

Key Clinical Message
Report a female diagnosed as type 1 Gaucher disease after a femoral pathologic fracture when she was 55 years old. Enzyme replacement therapy was started, and she achieved therapeutic goals. In 2015, a Ph’ CML with numerous pseudo‐Gaucher cells in bone marrow appears. BCR/ABL was not present at GD diagnosis.
Keywords: Chronic myeloid leukemia, enzymatic replacement therapy, Gaucher disease, tyrosine kinase inhibitors
Introduction
Gaucher disease (GD, OMIM230800) is a rare disease with an incidence ~1:70,000 live births in general population and around 1:110,000 in Spain (http://www.feeteg.org) 1, 2. GD is an autosomal recessive lysosomal storage disorder caused by a deficiency of acid‐B‐glucosidase (glucocerebrosidase) 3 secondary to a mutation in the encoding gene localized in the chromosome region 1q21. The glucocerebrosidase enzyme (GCase) is responsible for the cleavage of the glucosylceramide (GC), a common substrate from the degradation of cell membrane from aged blood cells. This deficiency leads an accumulation of GC into the macrophages; these GC engorged macrophages know as Gaucher's cells can be founded in the spleen, liver, and bone marrow 1. The disease manifestation is widely heterogeneous 4 and include cytopenias, spleen and liver enlargements, bone affectation, and, in some patients, neurological symptoms. In Western countries, majority of patients are type 1 GD (GD1), defined by the absence of neurological involvement 1, 2.
There are two main therapy approaches, the enzyme supplementation and the decrease the substrate formation rate. The enzyme replacement therapy (ERT) is not a curative therapy, but has been demonstrated to be a cost‐effective therapy to control hematological parameters, biomarkers, visceral, and bone infiltration and avoid complications in treated patients 5, 6, 7. The substrate reduction therapy (SRT) is based in the inhibition of the enzyme glucosylceramide synthase avoiding the formation of the GC; it is also a noncurative therapy with few patients’ restrictions depending on the used medication but with good results controlling the disease manifestations 8, 9, 10.
GD1 patients and mutations carriers have been associated with more risk to developed Parkinson disease, and GD1 patients can present pulmonary hypertension, cholelitiasis, and hypergammaglobulinemia. Also, an increased risk of clonal hematological process, specially multiple myeloma and monoclonal gammopathy of undetermined significance (MGUS) and other malignancies, has been described 11, 12.
Chronic myeloid leukemia (CML) (OMIM608232) is a myeloproliferative neoplasm secondary to an acquired translocation between chromosomes (9;22) that causes an small chromosome called Philadelphia (Ph), in more than 95% of cases, this translocation encoding the BCR‐ABL fusion gene that produces an endogenous activated tyrosine kinase enzyme 13.
The CML is also a rare disease, with an estimated incidence of 1–2 cases per 100,000 inhabitants. It was the first neoplasm with a unique genetic basis and with an effective oral target therapy; the BCR‐ABL tyrosine kinase inhibitor (TKI) imatinib, the second‐generation TKI inhibitors have changed the prognosis of the disease permitting to achieve a cytogenetic and molecular response, improving the patients’ survival. The achievement of the complete cytogenetic response (CCyR) has associated with similar survival to general population, and it becomes the most important achieved goal. Most of patients whom achieve a CCyR maintain their response, but eventually, some patients required changes in TKI therapy due to toxicities, loss of response or progression to accelerate, and/or blastic phases 14, 15.
There are only two CML cases reported on GD1 population in PubMed (accessed 04 January 2018), both of them occurred previous to the imatinib era 16, 17. Here, the first case of a chronic phase CML diagnosis in a patient with GD1 who receive both ERT and TKI therapy is presented with the aim to provide new information regarding safety and management issues when both conditions require simultaneously therapy.
Case Report
Gaucher disease diagnosis and therapy
A woman was diagnosed when she was 55 years old as type 1 Gaucher disease due to a femur pathologic fracture. At diagnosis, the patient presented with low platelets count (Hb 11.8 g/dL, platelets 49.0 × 109/L), and moderate visceral enlargement (liver 4 cm and spleen 5 cm palpable bellow costal margin). Bone assessment by MRI revealed a diffuse infiltration on dorsal and lumbar vertebrae, pelvis and femurs, and also multiple focal lesions on spleen and marked osteoporosis. Bone marrow biopsy showed infiltration by Gaucher's cells, and the enzymatic assay confirmed a lower GCase activity (0.93 nmol/L/mgprot.h, 12% respect to control); the biomarkers were highly elevated: chitotriosidase (CHT) 16,608 nmol/L/mL/h and CCL18/PARC 1969 ng/mL. The genotype was [N370S]+[L444P], the most common on Spanish population 1.
GD therapy and follow‐up
Following the diagnosis, the patient initiated ERT (60 U/kg/every other week (eow) during the first year, then 40 U/kg/eow) normalizing her blood counts at 6 month, decreasing the biomarkers (CCL18: 722 ng/mL and CHT: 1262 nM/mL/h). She get normalizing the visceral size after 1 year on ERT; the bone marrow infiltration progressively decreased until a fully clearance of vertebral and pelvis after 2 years on ERT. At present, the patient with minimal Gaucher disease activity is receiving ERT during almost 10 years with persistence of osteopenia specially in spine.
CML diagnosis
In May 2015, at 65 years, the patient was asymptomatic, but she was presenting a persistent leukocytosis with myelocytosis and basophilia (Fig. 1), WBC: 23.43 × 109/L hemoglobin 14.8 g/dL, Platelets 202 × 109/L. LDH 1874.0 U/L (219.0–439.0), ALT 59.0 U/L (5.0–40.0), AST 79.0 U/L(5.0–40.0), GGT 323.0 U/L (5.0–36.0). The bone marrow examination was observed granulocyte hyperplasia and numerous pseudo‐Gaucher's cells (Fig. 2). The genetic study reveals the presence of Ph chromosome in 95% of analyzed metaphases and the molecular study BCR‐ABL p210 transcript with negativity for JAK2 mutation. A bone marrow MRI was performed showing an infiltrative patron with hypointensity signal in T2 (Fig. 3). With all these data, the diagnosis of CML on chronic phase was made (Sokal score 0.7, low risk). A molecular study has been performed in the stored samples at diagnosis of GD showed negativity for BCR/ABL transcripts at this time.
Figure 1.
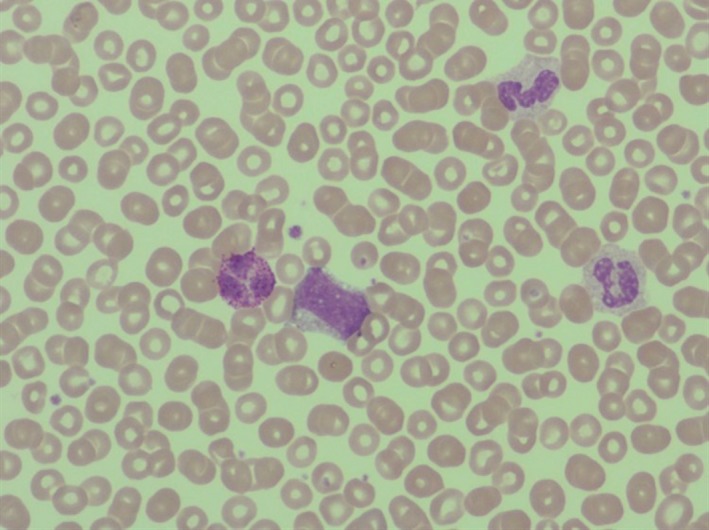
Peripheral blood smear: leukocytosis with myelemia and basophyilia: myelocytes 8%, metamyelocytes 6%, bands 1%, neutrophils 695, lymphocytes 8%, monocytes 6%, basophils 2%.
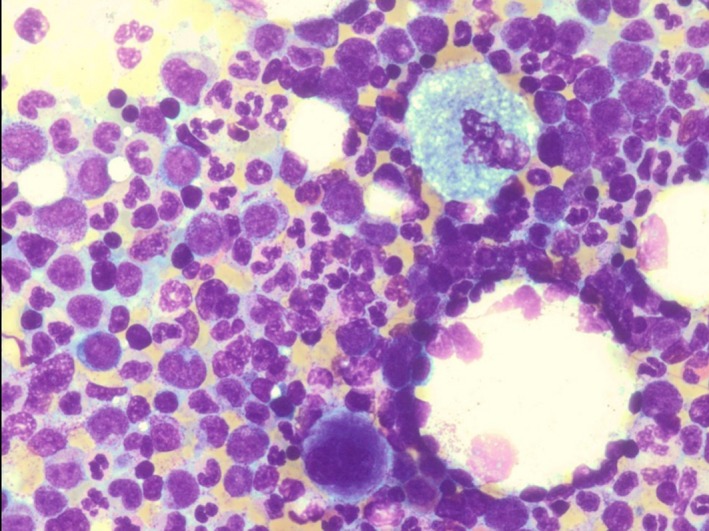
Figure 2.
Bone Marrow aspiration. W‐G. 20×. Gaucher cells and hypercellularity with granulocytic hyperplasia.
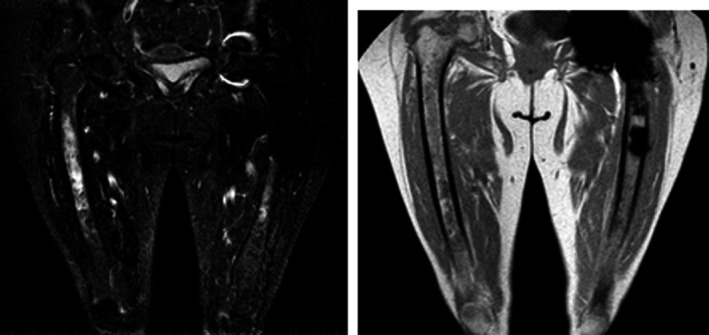
Figure 3.
Bone Marrow image examination. MRI (T1–T2). Infiltration by Gaucher cells and hematopoietic hyperplasia. Joint left hip replacement.
CML therapy
In July 2015, patient started imatinib therapy (400 mg/daily); at the same time, she was continuing on ERT at 40 U/kg/eow. The key optimal milestones used during her follow‐up were based ELN recommendations 2013; they were as follows: complete hematologic response (CHR) at 1 month; complete cytogenetic response (CCyR, <1% of Ph+) at 6 months; and major molecular response or MR3 (<0.01% transcript) at 12 months.
Despite these good initial outcomes after 18 months of therapy, a mutation in ABL (p.Tyr342His (Y342H) with resistance to Imatinib was detected. In January 2017, she changed TKI therapy to dasatinib 100 mg/day, and her ratio BCR/ABL after 1 year of therapy is 0.05%. The detailed cytogenetic, molecular response, and lysosomal biomarkers are detailed in Table 1.
Table 1.
Cytogenetic, molecular, and GD biomarkers evolution
| Follow‐up | Cytogenetic/molecular response | CML therapy | GD biomarkers CHT (nmol/mL/h)/CCL18 (ng/mL) |
|---|---|---|---|
| CML Dx | 95% Ph+ | 1692/722 | |
| 1 month | CHR | Ima 400 mg/day | |
| 3 months | 12%, PCyR; 19% | Ima 200 mg/day, restarted 400 mg/day | |
| 6 months | CCyR; 0.3%, MR2 | Ima 400 mg/day | 361/492 |
| 9 months | CCyR; 0.18%, MR2 | Ima 400 mg/day | |
| 12 months | CCyR; 0.15%, MR2 | Ima 400 mg/day | 400/478 |
| 18 months | Mutation: p.Tyr342His (Y342H). | Dasatinib 100 mg/day | 668/256 |
| 24 months | CCyR; 0.09% MR3 | Dasatinib 100 mg/day | 412/200 |
| 30 months | CCyR; 0.05% MR3 | Dasatinib 100 mg/day | 563/209 |
Dx, diagnosis; CML, chronic myeloid leukemia; CHT, chitotriosidase; PCyR, partial cytogenetic response, CCyR, complete CyR; MR2, ‐2 log reduction on BCR‐ABL/ABL transcript level.
Discussion
Preliminary studies in GD population reported in 1982 18 and 1993 19 have found that 54% of the deaths were secondary to cancer. Compared to controls, GD patients showed an overall 3.6‐fold increase risk to develop any malignancy with 14.7‐fold increase for hematological malignancies. This observation is supported by 15 cross‐sectional cohort studies and 65 case reports and series 11, 20. These different works detected a general increased risk for cancer in GD population (pooled relative risk of 1.7–1.27–2.31‐) 21, 22, 23 with special mention to hepatocellular and renal carcinomas and also reported an increased risk for multiple myeloma (estimated relative risk: 25.0–51.1) and for hematological malignancies in general 21, 22 with an estimated relative risk (RR) of 3.5–12.7 22, 23. Furthermore, the report from the international collaborative Gaucher group registry (ICGGR) has also reported an increased risk for multiple myeloma (RR = 5.9) but not for overall cancer (RR = 0.79) 24.
The interest on myeloid malignancies incidence of Gaucher disease patients has generated few case reports in PubMed 19, 23, and only two described a CML case, one in 1982, previous to the availability of ERT and TKI in a patient who progress to blastic crisis 16, the second case was published in 1998 from a non‐Ashkenazy patient diagnosed of CML in 1988, after diagnosis, the patient started busulfan therapy for CML, but an increase on bone symptoms was recorded and due to the availability of ERT the patient started ERT with good outcomes 17; in both reports, the increased turnover rate of leukocytes increased the accumulation of GC, the number of Gaucher's/pseudo‐Gaucher cells, and a worsening on GD symptoms. In the case here presented, a key classical diagnostic feature of CML was founded during the initial assessment, the presence of pseudo‐Gaucher cells. The pseudo‐Gaucher cells are morphologically similar to those seen in the hereditary GD. The accumulation of GC in CML patients (not suffering from GD) may be the result from a markedly increase in the granulocyte production with a shorter life span, this high turnover of the myeloid population leads to an overproduction of GC which results in an insufficiency of the macrophages to metabolize it.
The cellular and molecular bases to understand the pathogenesis of cancer in GD population are not elucidated 25, 26. Some authors suggest this association is product of an intrinsic feature of GD 23 while other also implicates the used (ERT) therapy 27. However, the impact of ERT on this cancer—GD association has not been established 26. The diagnosis of cancer preceded ERT in 70% of reported patients, but as the use of ERT, the incidence of cancer in patients exposed or no to ERT is similar (4.6% and 4.4%) 24, this made unlikely that ERT per se increase (or decrease) the overall risk for cancer.
The age is an important general risk factor for cancer development 25. The median age at general cancer diagnosis is 53 years (range 10–76) 22, and about 65% of all reported cancers occurs in patients older than 50 years 24, also, the incidence of gammopathies, specially Multiple Myeloma is highly related with elderly, one report mention that in people over 70 years, MM was the diagnosis in almost 31% of them.
Other identified risk factors for cancer in GD patients include splenectomy, chronic inflammation status, and an activated B‐cell function as result of substrate accumulation 11, 27, deregulated T‐cell function, augmented macrophage activation with disruption of antigen presentation process, and other modifiers genes 28. The mutation N370S is the most commonly reported in the ICCG registry 4, in homozygosis is associated with a mild GD, but also an association with a high risk of cancer has been mentioned 23, 29.
The therapy with TKI is the current gold standard for chronic phase CML as the presented case. Imatinib is the first developed TKI; the acquired advantage on survival and rate of progression has permitted that CML patients exhibit a near normal life span 30. However, cases with primary resistance have been described; one of the mechanism for this primary resistance is mediated by glycoprotein binding that block the imatinib linkup to the kinase, at this point glucosylceramide synthase (GCS), the enzyme that produces the glucocerebroside, may be implicated 31, 32.
The proposed role of glycosphingolipids in carcinogenesis is based on the ability of sphingolipids to promote DNA/chromosomal damage, activate proliferative pathways, and upregulate some drug resistance genes 33, 34, 35. Two main sphingolipids, ceramide and sphingosine, are involved in the pathways of proliferation and apoptosis 35; ceramide has been described as proapoptotic mediator while sphingosine has been implicated in cell survival and proliferation process 35, 36.
Some reports indicate that the cell clearance of ceramide is related with the drug resistance, and few ceramide‐based therapies are emerging as potent anticancer agents 34. Ceramide is a precursor to many complex sphingolipids such as glucosylceramide (GC). Therefore, therapies that increase the ceramide level in the cells, as ERT, could be useful and even speculatively the inhibition of the GCS by substrate reduction therapy also could be useful, based on this observation. The reported patient is receiving ERT with good outcomes for GD management, and based on the literature reviewed; there are no reasons to stop the therapy during TKI treatment.
The case here described, is the first case of a type 1 Gaucher Disease patient receiving concomitantly TKIs and enzyme replacement therapy.
Conclusion
Patient with Gaucher disease exhibits an increased overall risk for cancer, of them, multiple myeloma and B‐cell neoplasms are the most diagnosed while myeloid malignancies, like chronic myeloid leukemia, are rare conditions, actually (30 December 2017), there are only two entries in PubMed, this is the first CML case in a GD patient diagnosed in the TKI era.
There are many unmeet needs to understand the implication of sphingolipids on cancer development and their future implication for target therapy in especial the strategies to increase the ceramide level on cancer cells offers good approaches.
The presented case has receiving enzymatic replacement therapy, and TKIs for treat both conditions simultaneously during the combination of both therapies do not implicates an increase on side effects, the management needs to be careful to achieve the best outcome in both condition.
Conflict of Interest
None declared.
Authorship
MN: performed study design, drafted the manuscript, approved the final version of manuscript. MA: revised manuscript content, English editing, approved final version of manuscript. PI: carried out genetic GBA and CHIT1 sequencing, biomarker analysis and follow‐up, approved final version of manuscript. Laura López de Frutos: performed molecular BCR/ABL and ABL variants study, approved final version of manuscript. ML: revised manuscript content, Approved the final version of manuscript. PG: performed study design, Revised the manuscript content, approved the final version of manuscript.
Clinical Case Reports 2018; 6(5): 887–892
References
- 1. Giraldo, P. , Alfonso P., Irún P., Gort L., Chabás A., Vilageliu L., et al. 2012. Mapping the genetic and clinical characteristics of Gaucher disease in the Iberian Peninsula. Orphanet J. Rare Dis. 7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pastores, G. M. , and Hugues D. A.. 2000. Gaucher disease in Pagon R. A., Adam M. P., Ardinger H. H., et al., eds. GeneReviews®. University of Washington, Seatle, WA: (Updated 2013 Sep 19). [Google Scholar]
- 3. Beutler, E. 1991. Gaucher's disease. N. Engl. J. Med. 325:1354–1360. [DOI] [PubMed] [Google Scholar]
- 4. Grabowski, G. A. 2008. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 372:1263–1271. [DOI] [PubMed] [Google Scholar]
- 5. Mistry, P. K. , Cappellini M. D., Lukina E., Özsan H., Mach Pascual S., Rosenbaum H., et al. 2011. A reappraisal of Gaucher disease‐diagnosis and disease management algorithms. Am. J. Hematol. 86:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hughes, D. , Cappellini M. D., Berger M., Droogenbroeck J. V., De Fost M., Janic D., et al. 2007. Recommendations for the management of the haematological and onco‐haematological aspects of Gaucher disease. Br. J. Haematol. 138:676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Dussen, L. , Biegstraaten M., Hollak C. E., and Dijkgraaf M. G.. 2014. Cost‐effectiveness of enzyme replacement therapy for type 1 Gaucher disease. Orphanet J. Rare Dis. 14(9):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elstein, D. , Hollak C., Aerts J. M. F. G., Van Weely S., Maas M., Cox T. M., et al. 2004. Sustained therapeutic effects of oral miglustat (Zavesca, N‐butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J. Inherit. Metab. Dis. 27:757–766. [DOI] [PubMed] [Google Scholar]
- 9. Giraldo, P. , Alfonso P., Atutxa K., Fernández‐Galán M. A., Barez A., Franco R., et al. 2009. Real‐world clinical experience with long‐term miglustat maintenance therapy in type 1 Gaucher disease: The ZAGAL project. Haematologica 94:1771–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cox, T. M. , Drelichman G., Cravo R., Balwani M., Burrow T. A., Martins A. M., et al. 2015. Eliglustat compared with imiglucerase in patients with Gaucher's disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open‐label, non‐inferiority trial. Lancet 385:2355–2362. [DOI] [PubMed] [Google Scholar]
- 11. Arends, M. , Dussen L., Biegstraaten M., and Hollak C. E.. 2013. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 161:832–842. [DOI] [PubMed] [Google Scholar]
- 12. Mistry, P. K. , Belmatoug N., vom Dahl S., and Giugliani R.. 2015. Understanding the natural history of Gaucher disease. Am. J. Hematol. 90:S6–S11. [DOI] [PubMed] [Google Scholar]
- 13. Kantarjian, H. , Sawyers C., Hochhaus A., Guilhot F., Schiffer C., Gambacorti‐Passerini C., et al. 2002. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 346:645–652. [DOI] [PubMed] [Google Scholar]
- 14. Baccarani, M. , Deininger M. W., Rosti G., Hochhaus A., Soverini S., Apperley J. F., et al. 2013. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 122:872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Brien, S. , Radich J. P., Abboud C. N., Akhtari M., Altman J. K., Berman E., et al. 2014. Chronic myelogenous leukemia, version 1. 2015. J. Natl. Compr. Canc. Netw. 12:1590–610. [DOI] [PubMed] [Google Scholar]
- 16. Shinar, E. , Leibovitz Gershon Z., Leiserowitz R., Matzner Y., Yatziv S., and Polliack A.. 1982. Coexistence of Gaucher Disease and Philadelphia positive chronic granulocytic leukemia. Am. J. Hematol. 12:199–202. [DOI] [PubMed] [Google Scholar]
- 17. Petrides, P. E. , leCoutre P., Müller‐Höcker J., Magin E., Harzer K., Demina A., et al. 1998. Coincidence of Gaucher's disease due to private mutation and Ph’ positive chronic myeloid leukemia. Am. J. Hematol. 59:87–90. [DOI] [PubMed] [Google Scholar]
- 18. Lee, R. E. 1982. The pathology of Gaucher disease. Prog. Clin. Biol. Res. 95:177–217. [PubMed] [Google Scholar]
- 19. Shiran, A. , Brenner B., Laor A., and Tatarsky I.. 1993. Increased risk of cancer in patients with Gaucher disease. Cancer 72:219–224. [DOI] [PubMed] [Google Scholar]
- 20. Cappellini, M. D. 2015. A rare condition in haematological practice‐Gaucher disease. Eur. Oncol. Haematol. 11:15–20. [Google Scholar]
- 21. Zimran, A. , Liphshitz I., Barchana M., Abrahamov A., and Elstein D.. 2005. Incidence of malignancies among patients with type I Gaucher disease from a single referral clinic. Blood Cell Mol. Dis. 34:197–200. [DOI] [PubMed] [Google Scholar]
- 22. de Fost, M. V. D. S. , Vom Dahl S., Weverling G. J., Brill N., Brett S., Häussinger D., et al. 2006. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cell Mol. Dis. 36:53–58. [DOI] [PubMed] [Google Scholar]
- 23. Taddei, T. H. , Kacena K. A., Yang M., Yang R., Malhotra A., Boxer M., et al. 2009. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 84:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosenbloom, B. E. , Weinreb N. J., Zimran A., Kacena K. A., Charrow J., and Ward E.. 2005. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood 105:4569–4572. [DOI] [PubMed] [Google Scholar]
- 25. Mistry, P. K. , Taddei T., Vom Dahl S., and Rosenbloom B. E.. 2013. Gaucher disease and malignancy: a model for cancer pathogenesis in an inborn error of metabolism. Crit. Rev. Oncog. 18:235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cox, T. , Rosenbloom B., and Barker R.. 2015. Gaucher disease and comorbidities: B‐cell malignancy and parkinsonism. Am. J. Hematol. 90(S1):25–28. [DOI] [PubMed] [Google Scholar]
- 27. Choy, F. , and Campbell T.. 2011. Gaucher disease and cancer: concept and controversy. Int. J. Cell Biol. 2011:150450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lo, S. M. , Stein P., Mullaly S., Bar M., Jain D., Pastores G. M., et al. 2010. Expanding spectrum of the association between type 1 Gaucher disease and cancers: a series of patients with up to 3 sequential cancers of multiple types – correlation with genotype and phenotype. Am. J. Hematol. 85:340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grabowski, G. A. , Petsko G. A., and Kolodny E. H.. 2010. Gaucher disease. The online metabolic and molecular bases of inherited disease. Pp. 1–139. McGraw‐Hill, Inc., New York, NY. [Google Scholar]
- 30. Cortes, J. , Quintas‐Cardama A., Jabbour E., O'Brien S., Verstovsek S., Borthakur G., et al. 2011. The clinical significance of achieving different levels of cytogenetic response in patients with chronic phase chronic myeloid leukemia after failure to front‐line therapy: is complete cytogenetic response the only desirable endpoint? Clin. Lymphoma Myeloma Leuk. 11:421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lo, S. M. , Choi M., Liu J., Jain D., Boot R. G., Kallemeijn W. W., et al. 2012. Phenotype diversity in type 1 Gaucher disease: discovering the genetic basis of Gaucher disease/hematologic malignancy phenotype by individual genome analysis. Blood 119:4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baran, Y. , Bielawski J., Gunduz U., and Ogretmen B.. 2011. Targeting glucosylceramide synthase sensitizes imatinib‐resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. Cancer Res. Clin. Oncol. 137:1535–1544. [DOI] [PubMed] [Google Scholar]
- 33. Radin, N. S. 1999. Chemotherapy by slowing glucosphingolipid synthesis. Biochem. Pharmacol. 57:589–595. [DOI] [PubMed] [Google Scholar]
- 34. Barth, B. M. , Shanmugavelandy S. S., Tacelosky D. M., Kester M., Morad S. A., and Cabot M. C.. 2013. Gaucher's disease and cancer: a sphingolipid perspective. Crit. Rev. Oncog. 18:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gouazé‐Andersson, V. , Jing Y. Y., Kreitenberg A. J., Bielawska A., Giuliano A. E., and Cabot M. C.. 2007. Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochim. Biophys. Acta 1771:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gatt, S. , and Dagan A.. 2012. Cancer and sphingolipid storage disease therapy using novel synthetic analogs of sphingolipids. Chem. Phys. Lipids 165:462–474. [DOI] [PubMed] [Google Scholar]