

Key Clinical Message
In the context of leukocytosis due to increased number of neutrophils and their precursors, with significant dysgranulopoiesis and no or minimal basophilia and no or minimal monocytosis, the typical feature of “clumped” chromatin, in irregularly coarse compacted nuclei, should lead to suspect the diagnosis of atypical chronic myeloid leukemia.
Keywords: aCML, clinical case, hematology, leukocytosis, peripheral blood smear
A 68‐year‐old female patient presented to our emergency department with progressive fatigue, dyspnea, and generalized bone pain. Medical examination revealed fever, diffuse bruising, and hematomas. Total thyroidectomy due to unspecified cancer, poorly controlled essential hypertension and neurological hearing impairment were relevant issues in medical history. The complete blood count revealed hyperleukocytosis (WBC 319 × 109/L), severe normocytic anemia (Hb 5.6 g/dL, MCV 98 fl), thrombocytopenia (20 × 109/L), highly increased LDH (3241 U/L; n.v. 230–460), and normal chemistry and coagulation tests. The peripheral blood smear, shown in Figure 1, depicted a differential count (%) of neutrophils 62, eosinophils 0, basophils 0, lymphocytes 3, monocytes <1, promyelocytes 1, myelocytes 5, metamyelocytes 13, blasts 16. Moreover, an intense dysgranulopoiesis characterized by the distinctive feature of “clumped” chromatin, in irregularly coarse compacted nuclei, was also noticed (detail in Fig. 2). Leukocyte alkaline phosphatase score was markedly reduced. These characteristics were highly suspicious for atypical chronic myeloid leukemia (aCML). According to 2016 WHO 1, this condition is classified as a myeloproliferative/myelodysplastic syndrome characterized by (1) leukocytosis due to increased number of neutrophils and their precursors, with significant dysgranulopoiesis, which may include abnormal chromatin clumping, with no or minimal absolute basophilia and no or minimal absolute monocytosis; (2) no evidence of BCRABL1 fusion gene, PDGFRA, PDGFRB, FGFR1 rearrangement, or PCM1JAK2 translocation, and (3) a hypercellular bone marrow with granulocytic proliferation and granulocytic dysplasia, without blasts exceeding 20% in either bone marrow (BM) or peripheral blood. BM aspiration and trephine biopsy were performed, showing (Fig. 3) a nearly 100% cellularity with markedly reduced erythropoiesis and megakaryocytopoiesis, along with a hyperplastic and dysplastic granulocytic lineage; blast cells were accounted between 10% and 20% on BM smears. Results of standard cytogenetic and molecular analysis revealed a normal karyotype, and the absence of the above‐mentioned rearrangements mandatory for diagnosing aCML. Interestingly, within the standard common myeloid mutations panel, the only molecular marker we found was FLT3 ITD. The patient was treated with three subsequent leukapheretic procedures in association with hydroxycarbamide, obtaining reduction in leukocytosis to values near to 30 × 109/L. Nonetheless, clinical conditions rapidly worsened: the patient developed progressive confusion and generalized weakness. Head CT detected multiple intra‐parenchimal hemorrhagic lesions not suitable for surgical intervention. Moreover, diabetes mellitus was diagnosed and acute renal failure developed during hospital admission. Considering patient's significant comorbidities, she was discharged after clinical stabilization and continued supportive therapy only.
Figure 1.
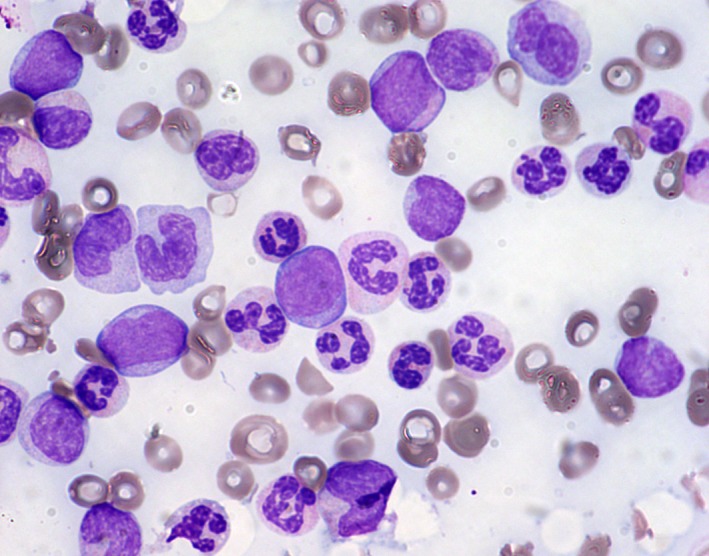
Peripheral blood smear, May‐Grünwald Giemsa stain, 600x. In detail: medium‐sized myeloblasts, promonocytes, and atypical neutrophils with a characteristic “clumped” chromatin, in irregularly compacted nuclei, and hypergranular cytoplasm can be noticed.
Figure 2.
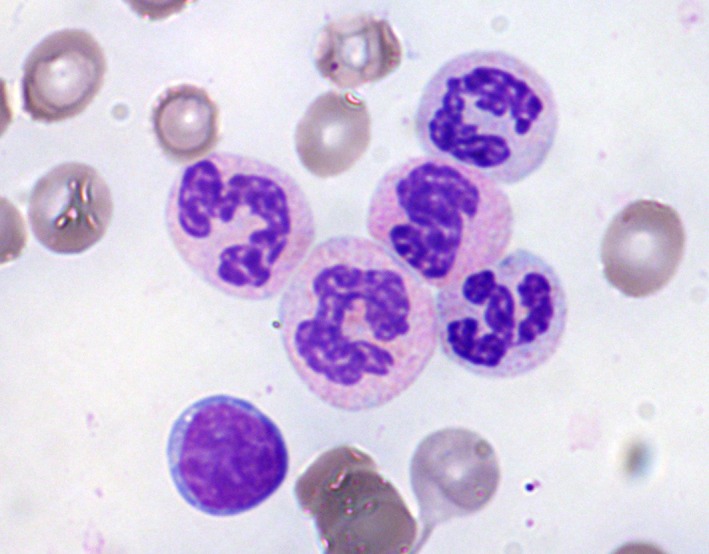
Peripheral blood smear, May‐Grünwald Giemsa stain, 1000x. Detail of atypical neutrophils with a characteristic “clumped” chromatin, in irregularly compacted nuclei, and hypergranular cytoplasm.
Figure 3.

Bone marrow trephine biopsy, hematoxylin and eosin stain, 200x. In detail: an extremely hypercellular bone marrow, with intense granulocytic hyperplasia showing dysplastic maturation, can be seen.
Authorship
GB, AP, and AG: observed the smears and wrote the diagnosis; AG: wrote the description and collected the pictures; ML: treated the patient and provided the clinical information reported.
Conflict of Interest
None declared.
Clinical Case Reports 2018; 6(5): 915–916
References
- 1. Arber, D. A. , Orazi A., Hasserjian R., Thiele J., Borowitz M. J., Le Beau M. M., et al. 2016. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405. [DOI] [PubMed] [Google Scholar]