

Key Clinical Message
Chromosomal microarray (CMA) can detect pathogenic copy number variations in 15–20% of individuals with intellectual disability and in 10% of patients with autism spectrum disorders. The diagnostic rate in specific learning disorders (SLD) is unknown. Our study emphasizes the usefulness of CMA in the diagnostic workout assessment of familial SLD.
Keywords: 17q duplication, 21q deletion, chromosomal microarray, developmental coordination disorder, dyspraxia, specific language impairment, specific learning disorder
Introduction
Specific learning disorders are defined as a failure to learn or use specific academic skills despite age‐appropriate learning opportunities and instruction, in the absence of intellectual disability or autism spectrum disorder 1, 2. SLD include a wide range of neurodevelopmental disorders, such as specific language impairment (SLI), including phonological and speech disorders; dyslexia, encompassing difficulties related to decoding and spelling words; developmental coordination disorder (DCD), affecting specifically fine motor performance; and dyscalculia, characterized by difficulties to acquire basic arithmetic skills 2. The prevalence of SLD differs significantly in the studies reported in the literature (1,2–20%), due to the heterogeneity of the definition criteria and diagnostic assessments 1, 2.
Genetic factors play a role in the etiology of a subset of SLD. Here, we report the clinical and molecular cytogenetic characterization of four children from two unrelated families presenting with a syndromic SLD, and we review the previously reported genetic causes of these specific neurodevelopmental disorders.
Clinical Report
Family 1
Patient 1
This patient was the first child of healthy unrelated parents. Family history was unremarkable. He was born at term, after an uneventful pregnancy. At birth, his occipital‐frontal circumference (OFC) was 35 cm (0 SD); his length was 48 cm (−2 SD); and his weight was 3140 g (10–25th centile). Apgar was 10/10 at 1 and 5 min, respectively.
The child had gastroesophageal reflux during infancy, treated until 2 years of age. Gross motor milestones were normally achieved: The boy started sitting at 8 months and walking unsupported at 13 months of age. He pronounced his first words at the age of 16 months but, subsequently, a severe speech delay was noticed: He was able to associate only two words at the age of 3 years and started making sentences at the age of 7 years. He had speech therapy and attended a mainstream school with specific educational support. Behavior disorders were noted including a significant anxiety, impulsivity, and emotional lability. He also had sleep disturbance with frequent awakening. Hearing test was normal, and ophthalmological examination revealed astigmatism and mild myopia.
He was referred for a genetic assessment at 10 years of age. His OFC was 55 cm (+0.5 SD); his stature was 142.7 cm (+0.5 SD); his weight was 38.4 kg (between 75th and 90th centile); and his body mass index (BMI) was 18.8 (+1.5 SD). Dysmorphic features were noted (Fig. 1), including frontal upsweep, widely spaced and sparse eyebrows, edematous eyelids, narrow palpebral fissures, bilateral epicanthus, anteverted nares, and mild micrognathia. Spatulate fingertips, camptodactyly of the 5th left finger and clinodactyly of the 3rd right toe were also noted (Table 1). Neurological examination was normal. A neuropsychological assessment was performed (Table 2). Raven's Progressive Matrices and Peabody Pictures Vocabulary Test ruled out ID, whereas a SLD was diagnosed, characterized by severe SLI and DCD.
Figure 1.
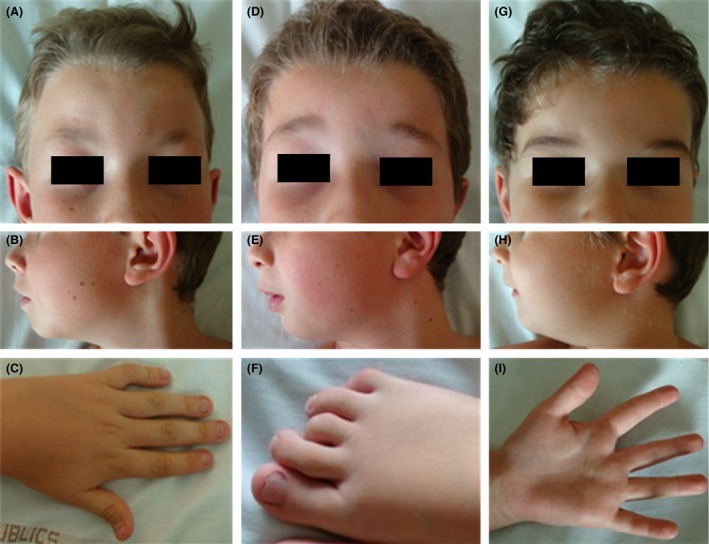
Clinical features of patients 1 (A–C), 2 (D–F), and 3 (G–I). Note frontal upsweep, widely spaced and sparse eyebrows, edematous eyelids, narrow palpebral fissures, epicanthus, mild micrognathia, and limb abnormalities including spatulate fingertips, camptodactyly of the 5th left finger in patient 1 (C), clinodactyly of the 3rd and 4th toes in patient 2 (F), and a deep web space between the 2nd and 3rd fingers in patient 3 (I).
Table 1.
Main clinical features of patients 1–4
| Patients | Family 1 | Family 2 | ||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Dysmorphic facial features | + | + | + | + |
| Frontal upsweep | + | + | + | − |
| Edematous eyelids | + | + | + | + |
| Wide‐spaced eyebrows | + | + | + | + |
| Narrow palpebral fissures | + | + | + | − |
| Epicanthus | + | + | + | + |
| Anteverted nares | + | − | − | − |
| Bulbous nasal tip | − | − | − | + |
| Micrognathia | + | + | + | − |
| Limb abnormalities | + | + | + | − |
| Spatulate fingertips | + | + | + | − |
| Camptodactyly of fingers | + | − | − | − |
| Clinodactyly of toes | + | + | − | − |
| Gastroesophageal reflux | + | + | + | − |
| BMI (SD) | +1.5 | +2 | +1.5 | +2 |
| Congenital heart disease | − | − | − | + |
SD, standard deviations.
Table 2.
Neuropsychological features of patients 1–4
| Patients | Family 1 | Family 2 | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| Raven's progressive matrices |
31 (25–50th percentile) |
26 (10–25th percentile) |
17 (50th percentile) |
NE | Pathological threshold < 10th percentile |
| Peabody pictures vocabulary test | 91 | 83 | 104 | NE | Pathological threshold < 70 |
| WISC IV/WPPSI III | NE | NE | Pathological threshold < 70 | ||
| Verbal comprehension index | 71 | 81 | |||
| Perceptual reasoning index | 56 | 65 | |||
| Specific language impairment | + | + | + | + | |
| Developmental coordination disorder | + | + | + | + | |
| Attention deficit hyperactivity disorder |
− (impulsivity) |
+ |
− (impulsivity) |
+ | |
NE, not evaluated.
Brain magnetic resonance imaging (MRI) was normal. A large genetic and metabolic assessment was normal, including thyroid and liver function tests, CK level, full blood count, blood ammonia, plasma amino acids, screening for CDG syndromes, blood acylcarnitines, screening for creatine deficiencies, standard karyotype, molecular analysis of FMR1, MECP2, and ARX.
Patient 2
Patient 2 was the younger brother of patient 1. He was born at term after an uneventful pregnancy. At birth, his OFC was 34.5 cm (−1 SD); his length was 49.5 cm (−1 SD); and his weight was 3150 g (10–25th centile). Apgar score was 9 and 10 at 1 min at 5 min, respectively. The child had gastroesophageal reflux during infancy. During the first year of life, he had laryngeal stridor which, subsequently, progressively disappeared. He started walking independently at 18 months of age and showed speech delay, learning difficulties, and hyperactivity requiring speech and occupational therapy. He attended a mainstream school with specific educational support. Hearing test was normal, and ophthalmological examination showed hyperopia.
At the age of 9 years, his OFC was 55 cm (+ 1.5 SD); his stature was 139.3 cm (+1.5 SD); his weight was 37 kg (between 90th and 97th centile); and BMI was 19.1 (+2 SD). Clinical examination showed a long face, frontal upsweep, wide‐spaced and sparse eyebrows, narrow and down‐slanting palpebral fissures, edematous eyelids, left epicanthus, micrognathia, and a long philtrum (Fig. 1). He also had spatulate fingertips, bilateral clinodactyly of the 3rd toes, and overriding 4th toes (Table 1). Neurological examination was normal. Neuropsychological assessment was consistent with a SLD, including SLI and DCD, associated with attention deficit/hyperactivity disorder (ADHD; Table 2).
Patient 3
The youngest sibling of the family was born at term after an uneventful pregnancy. At birth, his OFC was measured at 34 cm (−1 SD); his length was 52 cm (−0.5 SD); and his weight was 3210 g (50th centile). Apgar score was 10/10 at 1 and 5 min, respectively. As his two brothers, he had gastroesophageal reflux requiring treatment until the age of 2 years. He started walking independently at the age of 18 months. He was not able to pronounce any word at the age of 2 years; he had speech and occupational therapy with progressive speech improvement and he started attending a mainstream school.
He was referred to the genetic department at the age of 5 years. His OFC was 51 cm (−0.5 SD); his stature was 112.9 cm (+0.5 SD); his weight was 22 kg (90th percentile); and his BMI was 17.2 (+1.5 SD). Clinical examination revealed dysmorphic features similar to his brothers (Fig. 1), including frontal upsweep, wide‐spaced eyebrows, edematous eyelids, narrow palpebral fissures, bilateral epicanthus, right convergent strabismus, a long philtrum, low posterior hairline, and micrognathia. Clubbing of fingers was noted and there was a deep web space between the 2nd and the 3rd fingers, bilaterally (Table 1). Neurological examination was normal. Neuropsychological assessment revealed, as his brothers, a SLD including SLI and DCD (Table 2). Ophthalmological examination showed hyperopia. On the basis of the pedigree, an X‐linked disorder was initially suspected.
Family 2
Patient 4
This girl was born at term after an uneventful pregnancy. Parents were in good health; there was no consanguinity. A maternal uncle had learning difficulties, epilepsy, and overweight but it was not possible to have more detailed information about his clinical picture.
At birth, patient 4's OFC was 33 cm (−1 SD); her length was 48 cm (−0.5 SD); and her weight was 2890 g (25–50th centile). She had recurrent otitis in the first year of life. She started walking independently at 16 months of age and she developed behavioral problems including stereotypic movements, echolalia, and an oppositional attitude, as well as learning difficulties. Overweight was also noted. She had speech and occupational therapy, and she attended a mainstream school with educational support.
She was examined at the age 3 years and 9 months. Her OFC was 48.4 cm (−1 SD); her stature was 98 cm (0 SD); her weight was 17 kg (90th percentile); and her BMI was 17.7 (+2 SD). Clinical examination showed dysmorphic features (Fig. 2) including edematous eyelids, bilateral epicanthus, apparent hypertelorism, a bulbous nasal tip, and thick lips (Table 1). Hands and feet were normal as was neurological examination. Neuropsychological assessment was consistent with a SLD including SLI, DCD, and ADHD (Table 2).
Figure 2.
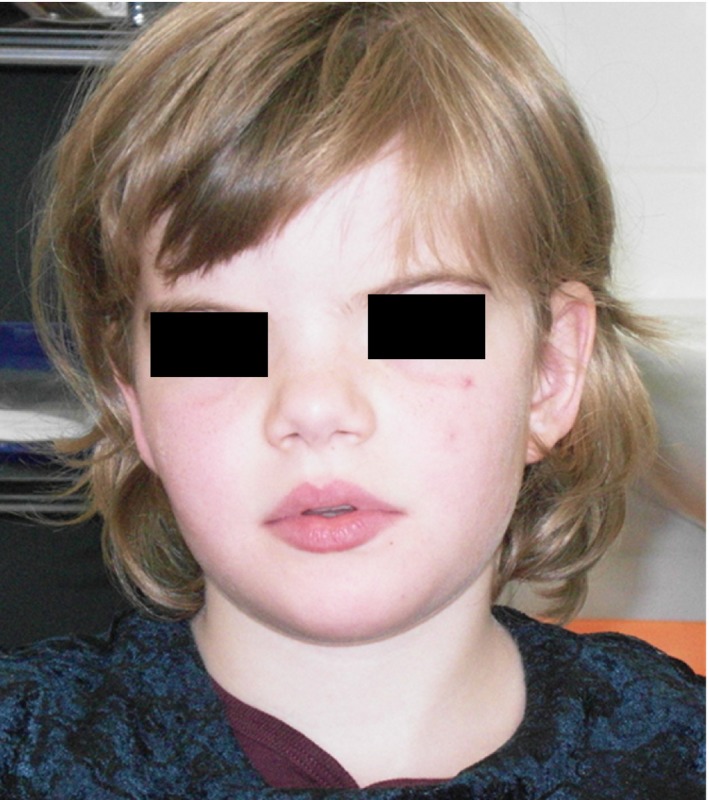
Clinical features of patients 4. Note edematous eyelids, bilateral epicanthus, apparent hypertelorism, a bulbous nasal tip, thick alae nasi, and thick lips.
Brain MRI was normal as was hearing test. Echocardiography, performed at birth because of a heart murmur, showed patent ductus arteriosus (PDA) and atrial septum defect. Repeated echocardiography at 6 years of age showed a persistent PDA with spontaneous resolution of the atrial septum defect.
Material and Methods
Chromosomal microarray was performed on a custom 244,000 (244k) or a 180,000 (180k) Agilent catalogue‐oligonucleotide microarray (Human Genome CGH Microarray Kit 244A or SurePrint G3 Human CGH Microarray Kit, 4 × 180k; Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer protocol in patients 1 and 4, respectively. Patients’ DNA was compared with a control DNA on the custom 244k microarray or against DNA from two other patients with different diseases, according to the loop model for the catalogue 180k microarray. Concerning patient 1, given the first hypothesis of an X‐linked disorder, the custom array was enriched in oligonucleotides on the X chromosome: The overall mean probe spacing was about 1530 bp and spacing between two probes on an X‐linked ID gene was approximately 100 bp. The overall mean probe spacing on autosomes was around 28.5 kb.
Concerning patient 4, the catalogue 180K microarray had about 13 kb of mean probe spacing (http://www.genomics.agilent.com). Arrays were analyzed with Feature Extraction 9.5.3.1. Results were interpreted with DNA Analytics 4.0.85. The following parameters were used for interpretation: ADM‐2, threshold: 5.0, window: 0.2 Mb, and cutoff: 0.25. A CNV was considered to have occurred if at least five contiguous oligonucleotides had an abnormal log2 ratio for the 244k custom array or three contiguous oligonucleotides for the 180k catalogue array.
Results
Family 1
Chromosomal microarray, performed in patient 1, did not show pathogenic CNV on the X chromosome but revealed an unbalanced translocation between the long arms of chromosomes 17 and 21, leading to a 2.54 Mb 17q25.3qter duplication (78,502,910–81,060,000 bp) and a 3.65 Mb 21q22.3qter deletion (44,376,016–48,080,911 bp) (hg 19) (Fig. 3). Fluorescence in situ hybridization (FISH) analysis confirmed this result in patient 1 and showed the same unbalanced translocation in patients 2 and 3. FISH analysis revealed that the mother, the maternal grandfather, and a maternal aunt carried a balanced translocation (17q;21q); the father had normal chromosomes.
Figure 3.
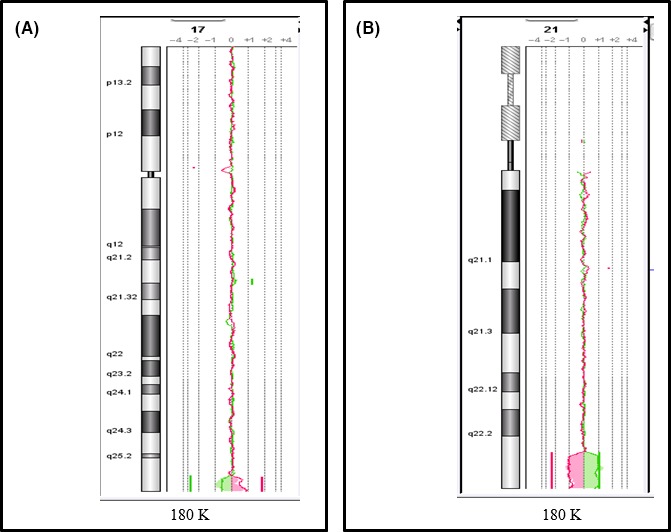
Cytogenetic features of patient 1, carrying an unbalanced translocation leading to 2.54 Mb 17q25.3 duplication spanning from 78,502,910 to 81,060,000 bp (hg19) (A), and a 3.65 Mb 21q22.3 deletion spanning from 44,376,016 to 48,080,911 bp (hg 19) (B).
Family 2
Chromosomal microarray performed in patient 4 showed an unbalanced translocation between the long arms of chromosomes 17 and 21, leading to a 5.15 Mb 17q25.3qter duplication (7,611,654–81,060,000 bp) and a 1.66 Mb 21q22.3qter deletion (46,460,868–48,090,458 bp) (hg 19) (Fig. 4). FISH analysis confirmed the unbalanced translocation in the child and revealed that the mother and the maternal grandmother carried a balanced translocation (17q;21q); the father had normal chromosomes. The maternal uncle carried the same unbalanced translocation found in the child.
Figure 4.
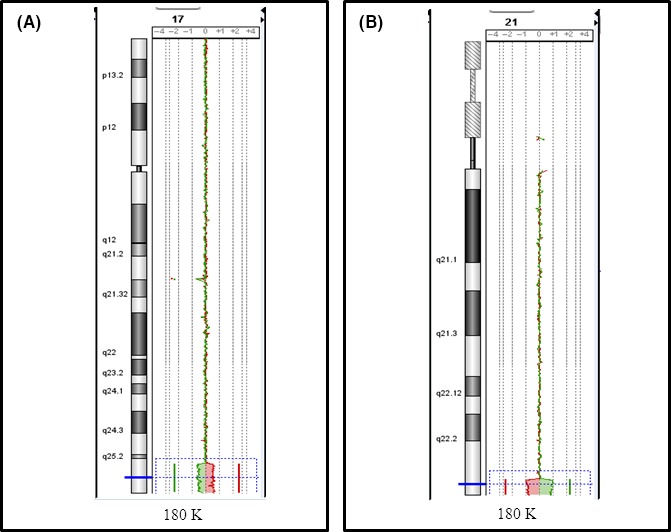
Cytogenetic features of patient 4, carrying an unbalanced translocation leading to a 5.15 Mb 17q25.3ter duplication, spanning from 7,611,654 to 81,060,000 bp (hg19) (A), and a 1.66 Mb 21q22.3ter deletion, spanning from 46,460,868 to 48,090,458 bp (hg 19) (B).
Discussion
We report the clinical and cytogenetic characterization of four children from two unrelated families with syndromic SLD, due to (17q;21q) unbalanced translocations resulting in partial 21q monosomy and partial 17q trisomy.
There is poor information available in the literature about this chromosomal abnormality. While several individuals have been reported carrying terminal 17q duplications associated with concomitant monosomy or trisomy of other chromosomes 3, 4, 5, 6, 7, 8, 9, the phenotype of pure 17q25.3 duplications has been reviewed only recently 10. Deletions and duplications involving this chromosomal region appeared to be frequently associated with congenital heart diseases and neurological involvement 10. It is interesting to notice that, among three patients carrying 17q25.3 duplications previously reported, one showed PDA as patient 4, and one had ADHD as patients 2 and 4 10.
21q22 deletions have been reported in patients showing a wide phenotypic spectrum, including midline defects (frontonasal dysplasia, encephalocele, and corpus callosum agenesis) and learning disability 11; syndromic hypoplastic left heart 12; and MCA/ID including microcephaly and ocular coloboma 13. Thus, it is difficult to evaluate the effect of the 21q partial monosomy on the clinical phenotype observed in the patients here reported.
We reviewed the literature concerning the previously reported cytogenetic causes of SLD. Sex chromosome aneuploidies can be associated with SLI and dyslexia 14, 15. The 7q11.23 duplication syndrome is frequently associated with speech and language difficulties, including speech delay and childhood apraxia of speech; interestingly, median IQ is 85, about 20% of patients have borderline intellectual abilities and only a minority of patients show ID (18%) 14, 16. Velocardiofacial syndrome (“classical” 22q11.2 microdeletion syndrome), distal 22q11.2, and 16p11.2 microdeletions have been reported in association with a wide range of speech and language impairment 14, 17. Among rarer chromosomal abnormalities, Cri‐du‐Chat syndrome, due to deletions of the short arm of chromosome 5, and 12p13.33 deletions have been reported to cause childhood apraxia of speech 18, 19; microdeletions of 1p21.3, 10q22q23, 12p12.1, 15q11.2 and microduplications of 15q11.2q13 and 17p11.2p11.2 have been reported in association with SLI 14; 2q37.3 deletions have been reported in association with DCD 20. Monogenic causes of SLD are rare. In particular, mutations in FOXP2 (OMIM: 605317), FOXP1 (OMIM: 605515), GRIN2A (OMIM: 138253), NRXN1 (OMIM: 600565), and SETBP1 (OMIM 611060) have been reported in patients presenting with speech disorders as well as other neurobehavioral abnormalities 14, 21, 22, 23, 24. A few other loci for SLI have been mapped, such as SLI1 (OMIM: 606711) on chromosome 16q 25, 26, 27; SLI2 (OMIM: 606712) on chromosome 19q 25; SLI3 (OMIM: 607134) on chromosome 13q21 28; SLI4 (612514) on chromosome 7q35‐36, including CNTNAP2 (OMIM: 604569) 29, 30; and SLI5 (OMIM: 615432) on chromosome 2q36.3, including TM4SF20 (OMIM: 615404) 29, 31. To the best of our knowledge, this is the first report of SLI and DCD associated with a chromosomal abnormality involving 21q22.3 and 17q25.3.
It is interesting to notice that a very small 21q22.3 deletion, including only four genes (PCNT, OMIM: 605925; DIP2A, OMIM: 607711; S100B, OMIM: 176990; and PRMT2, OMIM: 601961), has been reported in association with dyslexia; in particular, molecular variants in DIP2A and S100B have been reported in association with an increased risk of developmental dyslexia 32, 33, 34. It might be possible to speculate that these genes might play a role in the SLD observed in the reported patients.
In conclusion, we report the clinical and molecular cytogenetic characterization of four children from two unrelated families, presenting with a SLD caused by cryptic unbalanced translocations leading to 21q22.3 deletion and 17q25.3 duplication, thus expanding the spectrum of chromosomal abnormalities associated with these specific neurodevelopmental disorders.
From a practical point of view, CMA can detect pathogenic CNV in 15% to 20% of individuals with ID and/or multiple congenital abnormalities and in 10% of patients with autistic spectrum disorders (ASD) but the diagnostic rate in SLD is currently unknown, given their heterogeneity. Our study emphasizes the usefulness of CMA in the diagnostic workout assessment of familial SLD.
Authorship
JC and MR: collected the clinical and molecular cytogenetic information about the patients, reviewed the literature, and wrote the manuscript. DH and VdP: referred the patients and provided clinical data. GB and SKP: provided the neuropsychological data. AL, MT, and DS: provided the molecular cytogenetic data. VdP, GL, and PE: critically reviewed the manuscript.
Conflict of Interest
All the authors declare no conflict of interest.
Acknowledgment
We are very grateful to the patients and their family for their cooperation.
Clinical Case Reports 2018; 6(5): 827–834
References
- 1. Fortes, I. S. , Paula C. S., Oliveira M. C., Bordin I. A., de Jesus Mari. J., and Rohde L. A.. 2016. A cross‐sectional study to assess the prevalence of DSM‐5 specific learning disorders in representative school samples from the second to sixth grade in Brazil. Eur. Child Adolesc. Psychiatry 25:195–207. [DOI] [PubMed] [Google Scholar]
- 2. Tannock, R. 2013. Rethinking ADHD and LD in DSM‐5: proposed changes in diagnostic criteria. J. Learn. Disabil. 46:5–25. [DOI] [PubMed] [Google Scholar]
- 3. Bacino, C. A. , Kashork C. D., Davino N. A., and Shaffer L. G.. 2000. Detection of a cryptic translocation in a family with mental retardation using FISH and telomere region‐specific probes. Am. J. Med. Genet. 92:250–255. [PubMed] [Google Scholar]
- 4. Brisset, S. , Kasakyan S., L'Hermine A. C., Mairovitz V., Gautier E., Aubry M. C., et al. 2006. De novo monosomy 9p24.3‐pter and trisomy 17q24.3‐qter characterised by microarray comparative genomic hybridisation in a fetus with an increased nuchal translucency. Prenat. Diagn. 26:206–213. [DOI] [PubMed] [Google Scholar]
- 5. Fryns, J. P. , Parloir C., and Van den Berghe H.. 1979. Partial trisomy 17q. Karyotype: 46, XY, der(21), t(17;21)(q22;p13). Hum. Genet. 49:361–364. [DOI] [PubMed] [Google Scholar]
- 6. Marques, F. , Heredia R., de Oliveira C., Cardoso M. T., Mazzeu J., and Pogue R.. 2015. Partial trisomy 17q and partial monosomy 20q in a boy with craniosynostosis. Am. J. Med. Genet. Part A 167A:412–416. [DOI] [PubMed] [Google Scholar]
- 7. McCann, E. , Sweeney E., Sills J., May P., and Smith S.. 2006. Pfeiffer‐type cardiocranial syndrome: a patient with features of this condition and with an unbalanced subtelomeric rearrangement involving chromosomes 1p and 17q. Clin. Dysmorphol. 15:81–84. [DOI] [PubMed] [Google Scholar]
- 8. Sarri, C. , Gyftodimou J., Avramopoulos D., Grigoriadou M., Pedersen W., Pandelia E., et al. 1997. Partial trisomy 17q22‐qter and partial monosomy Xq27‐qter in a girl with a de novo unbalanced translocation due to a postzygotic error: case report and review of the literature on partial trisomy 17qter. Am. J. Med. Genet. 70:87–94. [DOI] [PubMed] [Google Scholar]
- 9. Velagaleti, G. V. , Jalal S. M., Michaelis R. C., Rowe T. F., Nichols J. R., and Lockhart L. H.. 2003. Molecular cytogenetic characterization of a de novo unbalanced translocation leading to trisomy 17q25–>qter and monosomy 18p11.3–>pter in a girl with dysmorphic features. Clin. Dysmorphol. 12:29–33. [DOI] [PubMed] [Google Scholar]
- 10. Probst, F. J. , James R. A., Burrage L. C., Rosenfeld J. A., Bohan T. P., Ward Melver C. H., et al. 2015. De novo deletions and duplications of 17q25.3 cause susceptibility to cardiovascular malformations. Orphanet J. Rare Dis. 10:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guion‐Almeida, M. L. , Richieri‐Costa A., Jehee F. S., Passos‐Bueno M. R., and Zechi‐Ceide R. M.. 2012. Frontonasal dysplasia, callosal agenesis, basal encephalocele, and eye anomalies syndrome with a partial 21q22.3 deletion. Am. J. Med. Genet. Part A 158A:1676–1679. [DOI] [PubMed] [Google Scholar]
- 12. Ciocca, L. , Digilio M. C., Lombardo A., D'Elia G., Baban A., Capolino R., et al. 2015. Hypoplastic left heart syndrome and 21q22.3 deletion. Am. J. Med. Genet. Part A 167A:579–586. [DOI] [PubMed] [Google Scholar]
- 13. Fukai, R. , Hiraki Y., Nishimura G., Nakashima M., Tsurusaki Y., Saitsu H., et al. 2014. A de novo 1.4‐Mb deletion at 21q22.11 in a boy with developmental delay. Am. J. Med. Genet. Part A 164A:1021–1028. [DOI] [PubMed] [Google Scholar]
- 14. Barnett, C. P. , and van Bon B. W.. 2015. Monogenic and chromosomal causes of isolated speech and language impairment. J. Med. Genet. 52:719–729. [DOI] [PubMed] [Google Scholar]
- 15. Simpson, N. H. , Addis L., Brandler W. M., Slonims V., Clark A., Watson J., et al. 2014. Increased prevalence of sex chromosome aneuploidies in specific language impairment and dyslexia. Dev. Med. Child Neurol. 56:346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mervis, C. B. , Morris C. A., Klein‐Tasman B. P., Velleman S. L., and Osborne L. R.. 2015. 7q11.23 duplication syndrome In: Pagon R. A., Adam M. P., Ardinger H. H., Wallace S. E., Amemiya A., Bean L. J. H., Bird T. D., Fong C. T., Mefford H. C., Smith R. J. H., Stephens K., eds. GeneReviews® [Internet], Seattle, WA. [Google Scholar]
- 17. Raca, G. , Baas B. S., Kirmani S., Laffin J. J., Jackson C. A., Strand E. A., et al. 2013. Childhood apraxia of speech (CAS) in two patients with 16p11.2 microdeletion syndrome. Eur. J. Hum. Genet. 21:455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marignier, S. , Lesca G., Marguin J., Bussy G., Sanlaville D., and des Portes V.. 2012. Childhood apraxia of speech without intellectual deficit in a patient with cri du chat syndrome. Eur. J. Med. Genet. 55:433–436. [DOI] [PubMed] [Google Scholar]
- 19. Thevenon, J. , Callier P., Andrieux J., Delobel B., David A., Sukno S., et al. 2013. 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. Eur. J. Hum. Genet. 21:82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ogura, K. , Takeshita K., Arakawa C., Shimojima K., and Yamamoto T.. 2014. Neuropsychological profiles of patients with 2q37.3 deletion associated with developmental dyspraxia. Am. J. Med. Genet. Part B 165B:684–690. [DOI] [PubMed] [Google Scholar]
- 21. Horn, D. , Kapeller J., Rivera‐Brugues N., Moog U., Lorenz‐Depiereux B., Eck S., et al. 2010. Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum. Mutat. 31:E1851–E1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lai, C. S. , Fisher S. E., Hurst J. A., Vargha‐Khadem F., and Monaco A. P.. 2001. A forkhead‐domain gene is mutated in a severe speech and language disorder. Nature 413:519–523. [DOI] [PubMed] [Google Scholar]
- 23. Lesca, G. , Rudolf G., Bruneau N., Lozovaya N., Labalme A., Boutry‐Kryza N., et al. 2013. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat. Genet. 45:1061–1066. [DOI] [PubMed] [Google Scholar]
- 24. Turner, S. J. , Morgan A. T., Perez E. R., and Scheffer I. E.. 2015. New genes for focal epilepsies with speech and language disorders. Curr. Neurol. Neurosci. Rep. 15:35. [DOI] [PubMed] [Google Scholar]
- 25. SLI Consortium (SLIC) . 2004. Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected by specific language impairment. Am. J. Hum. Genet. 74:1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li, N. , and Bartlett C. W.. 2012. Defining the genetic architecture of human developmental language impairment. Life Sci. 90:469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Newbury, D. F. , Winchester L., Addis L., Paracchini S., Buckingham L. L., Clark A., et al. 2009. CMIP and ATP2C2 modulate phonological short‐term memory in language impairment. Am. J. Hum. Genet. 85:264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bartlett, C. W. , Flax J. F., Logue M. W., Vieland V. J., Bassett A. S., Tallal P., et al. 2002. A major susceptibility locus for specific language impairment is located on 13q21. Am. J. Hum. Genet. 71:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bartlett, C. W. , Flax J. F., Logue M. W., Smith B. J., Vieland V. J., Tallal P., et al. 2004. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum. Hered. 57:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Centanni, T. M. , Sanmann J. N., Green J. R., Iuzzini‐Seigel J., Bartlett C., Sanger W. G., et al. 2015. The role of candidate‐gene CNTNAP2 in childhood apraxia of speech and specific language impairment. Am. J. Med. Genet. Part B 168:536–543. [DOI] [PubMed] [Google Scholar]
- 31. Wiszniewski, W. , Hunter J. V., Hanchard N. A., Willer J. R., Shaw C., Tian Q., et al. 2013. TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am. J. Hum. Genet. 93:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kong, R. , Shao S., Wang J., Zhang X., Guo S., Zou L., et al. 2015. Genetic variant in DIP2A gene is associated with developmental dyslexia in Chinese population. Am. J. Med. Genet. Part B 171:203–208. https://doi.org/10.1002/ajmg.b.32392. [DOI] [PubMed] [Google Scholar]
- 33. Matsson, H. , Huss M., Persson H., Einarsdottir E., Tiraboschi E., Nopola‐Hemmi J., et al. 2015. Polymorphisms in DCDC2 and S100B associate with developmental dyslexia. J. Hum. Genet. 60:399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Poelmans, G. , Engelen J. J., Van Lent‐Albrechts J., Smeets H. J., Schoenmakers E., Franke B., et al. 2009. Identification of novel dyslexia candidate genes through the analysis of a chromosomal deletion. Am. J. Med. Genet. Part B 150B:140–147. [DOI] [PubMed] [Google Scholar]