vacuum environment of the instrument sample chamber. The agar dehydration process can introduce artifacts in biofilm morphology and chemical distribution. Although there have been several excellent applications of matrix-assisted laser desorption/ionization (MALDI)31–35 and desorption electrospray ionization (DESI)36– 38 to study agar-based microbial samples, to our knowledge there are no reports describing the direct application of SIMS imaging to microbial communities while they are still affixed to the agar substrate. Other successful examples of microbial SIMS imaging include analysis of imprint-transferred surfactants from Bacillus swarming communities grown on agar,18 imaging of quinolone signaling molecules and rhamnolipid surfactants produced by Pseudomonas aeruginosa biofilms grown on silicon wafers,14–17 in situ examination of liquid biofilms through an SiN membrane,20,39 and ultra-high-resolution nanoSIMS imaging of nitrogen fixation by cyanobacteria.40,41 Another example of note is the use of SIMS to image quinolones and xenobiotics in P. aeruginosa biofilms cultivated on indium-tin oxide-coated glass slides and inside of an ex vivo pig lung model.19
As with other MS sampling modalities, the information generated by SIMS depends on both the properties and settings of the instrument, as well as the molecular properties of the analyte, such as gas-phase basicity and size. Furthermore, despite substantial improvements in modern cluster and polyatomic ion sources, SIMS remains a highly energetic ionization process, leading to hard ionization that can fragment biomolecules into smaller ions prior to detection. These factors prohibit direct comparison of the distribution or intensity of one analyte to that of another. Therefore, most SIMS experiments generate qualitative rather than quantitative information.
Several clever methods for quantitative MSI have been demonstrated. These include the use of a mimetic tissue model for brain imaging with MALDI,42 the addition of a standard into the DESI solvent,43 and the metabolic incorporation of isotopically labeled internal standards into bacterial and mammalian cells.40,44 Application of an isotopically labeled internal standard on top or beneath a sample has also been successfully employed for quantitative MALDI imaging of tissue.45,46 In comparison to other ionization modalities, the challenges associated with quantitative MSI are even more prominent for SIMS. In static SIMS, the primary ion beam only accesses the first few molecular layers of the sample, therefore an exogenously applied external standard is liable to influence or obscure the native surface composition.
Here we develop a workflow for quantitative SIMS imaging with a focus on 2-alkyl-4(1H)-quinolone (AQ) and 2-alkyl-4- hydroxyquinoline N-oxide (AQNO) structural isomers in static P. aeruginosa colony biofilms. In a process similar to the initial steps demonstrated for DESI37 and MALDI,34 we cultivate the microbial communities on thin agar and dry them under a gentle stream of nitrogen. The microbial samples, and a series of external calibration samples, are examined using SIMS product ion imaging. Quadratic calibration with an adjustment for common (interfering) fragmentation provides the surface density of each analyte on a pixel-by-pixel basis, enabling quantitative comparisons within and between samples. Our results indicate that the described methodology for quantitative SIMS imaging could be extended to examine small molecules in biological samples of diverse composition and origin.
EXPERIMENTAL SECTION
Materials and Reagents
LC-MS grade methanol, ethanol, fluorescein sodium salt, HHQ (2-heptyl-4-quinolone), and PQS (“Pseudomonas quinolone signal”; 2-heptyl-3-hydroxy- 4(1H)-quinolone) were purchased from Sigma-Aldrich (St Louis, MO). HQNO (2-heptyl-4-hydroxyquinoline N-oxide), NQNO (2-nonyl-4-hydroxyquinoline N-oxide), NHQ (2- nonyl-4-quinolone), and C9-PQS (2-nonyl-3-hydroxy-4(1H)-quinolone) were purchased from Cayman Chemical (Ann Arbor, MI). All standards were acquired at 96% purity or better and used without further purification.
Custom aluminum SIMS sample plates were machined by the expert craftsman in the University of Illinois School of Chemical Sciences’ Machine Shop. Double sided conductive copper tape, 3M 1182, was purchased from Ted Pella, Inc. (Redding, CA) and Structure Probe, Inc. (West Chester, PA).
Colony Biofilm Cultivation
Two P. aeruginosa strains, PAO1C (“wild-type” lab strain) and FRD1 (alginate overproducing cystic fibrosis lung isolate),47 were used in this study. Cell cultures were grown for 18 h at 37 °C with shaking at 240 rpm in modified fastidious anaerobe broth (FAB) culture medium supplemented with 30 mM filter-sterilized glucose as the source of carbon. Growth plates were prepared by aliquoting 7.5 mL of sterile FAB-noble agar (1% agar supplemented with 12 mM glucose) into 60 mm diameter Petri dishes. The agar plates were inoculated with 5 μL cell culture (OD600 = 1) and incubated at 37 °C for 72 h. More than 25 single and co-culture colony biofilms were used for optimization and verification of the methods developed here. Culture handling and manipulation were performed in accordance with institutional and CDC laboratory and biological safety guidelines,48 and included the use of aseptic techniques, personal protective equipment, and disinfection and sterilization procedures specific to the biosafety level of the organism.
Optical Imaging and Analysis
Macroscopic optical images of the hydrated and dried samples were obtained using a Nikon D3300 camera equipped with a Nikon AF-S DX NIKKOR lens (18–55 mm 1:3.5–5.6 VR II, ∞-0.28m/0.92ft ø52) (Nikon, Melville, NY). Images were processed using either MATLAB (MathWorks, Natick, MA) or ImageJ (64 bit) as previously described.49 Microscopy images of AQ aggregates within the colony biofilms were acquired using a Nikon Eclipse 90i confocal microscope equipped with a 10× objective, and the images processed with the NIS-Elements AR Imaging Software (Nikon).
Chemical Printing
Two pooled solutions were prepared in 50% ethanol (50% water, v/v) containing equimolar quantities of HHQ, PQS, and NQNO, or NHQ, C9-PQS, and HQNO. Direct dilutions were made into 50% ethanol to create a series of nine of each of the two pooled solutions ranging in concentration from 1.0–200 μM. The solutions were deposited on dry FAB agar using a chemical inkjet printer (CHIP 1000, Shimadzu Corp., Kyoto, Japan) in multi-pass iterative mode. Before printing and between solutions, the CHIP was rinsed with 250 μL isopropyl alcohol followed by 250 μL 50% ethanol. Dwell voltages and dwell times were initially set to 25 V and 35 μs, respectively, and adjusted as needed throughout printing to obtain uniform droplets. Each deposition area was constructed with two parallel lines containing 10 spots each, with an intraspot spacing of 200 μm, to form uniform regions of approximately 2.2 mm × 0.6 mm. Four iterative passes of 500 pL per droplet (2 s pause between each pass) resulted in a total deposition quantity of 40–8000 fmol of each analyte at a surface density range of 3.0–610 pmol/cm2.
For confocal fluorescence microscopy control experiments, solutions containing either 20 μM fluorescein or 20 μM fluorescein, and 100 μM each of HHQ, PQS, and NQNO, were deposited onto agar using the CHIP 1000 in spot mode. Each deposition spot was composed of 10, 100 pL drops for a total deposition quantity of 20 fmol fluorescein and 100 fmol HHQ, PQS, and NQNO. The dwell voltage and dwell time were set to 35 V and 35 μs respectively. Prior to solution deposition, the agar was prepared by dehydrating 1 cm × 1 cm squares on glass microscopy slides.
Confocal Fluorescence Microscopy
Fluorescein and AQ spots were examined using a Leica SP8 fluorescence confocal microscope (Buffalo Grove, IL) with a 10× dry objective (NA = 0.4). Both transmission and fluorescence channels were used with a Z-stacking range confined to the observable fluorescence signal. A stage step-size of 2.41 μm was used for the Zstacking, and the field of view was set to 512 × 512 pixels. The Ar laser excitation wavelength was 488 nm and emission was collected from 494–618 nm. The resulting data were processed in ImageJ using the Bio Formats toolbox.50,51 Fluorescent spots were selected with the ROI selection tool and the average intensity values of each Z-stack were exported into MATLAB for further analysis.
Scanning Electron Microscopy (SEM)
Dehydrated samples were sputter coated in Au/Pd using a Denton Vacuum Desk II (Moorestown, NJ) operated with 64 mTorr Ar and 40 mA current for 70 s. SEM was performed on dehydrated samples using a Philips XL30 ESEM-FEG (Hillsboro, OR) operated under high-vacuum conditions at 5 kV and an approximate working distance of 10 mm.
SIMS Imaging
Mass spectrometric measurements were performed with a modified quadrupole time-of-flight mass spectrometer, described in detail elsewhere.6 The instrument is a QSTAR XL (AB SCIEX, Framingham, MA) equipped with a Buckminsterfullerene (C60) ion source (Ionoptika Ltd., Hampshire, UK) for SIMS. The ion source was adjusted to produce a continuous beam of 20 keV C60 + primary ions at a 35 μm spot size with 500 pA sample current. The approximate primary ion dose was held to 5 × 1013 ions/cm2, which exceeds the static limit of 1 × 1013 primary ions/cm2, and represents an estimated sampling depth of 40 nm based on measurements from 20 keV C60 2+ bombardment of HeLa cells.9 SIMS sputtering rates depend on both chemical composition52 and surface morphology,53 and biofilms are both morphologically and chemically heterogeneous. Accordingly, the actual sampling depth will fluctuate from pixel-to-pixel.
Each sample was raster imaged three times with a 25 μm vertical offset between images to present a predominantly unperturbed sampling region for each acquisition. The first image produced secondary ions from m/z 60–850 with a pixel size of 100 μm × 100 μm and a sputtering time of 500 ms/pixel. The quadrupole bias was set to 15%, 25%, and 60% at m/z 100, 200, and 400, respectively. The second and third images were collected in product ion mode with a unit resolution precursor mass transmission for product of m/z 288 and m/z 260, 30 eV argon collision induced dissociation (CID), and “Enhance All” mode activated to selectively enhance transmission of product ions in the low mass range. The quadrupole bias was set to 5%, 47.5%, and 47.5% at m/z 40, 90, and 180, respectively; the pixel size was 200 μm × 200 μm, and the sputtering time was 1 s/pixel.
SIMS Data Analysis
AB SCIEX wiff data files produced by Analyst v1.2 and oMALDI Server v5.1 (AB SCIEX) were converted to mzML with the ProteoWizard msconvert function, 54 to imzML using imzMLConverter,55 and imported into MSiReader v0.09.56 For standard curve creation and quantitation, the m/z intensity data for each sample or region of interest was exported to Excel using the MSiReader ROI tool (‘sum of window’ over 0.1 Da). Further processing, which was performed using either Excel or MATLAB, is detailed in the Results and Discussion section.
RESULTS AND DISCUSSION
Dehydration Causes Agar Contraction but Preserves the Overall Morphology of the Colony Biofilm
The dehydration- induced changes in sample morphology and analyte distribution were assessed by evaluating optical images of biofilms before and after desiccation. Images from one representative sample, a 72 h co-culture of two P. aeruginosa strains, are provided in Figure 1. These two strains were chosen because of their differing attributes: PAO1C (left) is a common laboratory strain that readily spreads in motility agar assays,57 whereas FRD1 (right) is a mucoid cystic fibrosis isolate strain with limited motility that produces the exopoly-saccharide alginate in great abundance.47
Figure 1.

Evaluation of biofilm dehydration in preparation for SIMS imaging. A 72 h P. aeruginosa PAO1C (left) and FRD1 (right) co-culture: (a) as cultivated in a Petri dish; (b) excised and transferred to a SIMS sample plate with double-sided copper tape; (c) after dehydration with N2; (d) overlay of the same sample before (green) and after (purple) dehydration; (e) quiver plot with vector length and color showing the magnitude and direction of dehydration- induced contraction; and (f) inset of the quiver plot shown in e. The scale bars represent 1 cm in a–d and 2.5 cm in f.
The biofilms were cultivated on thin agar (Figure 1a), transferred to a 250 μm recessed aluminum SIMS sample plate covered with 88-μm-thick double-sided conductive copper tape (Figure 1b), and dehydrated under a gentle stream of nitrogen (Figure 1c). An overlay of the hydrated (green) and dehydrated sample (blue) shows a slight contraction of the agar during dehydration (Figure 1d), and measurements of four independent biofilms show that the agar area is reduced by 26% (± 2 standard deviation (SD)) during dehydration.
Changes in sample morphology were also evaluated in a spatially resolved fashion. Images of biofilms acquired before and after dehydration were overlaid and registered using an affine transformation to account for variance in angle, distance, and camera position. Approximately 50 teach points present on both images were utilized to perform a non-affine polynomial transform to dynamically “stretch” the dehydrated sample to match the shape of its hydrated counterpart. The magnitude of contraction was calculated for an equally spaced array, and is represented as a rainbow quiver plot (Figure 1e and 1f). The magnitude of distortion at a given point on the quiver plot is represented both by the length of the vector and its color, with long yellow vectors indicating regions of greater contraction, and short blue vectors indicating regions with less contraction. The inset image (Figure 1f) shows that the sample contracts by over 2 mm at the agar edge and converges to a point of zero contraction near the intersection of the two biofilms. We surmise that the point of least contraction is shifted slightly towards the FRD1 biofilm due to the thicker biofilm structure, which results in slower drying for the under-lying agar.
Overall these results indicate that although the agar contracts during drying, it does so in a uniform and predictable manner, with the greatest contraction occurring near the agar edges. The macroscopic biofilm morphology remains intact and suitable for follow-up analysis. In our experience, the best results are obtained by starting with a thin agar of uniform thickness and unfurling it gradually onto the SIMS substrate with a steel spatula or a razor blade. Care must be taken to avoid distorting the agar or trapping air bubbles during the transfer process. We initially experimented with several different preservation procedures, including vacuum desiccation, freeze-drying, and oven drying; we found that a simple nitrogen- assisted dehydration provided the best retention of biofilm morphology and the most consistent results. Attempts were also made to deposit the agar directly onto aluminum sample plates or onto silicon wafers, thus avoiding the use of copper tape and the possible introduction of chemical contaminants, but the agar tended to separate from the substrate during dehydration so the tape was found to be essential for adhesion.
To evaluate changes in analyte distribution, endogenous aggregates in the hydrated and dried samples were visualized with optical microscopy (Figure S1). Previous Raman microscopy investigations into aggregates of this shape in P. aeruginosa biofilms revealed that they contain both AQs and AQNOs,14,58 which are the primary analytes of interest for this study. Representative microscopy images acquired near the center of the PAO1C biofilm show that the overall shape and distribution of the AQ and AQNO aggregates are largely retained during dehydration (Figure S1). These investigations suggest that the sample preservation was not a limiting factor in the ultimate imaging resolution, which was confined to 100 μm or greater in the current work.
Notably, this analysis does not account for hydrophilic analytes, which may partially absorb into the agar and migrate throughout the sample, or amphiphilic rhamnolipid biosurfactants, which could diffuse laterally during drying.59 The solubility of AQs and AQNOs is improved by the presence of rhamnolipids, so it is possible that these analytes can be distributed more widely across the surface if substantial rhamnolipid diffusion occurs.60
Optimization and Evaluation of Standard Deposition
The experiments described in this manuscript largely focus on two AQs and two AQNOs reported to be most abundant in P. aeruginosa,61 namely the saturated 7- and 9-carbon variants of the major subclass (i.e., PQS, C9-PQS, HQNO, and NQNO). In P. aeruginosa, PQS is a known cell-to-cell signaling molecule, 62 and both HQNO63 and NQNO64 are active against Staphylococcus aureus. The functional roles of C9-PQS and most of the other AQs and AQNOs produced by P. aeruginosa are currently unknown.65 Differing only by the location of a hydroxyl group, PQS and HQNO both produce a protonated molecular ion at m/z 260.1650. Similarly, the 9-carbon variants of these two molecules (i.e., C9-PQS and NQNO) are both found at m/z 288.1963. Differentiation of these analytes with mass spectrometry therefore requires fragmentation-based MS2 analysis. As previously reported, the two AQNOs (HQNO and NQNO) fragment under CID to yield a base peak of m/z 159.07, while AQs produce a base peak of m/z 175.07, which, along with several other distinguishing fragments, enables this differentiation (Figure S2).61 However, closer inspection reveals that each of these analytes also produces a competing product ion signal that directly interferes with the isomeric counterpart. In fact, for MS2 analysis of both m/z 260 and m/z 288, no single product ion can be used to completely separate the signal arising from either compound. Because of these complications, the standards were separated into two separate multicomponent dilution series, one containing HHQ, PQS and NQNO, and another containing NHQ, C9-PQS, and HQNO (HHQ and NHQ were included as part of the original study design but were not used for quantitation and therefore will not be discussed further).
The quantitative imaging method described herein requires analysis of standards on an agar surface; therefore, we sought to determine the extent to which the standards absorbed into the agar during deposition. Using similar conditions to the standard curve preparation, 1 nL aliquots of the 100 μM standard solutions were deposited onto dried agar via chemical inkjet printing, and the resulting spots were evaluated with SEM. As can be seen in Figure 2a and 2b, the deposited standards cover a circular region of approximately 200 μm in diameter and appear to form an even layer over the underlying agar. The features observable around and underneath the deposited standards in the 400× magnification electron micrograph (Figure 2b) show that the microstructure of the agar surface is retained during deposition and solvent evaporation, suggesting that the solution dries before dissolving surface salts or absorbing into the agar. Control spots, which contained only solvent, were unobservable by SEM.
Figure 2.

SEM and three-dimensional confocal fluorescence microscopy evaluation of standards deposited onto dehydrated agar via chemical inkjet printing. (a) Electron micrograph of five 1 nL depositions (as indicated by red arrows) containing 100 μM each of HHQ, PQS, and NQNO. (b) Higher magnification micrograph of a single standard spot from a. (c) Average fluorescence intensity depth profile for fluorescein in four spots containing either 20 μM fluorescein (blue squares) or 20 μM fluorescein and 100 μM each of HHQ, PQS, and NQNO (red circles). The integrated fluorescence intensity in the space below and above the agar surface is provided in blue and red text for the two deposition conditions. Shown are 23 Z-stacks from −26.4 μm below the surface to 26.4 μm above the surface. The scale bars in a and b represent 500 μm and 100 μm respectively, and the error bars in c represent standard deviation.
For further verification, 1 nL droplets of a solution containing either 20 μM fluorescein and 100 μM each of HHQ, PQS, and NQNO or only 20 μM fluorescein were deposited on dried agar and examined with three-dimensional confocal fluorescence microscopy. As can be seen in Figure 2c, the fluorescence intensity profile for fluorescein in the standard mixture (red circles) takes the form of an asymmetric Gaussian point spread function with a maximum intensity at the agar surface (depth = 0 ± 1.2 μm). Significant fluorescence signal bleed prevents the determination of analyte depth; however, the integrated fluorescence intensity is slightly greater above the surface than below (p = 0.02 for n = 4 spots). The control condition with spots containing only fluorescein (blue squares) exhibits less asymmetry and does not show a significant difference in integrated fluorescence intensity for regions below and above the surface (p = 0.4). This suggests that the observed surface excess is not an artifact of the refractive index difference between the air and the agar, but rather, arises due to the presence of AQs and AQNOs.
In combination with the low water solubility of AQs and AQNOs,66 as well as our observation of endogenous AQ and AQNO crystals on the surface of biofilms (Figure S1), the SEM and confocal fluorescence microscopy data support the conclusion that the standards largely remain on the agar surface following deposition. It is possible that components of the agar (e.g., salts) migrate during droplet drying to co-crystalize with the exogenous standards, or that a portion of the standards absorb into the agar. Both of these possibilities may affect the measured ion abundance.
We next optimized the chemical inkjet printing conditions to achieve reliable and repeatable standard curves and manageable analysis times. Initially, the multicomponent standard solutions were deposited as an ordered array of spots. However, following C60-SIMS product ion imaging, the image-toimage consistency was found to be poor, most likely because the spot diameter (~200 μm) was too small for the width of the stage raster (100 μm) and the diameter of the primary ion beam (35 μm). To overcome this issue, 20 aliquots of each solution were deposited evenly at a spot-to-spot interval of 200 μm over an area of 200 μm × 2000 μm. Due to the overlapping and pooling of the standard solutions, these deposition conditions resulted in the formation of 2.2 mm × 0.6 mm regions containing between 40–8000 fmol of each analyte at a surface density range of 3.0–610 pmol/cm2.
Relative Quantitation is Accomplished through Pixel-wise Normalization to External Nonlinear Least Squares Regression Curves
Representative standard images from the optimized line deposition conditions are provided in Figure 3a and 3b, and Figures S3 and S4. The average relative standard deviation of the ion intensity across all concentrations was 24%, 22%, 42%, and 24% for PQS, HQNO, C9-PQS, and NQNO, respectively.
Figure 3.
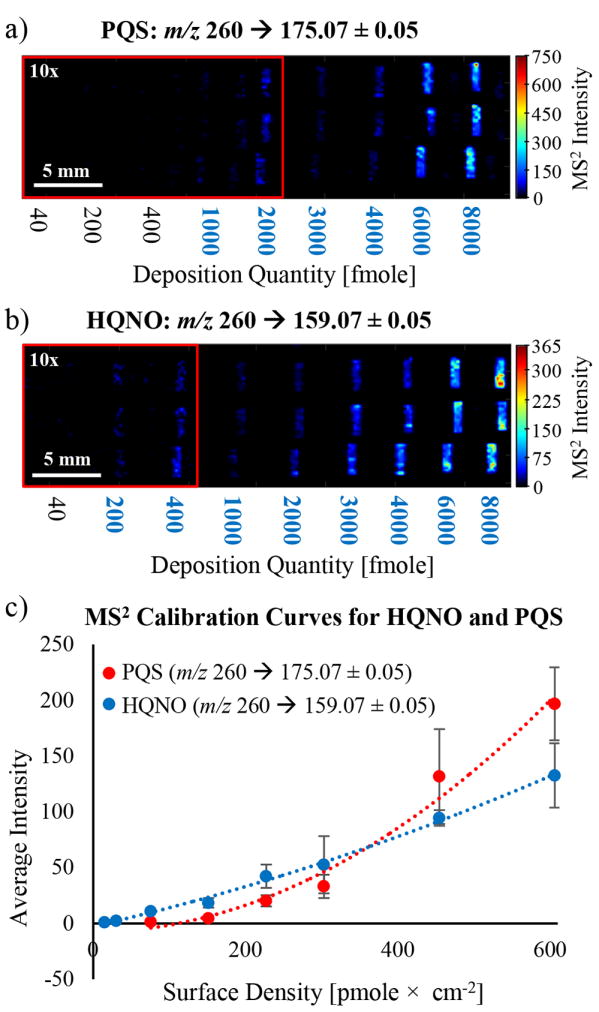
SIMS product ion quadratic calibration images, curves, and regression data: calibration images for (a) PQS (m/z 260 → 175.07) and (b) HQNO (m/z 260 → 159.07) printed on dry agar; and (c) calibration curves for PQS and HQNO. The color scale was magnified by a factor of 10 in the red boxes in a and b to illuminate the lower deposition quantities, and the blue quantities in the image indicate the quantitation range used. Additional calibration information, including calibration curves for C9-PQS and NQNO, can be found in Figures S3 and S4.
The product ion intensities for each analyte were used to create second order polynomial linear regression curves of the form of Equation 1:
| (1) |
where i is the measured ion intensity, ρ is the is surface density with units of mass × area−1, and a, b, and c are constants (Figures 3c, S3, and S4). We initially attempted to apply linear regression curves but they were found to be unreliable over a meaningful concentration range because of nonlinearity, which we speculate arise from second-order surface interactions (e.g., absorption, intercalation, or intermolecular forces) that suppress ionization at low concentrations, and surface saturation at high concentrations. Although a calculation in terms of three dimensional area (e.g., concentration of the first 40 nm) was considered in place of surface density, the veracity of this calculation is uncertain in the absence of an accurate sputtering depth for every pixel.
The quadratic equation can be used to solve the second order polynomial regression curves from Figure 3 to accommodate differences in ionization efficiency and directly calculate the analyte-specific surface density in each pixel. Using the product ion transition for PQS (m/z 260 → 175) as an example (Equation 2):
| (2) |
In instances where the discriminant is less than zero and no real solution is available, the free term (c) can be set equal to zero; however, manual interpretation is necessary to ensure the accuracy of the fit.67
As previously discussed, the measured surface density for PQS (ρPQS(means)) is an overestimate of the actual surface density due to the competing product ion transition for HQNO (m/z 260 → 175). Examination of the four compounds over a series of concentrations revealed that the relative intensities of m/z 159 and m/z 175 take on a constant value for each analyte: the 159/175 ratios for PQS and C9-PQS are 0.04 ± 40% (SD) and 0.020 ± 10%, respectively, and the 175/159 ratios for HQNO and NQNO are 0.13 ± 8% and 0.07 ± 20%, respectively. The corrected surface density for PQS (ρPQS175(corr)) can be determined by removing the contribution of m/z 175 arising from HQNO (ρHQNO175(exp)), as shown in Equation 3:
| (3) |
Of course ρHQNO175(exp) is still an unknown value and must be further defined by Equations 4–6:
| (4) |
| (5) |
| (6) |
where the experimentally derived relative fragmentation ratios for HQNO and PQS are delineated by fHQNO and fPQS respectively, and ρHQNO159(means) is given by Equation 2 solved for HQNO. Combining Equations 3 and 6 and solving for the ρPQS175(corr) term yields Equation 7, which is the final formula used to calculate the corrected surface density for PQS:
| (7) |
More generally, for any analyte n with an interfering compound m, Equation 8 can be used to calculate the corrected surface density of n:
| (8) |
By adding additional terms to Equation 3, this analysis can theoretically be extended to accommodate additional interfering species. It is therefore not limited to MS2 data, but can also be extended to MS1 in instances when the chemical composition and in-source fragmentation characteristics of the sample are known to a high degree of confidence.
Finally, the accurate interpretation of quantitative measurements requires information on the lower and upper limits of quantitation (LLOQ and ULOQ) for each analyte. Here the LLOQ is defined by two criteria. First, the initial (raw MS2) intensity of a given pixel must be greater than the lowest intensity on the calibration curve. Second, the initial intensity must be greater than the average background intensity from the sample. If the intensity of a given pixel is lower than either of these values, the surface density for this analyte is set to zero. The ULOQ for each analyte is defined as being the highest intensity value on the calibration curve. Intensity values that are greater than the ULOQ are adjusted to 110% of the ULOQ in the quantitative image, effectively saturating the intensity scale for this pixel. In this way, for any given pixel, analyte intensities that are lower than the LLOQ will display a surface density of zero, and analytes with an intensity greater than the ULOQ will yield a “saturated” surface density.
Full calibration data for the four analytes used in this study, including the three calibration constants (a, b, and c), the coefficient of determination values (R2), the ULOQs, and LLOQs, can be found in Table 1.
Table 1.
Quadratic Regression Data
| PQS | HQNO | C9-PQS | NQNO | |
|---|---|---|---|---|
| MS1m/z | 260 | 260 | 288 | 288 |
| MS2m/z | 175 | 159 | 175 | 159 |
| a | 5.6E-04 | 1.2E-04 | 3.6E-03 | 3.1E-03 |
| b | 7.5E-03 | 1.5E-01 | −5.0E-02 | 3.7E+00 |
| c | −7.8E+00 | −2.4E+00 | 0.0E+00 | −6.1E+01 |
| R2 | 0.980 | 0.996 | 0.998 | 0.998 |
| Fragmentation factor | 0.044 | 0.13 | 0.020 | 0.070 |
| LLOQ [intensity] | 1.4 | 0.99 | 0.96 | 12 |
| ULOQ [intensity] | 200 | 130 | 75 | 950 |
| LLOQ [pmol × cm−2] | 75 | 15 | 3.0 | 15 |
| ULOQ [pmol × cm−2] | 610 | 610 | 150 | 230 |
Quantitative SIMS Imaging of AQs and AQNOs in P. aeruginosa Colony Biofilm
To demonstrate the capability of our methods, we subjected a series of P. aeruginosa biofilms to the quantitative SIMS imaging protocols described above. An example imaging sequence for two AQs and two AQNOs in a 72 h PAO1C colony biofilm is shown in Figure 4. First, we show the raw images required in standard (MS1) mode (Figure 4a), which is how ion images are overwhelmingly reported in the literature. Ion images for m/z 260.17 (the protonated molecular ion for both PQS and HQNO) and m/z 288.20 (the protonated molecular ion for both C9-PQS and NQNO) show the four analytes to be primarily confined to a region near the center of the biofilm community (Figure 4a (iii)). In SIMS, prominent in source fragment ions arising from the analytes of interest can sometimes be more intense than the molecular ion; therefore, images for these ions are also commonly displayed. An ion image for an AQ fragment (m/z 175.07) appears with high intensity near the sample center, and with a small ring of lower intensity around the biofilm edge (Figure 4a (iii)). The common in source fragment of both AQNOs and HHQ-type AQs (m/z 159.07) has a distribution similar to both m/z 260.17 and m/z 288.20; however, the edge of the sample center shows up at a higher intensity than the internal region (Figure 4a (iv)). Unfortunately, because of common in source fragmentation, ion images for m/z 175.07 and 159.07 are not specific to any one molecule, or even a single class of molecules, and thus, cannot reliably be used to assign the distribution of an analyte without extensive follow-up analysis.
Figure 4.

SIMS imaging of a 72 h P. aeruginosa PAO1C colony biofilm. (a) Raw SIMS MS1 images for (a (i)) PQS and HQNO, (a (ii)) C9-PQS and NQNO, and AQ fragments (a (iii)) m/z 175.07 and (a (iv)) 159.07; (b–e) MS2 quantitative image progressions for (b) PQS, (c) HQNO, (d) C9-PQS, and (e) NQNO. Images in column b–e (i) show the uncorrected (raw) secondary ion intensity. Images in column b–e (ii) are corrected for ionization efficiency; column b–e (iii) are corrected for common (interfering) product ions; column b–e (iv) show the ULOQ and LLOQ corrections; and column b–e (v) are scaled to a common maximum surface density of 610 pmol × cm−2 to facilitate direct and quantitative analyte-to-analyte distribution comparisons. High intensity regions outside of the agar shown in a–d arose from the chemical background of the copper tape.
In contrast to MS1 images, product ion images have two criteria for discriminating the analyte from the chemical background— the precursor ion must be ionized to generate an m/z value within a certain range (± 0.5 Da in this case), and a product ion must be generated with a separate, specific m/z value within an additional selection window (± 0.05 Da in this case)—therefore product ion imaging is much more selective. Product ion images for PQS, HQNO, C9-PQS, and NQNO are provided in Figure 4 (i–v), 4c (i–v), 4d (i–v), and 4e (i–v), respectively, with each row (i–v) showing successive steps along the quantitation process.
The largest changes in both distribution and intensity are observed for PQS. The raw image (Figure 4b (i)), which is scaled to the maximum single-pixel ion intensity, shows a region of moderate signal intensity near the center with small regions (< 200–400 μm) containing high intensity pixels. Following correction for ionization efficiency by applying Equation 2 (Figure 4b(ii)), a similar signal distribution is observed; however, the nonlinear calibration has the effect of amplifying low intensity signals, revealing an additional ring of PQS bordering the biofilm center.
Following subtraction of the signal contribution from HQNO by using Equation 7 (Figure 4b (iii)), both the measured surface density and the perceived distribution change substantially, showing that, aside from a few isolated hotspots, the center of the sample has much less PQS than originally observed. In Figure 4b (iv) the surface density of pixels with an intensity less than the calibration and sample LLOQs are set equal to zero, and those with a surface density greater than the ULOQ are set to 110% of the ULOQ. In the case of PQS, no pixels have an intensity above the ULOQ, so the ULOQ adjustment has no effect. Finally, in Figure 4b (v), the maximum surface density of the image is scaled to a common value of 610 pmol × cm−2 to enable direct analyte-to-analyte comparisons.
Analogous operations were performed on the remaining three analytes (Figure 4c–e) to generate quantitative ion images. Note that the change in surface density brought on by fragmentation is minor, and largely unobservable within these images. It can be helpful to view the quantitative progressions as two-dimensional scatter plots, with each pixel plotted as a function of intensity or surface density, as shown in Figures S5–S8. This perspective shows, for example, the effect that fragmentation correction and LOQ filtration has on the perceived distribution of NQNO, which is actually quite significant at low concentrations (Figure S8). An additional quantitative imaging example is provided for a second 72 h PAO1C biofilm in Figure S9, showing changes in the AQ and AQNO distributions and surface densities that are similar to those observed in Figure 4. However, the surface densities are much higher, indicating a large degree of biological variability.
Although AQ and AQNO concentrations for agar-based PAO1C biofilms were not available in the literature, we can draw insight from quantitative analysis of P. aeruginosa liquid cultures. For example, Lépine and co-workers66 reported a maximum concentration of 1.7 μM PQS and 3.3 μM HQNO after 16 h of P. aeruginosa PAO1 planktonic growth in FAB media, a time point that corresponded to the early stationary phase. For 16 h PA14 planktonic cultures they measured PQS and HQNO concentrations of 5.0 μM and 6.94 μM, respectively. 66 In separate work, Lépine et al. measured PQS and HQNO concentrations of 4.8 μM and 11 μM, respectively, in planktonic cultures of PA14 grown for 8 h in LB.61
For the measurements done here, even if we assume that none of the analyte is contained within the bulk of the biofilm, and that the biofilms are 50-μm thick, which would be on the upper end of most estimates,68 the local concentrations for PQS and HQNO (Figure 4) are more than 10 times greater than those reported from previous measurements. Examined from a different perspective, the average concentrations of PQS and HQNO over the entire area of the biofilm (again assuming a thickness of 50 μm) are 30 μM and 17 μM, respectively, values that are closer to the Lépine measurements.61,66 Our quantitative ion images and the calculations performed above do not consider the drying-induced contraction of the biofilms; however, this factor would only account for a small portion of the concentration disparity. Because of differences in both the specific P. aeruginosa strain examined and the cultivation conditions used, it is difficult to make direct comparisons between our work and that of the prior studies, but does suggest that the concentration of AQs and AQNOs is much higher in agar-based biofilms than in planktonic cultures. Furthermore, the large differences in local surface density highlight the importance of performing imaging analyses over bulk measurements, as the local chemical microenvironment can be very different than an analysis of the bulk might suggest.
CONCLUSION
We demonstrate that a simple nitrogen-assisted dehydration is adequate for the preservation of agar-bound microbial communities for SIMS imaging. Although the agar contracts during dehydration, the macroscopic biofilm morphology remains largely unchanged. By following several aggregates through the drying process, it is apparent that the endogenous molecular distribution is also retained, at least for the analytes examined in this study. We also report a method for quantitative SIMS imaging that is capable of correcting for both analyte- specific differences in ionization efficiency and the presence of isomeric interference. This method was applied to image the surface density of two AQs and two AQNOs in P. aeruginosa biofilms, and appears effective in such applications.
This work does not fully address several of the remaining challenges for quantitative MSI. First, we did not completely account for spatially dependent variations in ionization efficiency brought on by heterogeneous matrix effects, topography, hardness, conductivity, salt concentration, and other factors. 69,70 These variations in ionization efficiency hinder accurate determination of analyte distribution, and can prohibit the direct comparison of the distribution or intensity of one analyte to others. Here, matrix effects were mitigated by creating calibration curves on agar, but the effects of the local microenvironment have not been accounted for. Second, to construct accurate images, it is necessary to quantitate over a large dynamic range of at least several orders of magnitude. While quadratic and higher order polynomial calibrations improve the dynamic range, meaningful ranges can be difficult to achieve for some analytes. Finally, one of greatest strengths of MSI is its capacity for multiplexed chemical analysis; it is often desirable to examine the distribution of tens or hundreds of compounds in a single image, which would be time consuming to achieve with our methodology. Despite these challenges, we anticipate that this method can be effectively applied to image the surface density of a wide variety of analytes in many different sample types.
Supplementary Material
Acknowledgments
The authors acknowledge the intellectual contributions of Dr. Troy Comi, Dr. Bin Li, Kyungwon Ko, Amit Patel, and Kisurb Choe. We also thank Robert Mowery and Stephanie Lozano for their assistance with data analysis as well as Dr. Mizuki Endo for his help with confocal fluorescence microscopy. Research funding was provided through the National Institutes of Health under Award No. R01 AI113219, and the Department of Energy through a subcontract from Oak Ridge National Laboratory (PTX-UT-Battelle) under Award No. ORNL-4000134575. SJBD and JFE are partially funded through the Springborn Endowment, and SJBD receives support through the NSF Graduate Research Fellowship Program.
Footnotes
Notes
The authors declare no competing financial interest.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Figures S1–S9, as noted in the text, provide the following information on analytes examined in this work: evaluation of changes in distributions, product ion spectra and images, calibration curves, and quantitative analyses. The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Vickerman JC, Winograd N. Int J Mass Spectrom. 2015;377:568–579. [Google Scholar]
- 2.Winograd N. Anal Chem. 2005;77:142–149 A. doi: 10.1021/ac051263k. [DOI] [PubMed] [Google Scholar]
- 3.Winograd N. Anal Chem. 2015;87:328–333. doi: 10.1021/ac503650p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fletcher JS, Rabbani S, Henderson A, Blenkinsopp P, Thompson SP, Lockyer NP, Vickerman JC. Anal Chem. 2008;80:9058–9064. doi: 10.1021/ac8015278. [DOI] [PubMed] [Google Scholar]
- 5.Carado A, Passarelli MK, Kozole J, Wingate JE, Winograd N, Loboda AV. Anal Chem. 2008;80:7921–7929. doi: 10.1021/ac801712s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanni EJ, Dunham SJB, Nemes P, Rubakhin SS, Sweedler JV. J Am Soc Mass Spectrom. 2014;25:1897–1907. doi: 10.1007/s13361-014-0978-9. [DOI] [PubMed] [Google Scholar]
- 7.Fisher GL, Bruinen AL, Ogrinc Potočnik N, Hammond JS, Bryan SR, Larson PE, Heeren RMA. Anal Chem. 2016;88:6433–6440. doi: 10.1021/acs.analchem.6b01022. [DOI] [PubMed] [Google Scholar]
- 8.Passarelli MK, Pirkl A, Moellers R, Grinfeld D, Kollmer F, Havelund R, Newman CF, Marshall PS, Arlinghaus H, Alexander MR, West A, Horning S, Niehuis E, Makarov A, Dollery CT, Gilmore IS. Nat Methods. 2017;14:1175. doi: 10.1038/nmeth.4504. [DOI] [PubMed] [Google Scholar]
- 9.Robinson MA, Graham DJ, Castner DG. Anal Chem. 2012;84:4880–4885. doi: 10.1021/ac300480g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bloom AN, Tian H, Winograd N. Biointerphases. 2015;11:02A306. doi: 10.1116/1.4939463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian H, Six DA, Krucker T, Leeds JA, Winograd N. Anal Chem. 2017;89:5050–5057. doi: 10.1021/acs.analchem.7b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Passarelli MK, Ewing AG, Winograd N. Anal Chem. 2013;85:2231–2238. doi: 10.1021/ac303038j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monroe EB, Jurchen JC, Lee J, Rubakhin SS, Sweedler JV. J Am Chem Soc. 2005;127:12152– 12153. doi: 10.1021/ja051223y. [DOI] [PubMed] [Google Scholar]
- 14.Baig NF, Dunham SJB, Morales-Soto N, Shrout JD, Sweedler JV, Bohn PW. Analyst. 2015;140:6544–6552. doi: 10.1039/c5an01149c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunham SJB, Comi TJ, Ko K, Li B, Baig NF, Morales-Soto N, Shrout JD, Bohn PW, Sweedler JV. Biointerphases. 2016;11:02A325. doi: 10.1116/1.4942884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanni EJ, Masyuko RN, Driscoll CM, Dunham SJB, Shrout JD, Bohn PW, Sweedler JV. Anal Chem. 2014;86:10885–10891. doi: 10.1021/ac5030914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanni EJ, Masyuko RN, Driscoll CM, Aerts JT, Shrout JD, Bohn PW, Sweedler JV. Anal Chem. 2014;86:9139–9145. doi: 10.1021/ac5020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debois D, Hamze K, Guérineau V, Le Caër JP, Holland IB, Lopes P, Ouazzani J, Séror SJ, Brunelle A, Laprévote O. Proteomics. 2008;8:3682– 3691. doi: 10.1002/pmic.200701025. [DOI] [PubMed] [Google Scholar]
- 19.Davies SK, Fearn S, Allsopp LP, Harrison F, Ware E, Diggle SP, Filloux A, McPhail DS, Bundy JG. mSphere. 2017;2:e00211–00217. doi: 10.1128/mSphere.00211-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding Y, Zhou Y, Yao J, Szymanski C, Fredrickson J, Shi L, Cao B, Zhu Z, Yu XY. Anal Chem. 2016;88:11244–11252. doi: 10.1021/acs.analchem.6b03909. [DOI] [PubMed] [Google Scholar]
- 21.Amaya KR, Sweedler JV, Clayton DF. J Neurochem. 2011;118:499–511. doi: 10.1111/j.1471-4159.2011.07274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angerer TB, Dowlatshahi Pour M, Malmberg P, Fletcher JS. Anal Chem. 2015;87:4305–4313. doi: 10.1021/ac504774y. [DOI] [PubMed] [Google Scholar]
- 23.Shon HK, Kim SH, Yoon S, Shin CY, Lee TG. Biointerphases. 2018;13:03b411. doi: 10.1116/1.5019611. [DOI] [PubMed] [Google Scholar]
- 24.Phan NTN, Fletcher JS, Sjövall P, Ewing AG. Surf Interface Anal. 2014;46:123–126. doi: 10.1002/sia.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phan NTN, Munem M, Ewing AG, Fletcher JS. Anal Bioanal Chem. 2017;409:3923–3932. doi: 10.1007/s00216-017-0336-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philipsen MH, Phan NNT, Fletcher JS, Malmberg P, Ewing AG. ACS Chemical Neuroscience. 2018 doi: 10.1021/acschemneuro.8b00046. [DOI] [PubMed] [Google Scholar]
- 27.The White House. 2016 https://obamawhitehouse.archives.gov/the-pressoffice/2016/05/12/fact-sheet-announcing-nationalmicrobiome-initiative.
- 28.Biteen JS, Blainey PC, Cardon ZG, Chun M, Church GM, Dorrestein PC, Fraser SE, Gilbert JA, Jansson JK, Knight R, Miller JF, Ozcan A, Prather KA, Quake SR, Ruby EG, Silver PA, Taha S, van den Engh G, Weiss PS, Wong GCL, et al. ACS Nano. 2016;10:6–37. doi: 10.1021/acsnano.5b07826. [DOI] [PubMed] [Google Scholar]
- 29.Miller MB, Bassler BL. Annu Rev Microbiol. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 30.Dunham SJB, Ellis JF, Li B, Sweedler JV. Acc Chem Res. 2017;50:96–104. doi: 10.1021/acs.accounts.6b00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Comi TJ, Si T, Dunham SJB, Sweedler JV. J Mass Spectrom. 2016;51:1030–1035. doi: 10.1002/jms.3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Si T, Li B, Zhang K, Xu Y, Zhao H, Sweedler JV. J Proteome Res. 2016;15:1955–1962. doi: 10.1021/acs.jproteome.6b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang JY, Phelan VV, Simkovsky R, Watrous JD, Trial RM, Fleming TC, Wenter R, Moore BS, Golden SS, Pogliano K, Dorrestein PC. J Bacteriol. 2012;194:6023–6028. doi: 10.1128/JB.00823-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffmann T, Dorrestein PC. J Am Soc Mass Spectrom. 2015;26:1959–1962. doi: 10.1007/s13361-015-1241-8. [DOI] [PubMed] [Google Scholar]
- 35.Anderton CR, Chu RK, Tolić N, Creissen A, Paša-Tolić L. J Am Soc Mass Spectrom. 2016;27:556– 559. doi: 10.1007/s13361-015-1324-6. [DOI] [PubMed] [Google Scholar]
- 36.Angolini CFF, Vendramini PH, Araújo FDS, Araújo WL, Augusti R, Eberlin MN, de Oliveira LG. Anal Chem. 2015;87:6925–6930. doi: 10.1021/acs.analchem.5b01538. [DOI] [PubMed] [Google Scholar]
- 37.Watrous J, Hendricks N, Meehan M, Dorrestein PC. Anal Chem. 2010;82:1598–1600. doi: 10.1021/ac9027388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen SN, Liyu AV, Chu RK, Anderton CR, Laskin J. Anal Chem. 2017;89:1131–1137. doi: 10.1021/acs.analchem.6b03293. [DOI] [PubMed] [Google Scholar]
- 39.Komorek R, Wei W, Yu X, Hill E, Yao J, Zhu Z, Yu XY. Journal of visualized experiments: JoVE. 2017:e55944. doi: 10.3791/55944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lechene CP, Luyten Y, McMahon G, Distel DL. Science. 2007;317:1563–1566. doi: 10.1126/science.1145557. [DOI] [PubMed] [Google Scholar]
- 41.Popa R, Weber PK, Pett-Ridge J, Finzi JA, Fallon SJ, Hutcheon ID, Nealson KH, Capone DG. ISME J. 2007;1:354–360. doi: 10.1038/ismej.2007.44. [DOI] [PubMed] [Google Scholar]
- 42.Groseclose MR, Castellino S. Anal Chem. 2013;85:10099–10106. doi: 10.1021/ac400892z. [DOI] [PubMed] [Google Scholar]
- 43.Lanekoff I, Stevens SL, Stenzel-Poore MP, Laskin J. Analyst. 2014;139:3528–3532. doi: 10.1039/c4an00504j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frisz JF, Lou K, Klitzing HA, Hanafin WP, Lizunov V, Wilson RL, Carpenter KJ, Kim R, Hutcheon ID, Zimmerberg J, Weber PK, Kraft ML. Proc Natl Acad Sci USA. 2013;110:E613–E622. doi: 10.1073/pnas.1216585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chumbley CW, Reyzer ML, Allen JL, Marriner GA, Via LE, Barry CE, Caprioli RM. Anal Chem. 2016;88:2392–2398. doi: 10.1021/acs.analchem.5b04409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pirman DA, Yost RA. Anal Chem. 2011;83:8575–8581. doi: 10.1021/ac201949b. [DOI] [PubMed] [Google Scholar]
- 47.Ohman DE, Chakrabarty AM. Infect Immun. 1981;33:142–148. doi: 10.1128/iai.33.1.142-148.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Centers for Disease Control and Prevention; U.S. Department of Health and Human Services, editor. 2009. pp. 1–405. [Google Scholar]
- 49.Morales-Soto N, Anyan ME, Mattingly AE, Madukoma CS, Harvey CW, Alber M, Deziel E, Kearns DB, Shrout JD. J Vis Exp. 2015 doi: 10.3791/52338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, MacDonald D, Tarkowska A, Sticco C, Hill E, Rossner M, Eliceiri KW, Swedlow JR. J Cell Biol. 2010;189:777. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW. BMC Bioinformatics. 2017;18:529. doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shard AG, Brewer PJ, Green FM, Gilmore IS. Surf Interface Anal. 2007;39:294–298. [Google Scholar]
- 53.Cook EL, Krantzman KD, Garrison BJ. Surf Interface Anal. 2013;45:93–96. doi: 10.1002/sia.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, et al. Nat Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Race AM, Styles IB, Bunch J. J Proteomics. 2012;75:5111–5112. doi: 10.1016/j.jprot.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 56.Robichaud G, Garrard KP, Barry JA, Muddiman DC. J Am Soc Mass Spectrom. 2013;24:718–721. doi: 10.1007/s13361-013-0607-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shrout JD, Chopp DL, Just CL, Hentzer M, Givskov M, Parsek MR. Mol Microbiol. 2006;62:1264–1277. doi: 10.1111/j.1365-2958.2006.05421.x. [DOI] [PubMed] [Google Scholar]
- 58.Baig NF, Polisetti S, Morales-Soto N, Dunham SJB, Sweedler JV, Shrout JD, Bohn PW. SPIE Proceedings. 2016;9930:993004–993008. doi: 10.1117/12.2236695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdel-Mawgoud AM, Lépine F, Déziel E. Appl Microbiol Biotechnol. 2010;86:1323–1336. doi: 10.1007/s00253-010-2498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Calfee MW, Shelton JG, McCubrey JA, Pesci EC. Infect Immun. 2005;73:878–882. doi: 10.1128/IAI.73.2.878-882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lépine F, Milot S, Déziel E, He J, Rahme LG. J Am Soc Mass Spectrom. 2004;15:862–869. doi: 10.1016/j.jasms.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 62.Rampioni G, Falcone M, Heeb S, Frangipani E, Fletcher MP, Dubern JF, Visca P, Leoni L, Cámara M, Williams P. PLoS Path. 2016;12:e1006029. doi: 10.1371/journal.ppat.1006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kogut M, Lightbown JW. Biochem J. 1962;84:368–382. doi: 10.1042/bj0840368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szamosvári D, Böttcher T. Angew Chem Int Ed. 2017;56:7271–7275. doi: 10.1002/anie.201702944. [DOI] [PubMed] [Google Scholar]
- 65.Fletcher MP, Heeb S, Chhabra SR, Diggle SP, Williams P, Cámara M. In: Pseudomonas: Volume 6: Molecular Microbiology, Infection and Biodiversity. Ramos JL, Filloux A, editors. Springer Netherlands; Dordrecht: 2010. pp. 29–57. [Google Scholar]
- 66.Lépine F, Déziel E, Milot S, Rahme LG. Biochimica et Biophysica Acta (BBA) - General Subjects. 2003;1622:36–41. doi: 10.1016/s0304-4165(03)00103-x. [DOI] [PubMed] [Google Scholar]
- 67.US Environmental Protection Agency. SW-846 Test Method 8000D: Determinative Chromatographic Separations. 2014;441 K:1–57. [Google Scholar]
- 68.Chopp DL, Kirisits MJ, Moran B, Parsek MR. Bull Math Biol. 2003;65:1053–1079. doi: 10.1016/S0092-8240(03)00057-0. [DOI] [PubMed] [Google Scholar]
- 69.Chughtai K, Heeren RMA. Chem Rev. 2010;110:3237–3277. doi: 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heeren RMA, Smith DF, Stauber J, Kükrer-Kaletas B, MacAleese L. J Am Soc Mass Spectrom. 2009;20:1006–1014. doi: 10.1016/j.jasms.2009.01.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.