

Amorphous forms serve as thermodynamic upper bounds on the free energy scale for synthesis of metastable crystalline polymorphs.
Abstract
Realizing the growing number of possible or hypothesized metastable crystalline materials is extremely challenging. There is no rigorous metric to identify which compounds can or cannot be synthesized. We present a thermodynamic upper limit on the energy scale, above which the laboratory synthesis of a polymorph is highly unlikely. The limit is defined on the basis of the amorphous state, and we validate its utility by effectively classifying more than 700 polymorphs in 41 common inorganic material systems in the Materials Project for synthesizability. The amorphous limit is highly chemistry-dependent and is found to be in complete agreement with our knowledge of existing polymorphs in these 41 systems, whether made by the nature or in a laboratory. Quantifying the limits of metastability for realizable compounds, the approach is expected to find major applications in materials discovery.
INTRODUCTION
Metastable materials, from hardened alloys to polymorphs of titania, silica, or alumina to carbon, can be obtained using a variety of techniques including rapid cooling, physical or chemical deposition, soft chemical or combinatorial synthesis, compression, and mechanical attrition (1–8). Discovery of these novel metastable materials for target applications has become one of the main pillars in advancing technologies (9–11), which has been revolutionized by high-throughput density functional theory (DFT) and the material databases that emerged with it (9, 12–14). The biggest challenge in this accelerated materials design paradigm is the lack of the capability to predict the synthesizability of materials (15, 16).
Prediction of synthesis pathways for a computer-designed material is a formidable task, if not impossible, because it requires computation of enthalpy and entropy functions and barriers to phase transformations between all competing phases under conditions pertaining to a chosen synthesis technique. Researchers instead use heuristic limits, chemical intuition, or rules of thumb to estimate the likelihood for successful synthesis of materials. Ab initio methods can be used to calculate the convex hull of a chemical space, which allows us to gauge the energy difference between the candidate compound and the ground-state phase(s). Large differences tend to correlate with increasing difficulty to realize and retain the candidate material. For example, a reasonable multiple of room temperature kBT (~25, 50, or up to 100 meV/atom) is usually cited as a soft criterion for synthesizability (11, 13, 16–18), with fair but crude presumptions including, for example, that such values are comparable to the magnitude of possible entropic contributions to free energy at finite temperatures. These numbers are often chosen conservatively, that is, they are low enough to justify the likelihood that those materials can be made. However, a recent analysis (16) in a curated set of ~30,000 inorganic materials obtained from the Materials Project (9) showed that the energy distance to the ground state at the 90th percentile of previously synthesized metastable polymorphs has considerable variation among different material classes such as oxides, nitrides, and others, with values ranging from ~0.05 to ~0.2 eV/atom.
To establish a fundamental energy limit for synthesizability, we draw inspiration from the process of synthesis itself, where precursors are decomposed to form new bonds and subsequently a new crystal structure. In this process, the free energy landscape provides a thermodynamic driving force, balanced by kinetic limitations depending on thermodynamic conditions, such as temperature, pressure, or other thermodynamic handles. If these conditions (for example, high temperature) can break the bonds (a prerequisite of synthesis), then an amorphous state can always be kinetically accessed as it represents a first “melt” of the precursors. Any crystalline polymorph will have to compete—both kinetically and thermodynamically—with a variety of noncrystalline states, represented, for simplicity, by the term amorphous. Here, we introduce the amorphous limit—a system-specific energetic upper bound for synthesizability of metastable crystalline polymorphs that can be calculated using ab initio methods—and demonstrate its application in a wide range of inorganic polymorphic systems.
RESULTS AND DISCUSSION
We hypothesize that if the enthalpy of a crystalline phase at T = 0 K is higher than that of an amorphous phase at the same composition, then that compound cannot be synthesized at any finite temperature, if all other conditions are kept constant. The thermodynamic argument behind this hypothesis is that the rate of Gibbs free energy decrease of a material with temperature at constant pressure is proportional to its entropy [(∂G/∂T)p = − S]. Because the entropy of the amorphous phase, as derived from the liquid, is almost invariably larger than that of a corresponding crystalline phase (19–22), the rate of decrease in G with T is the highest for the amorphous phase (and its extension to the supercooled liquid) among condensed phases. As illustrated in Fig. 1, a material with a higher zero-temperature free energy than the amorphous phase, such as polymorph A, cannot close this gap at finite temperatures and constant pressure. Because a phase transformation is only possible from a higher to a lower free energy, polymorph A cannot be stabilized via temperature control, for example, with a heat-anneal-quench route, or via crystallization from a precursor phase on this G-T domain. Conversely, polymorphs B and C, which have lower free energies than the amorphous phase at T = 0 K, have the thermodynamic requisites for synthesis within the G-T domain. These polymorphs can be accessed from the liquid/amorphous phase (polymorph B or C) or another crystal (polymorph C).
Fig. 1. A schematic Gibbs free energy (G) versus temperature (T) diagram typically used to explain polymorphic systems (52).
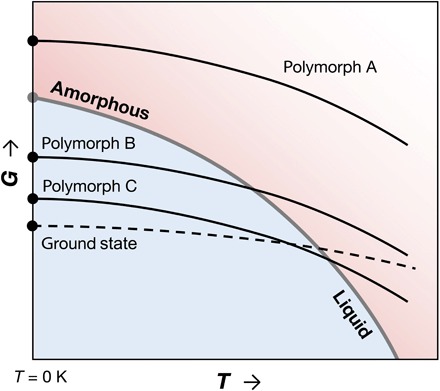
Free energies of three crystalline polymorphic phases (A, B, and C) and the amorphous phase are shown relative to the ground-state crystal. Although a deviation from the projection of liquid free energies to lower temperatures is expected, the amorphous phase is depicted as a continuation of the liquid phase as often assumed. At T = 0 K, G ≈ E (internal energy), as pressure-volume contributions to enthalpy are negligible near ambient pressure for condensed phases.
Temperature and pressure are naturally the most common thermodynamic handles pertaining to synthesis, but other handles such as electrochemical or mechanical forces or chemical potential can also play a role, for example, in deposition, ion exchange, irradiation, ion bombardment, or mechanical alloying, which may provide access to high-energy polymorphs. When these handles are released after synthesis, if such a polymorph has a higher free energy than the amorphous form in G-T (analogous to polymorph A in Fig. 1), and if crystallization kinetics are too slow for transformation to a lower energy crystal, then amorphization often occurs spontaneously or catastrophically (23–26). Underlying mechanisms include heterogeneous nucleation of an amorphous phase at ubiquitous two-dimensional defects, like grain boundaries, and mechanical instability of the crystal, in analogy with why non-negligible superheating of a crystal is rare (23, 25). On the basis of ample experimental evidence on the crystal-to-amorphous transformation, combined with such mechanisms, Johnson (23) pointed out that a metastable crystalline solid energetically less stable than the amorphous phase cannot survive.
Therefore, approaching zero temperature, one reaches an amorphous limit on the energy scale that can be used to establish a necessary condition for the synthesis and subsequent stabilization of polymorphs at finite temperatures, at a constant pressure. This limit can be estimated by sampling amorphous microstates approaching zero temperature, that is, by mapping out the potential energy landscape (PEL) of the amorphous system (27, 28). Laboratory time scales may allow systems to explore enough microstates to find the low-energy configurations that dominate at low temperatures, before the amorphous phase gets trapped in one (29). However, because of drastically shorter time scales in computations and limited sampling of low-lying basins by the fluctuations in local energy minima in high-temperature simulations of liquids (28, 30), simulations will unequivocally overestimate the energy of the amorphous phase approaching zero temperature. We can therefore adopt a practical definition for the amorphous limit as “the lowest energy among all ab initio sampled configurations.” Hence, the limit is fail-safe in a “variational” sense, that is, it can only decrease as we sample more configurations. By construction, it self-avoids false negatives, that is, it cannot classify any synthesizable material as nonsynthesizable, regardless of computational limitations in sampling. Although the limit is a function of pressure, and hence holds for any fixed finite pressure, we demonstrate its utility near zero pressure, which covers most synthesis conditions (low/ambient pressures) and is consistent with the energetics of crystalline materials obtained via typical high-throughput computations, such as in the Materials Project, allowing comparisons to polymorphs therein.
To test our hypothesis, we identified a set of 41 technologically important material systems from semiconductors to dielectrics, with a focus on well-studied oxide chemistries, elemental C and Si, and important compounds from other metal-anion chemistries such as nitrides. We approximate the energetics of corresponding amorphous states using a fully ab initio procedure commonly used in literature. Figure 2A shows the calculated energies of these amorphous structures and the corresponding crystalline polymorphs in the Materials Project, relative to the ground state. These polymorphs include all respective entries in the Materials Project database (9) and therefore nearly all ordered structures available in the Inorganic Crystal Structure Database (ICSD) (31) (which is composed mostly, but not exclusively, of experimental reports of synthesized materials) and hypothetical (non-ICSD) structures already existing in the database (such as those from high-throughput prototyping or ordering of disordered ICSD structures). In the corresponding probability distribution functions (PDFs) in Fig. 2B, the energies of crystalline polymorphs show a heavy-tailed negative exponential distribution similar to the trend observed by Sun et al. (16). On the other hand, amorphous materials show a broad PDF with a major peak near ~0.25 eV/atom, but with a strong positive skew toward lower energies. There is a significant overlap between the lower-energy tail of PDFs of amorphous materials and the PDF of crystals, including those in the ICSD. This overlap reveals a critical point that is frequently overlooked in materials discovery: Amorphous phases are, for some chemical systems, highly thermodynamically competitive.
Fig. 2. Assessing the crystalline synthesizability in 41 material systems in the “stability skyline” defined by the amorphous limits.
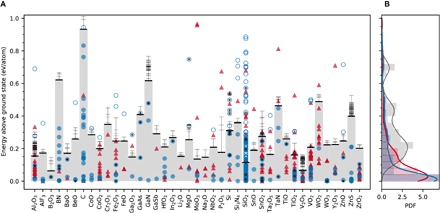
(A) Energies of inorganic amorphous materials (horizontal bars, amorphous limits in bold) are compared to the crystalline polymorphs available in the Materials Project. Synthesizability ranges defined by the amorphous limits are shaded in gray. Circles and triangles correspond to polymorphs with and without existing ICSD entries, respectively. A circle is open if the ICSD-acquired polymorph is above the amorphous limit and falls under at least one of the exception categories described in the text. (B) Corresponding PDFs for ICSD (in blue) and non-ICSD (in red) crystalline polymorphs, compared to amorphous polymorphs (histogram). ICSD structures that have been associated with “high-pressure” synthesis are further tagged with a solid black circle. Units of PDFs are atom per electron volt.
For each chemical system in Fig. 2A, the amorphous limit (table S1) splits the energy scale into two halves. Although many crystalline polymorphs are below their respective amorphous limit, a significant number of them (>150) are above it, and whether these are synthesized materials or not presents a rigorous test case for our hypothesis. Therefore, we carefully looked at the sources and references of the structures of these polymorphs above the amorphous limits and found that—without any exceptions—they fall under at least one of these categories: (i) hypothetical structure with no ICSD entry (for example, from prototyping), (ii) hypothetical structure listed in the ICSD (for example, zeolites), (iii) high-pressure structure listed in the ICSD, and (iv) erroneous ICSD entry or magnetic ordering (Supplementary Text). That is, within this set of 41 material systems, and more than 700 polymorphs, the amorphous limit resulted in zero false negatives when classifying experimentally known polymorphs as within the limit of synthesizability and has proven to be an accurate metric for quantifying accessible metastability.
We observe that the amorphous limits show strong chemical sensitivity. In the broadly explored class of metal oxides, the limits range from ~0.05 to ~0.5 eV/atom in Fig. 2A. Near the lower end of the scale are the glass- and network-forming oxides B2O3, SiO2, and V2O5. Glassy B2O3 is known for its inability to thermally crystallize under ambient pressure (32), in agreement with its low amorphous limit. A pronounced compositional dependence for the amorphous limit is observed among several oxides, for example, of Sn, Co, Ti, V, and W. The limits change significantly across material classes, for example, in B, Si, and Ta oxides versus nitrides, in Zn oxide versus sulfide, and among the four Ga chemical systems. Nitrides, which form metastable polymorphs in a much wider energy window compared to other chemistries (16), are consistently found to have high amorphous limits. Besides nitrides, C and Si are examples where a strong preference for covalently bonded structures leads to a high limit. Bucky-ball C60, for instance, a famous carbon allotrope, which is a molecular conformation that is above the ground-state graphite by almost half an electron volt per carbon atom (33), is within the amorphous limit.
The amorphous limits are controlled by a complex interplay between the character of the chemical bonding and its flexibility to conform to the packing in the amorphous phase. In accordance with the conventional understanding of glasses (34), radial and bond-angle distribution functions (figs. S1 to S42 and S43 to S83, respectively) hint that amorphous phases exhibiting rigid polyhedral units (sharper radial and angular distributions within units) consisted of smaller cations at the center and, with flexible polyhedral connections (for example, broader angular distributions for metal-anion-metal triplets), tend toward lower energies and hence provide lower amorphous limits, as in oxide systems like B2O3, SiO2, and V2O5. When the bonds are strong but lack the flexibility to conform to an efficient three-dimensional packing, the amorphous limits tend to increase significantly, as in many nitride systems, where these bonds can lock in very high energy metastable structures (16). However, there is still no universal description for the energetics of the amorphous materials beyond these observations. The ability to quantify these limits by ab initio methods paves the way for exploring the practical ranges of synthesizable metastability in inorganic materials without any a priori knowledge of the particular chemistry and the underlying complexity.
From a materials design perspective, successful synthesis is a major bottleneck for realizing new technological applications; hence, determining which novel functional materials are synthesizable or not is of vital importance. The amorphous limit shows remarkable accuracy and chemical sensitivity and is well positioned to replace and significantly improve widely used heuristic limits in joint experimental-computational materials discovery studies (11, 13, 16–18). Using rules of thumb or heuristic limits imposes arbitrary limitations, and a significant number of potentially useful, synthesizable materials may be discarded based on a low arbitrary limit, or vice versa. For example, a heuristic limit of 0.1 eV/atom for B2O3 would yield many false positives, whereas the same filter for BN would yield many false negatives. In Fig. 3, we show that in a target polymorph search in the systems studied in Fig. 2, heuristic limits such as 0.025, 0.05, and 0.1 eV/atom would, on average, exclude approximately 63, 39, and 26% of known synthesized polymorphs per system, whereas the corresponding amorphous limits exclude none. Although increasing the heuristic limit enables the capture of more synthesizable materials and the reduction of the number of “false negatives,” it would also inevitably enlarge the number of “false positives.” This latter metric is difficult to quantify because it is extremely challenging to experimentally exhaust every possible means of synthesis to label a hypothetical material as a true false positive. Nevertheless, given the available data, which indicate that the amorphous limit accurately labels materials above it as “unsynthesizable,” we expect that any excess energy window introduced by a heuristic limit above the corresponding chemically sensitive amorphous limits will exclusively result in false positives. As shown in Fig. 3, the magnitude of the unsynthesizable energy range, which contains no experimentally verified materials, increases quickly with the value of the heuristic limit. For example, if a heuristic limit is increased to 0.35 eV/atom to reach a sensitivity (capture rate of synthesized materials) of ~95%, then the upper ~0.15 eV/atom of that limit will consist exclusively of unsynthesizable materials for systems with amorphous limits smaller than the heuristic limit. Thus, it is not possible to find a single heuristic limit that works well across the broad range of chemistries and structures. On the other hand, the amorphous limit is system-specific and consistently identifies the narrowest energy range for synthesizability that is highly likely to exclude zero materials as false negatives.
Fig. 3. Performance of constant heuristic limits in capturing synthesized metastable materials (“sensitivity”) and excluding the “unsynthesizable ranges.”.
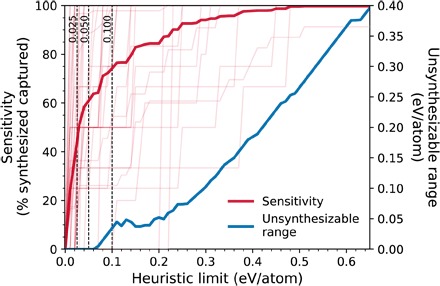
The sensitivity is defined as the percentage of known synthesized materials in a system that are within a given constant heuristic energy limit from the ground state. Sensitivities of heuristic limits for individual systems are also plotted in the background (thin lines) as a guide for the eye. The unsynthesizable range is defined for a system when a heuristic limit is greater than its amorphous limit, as the excess energy range between these two limits, averaged over these systems at each heuristic limit value.
Although the amorphous limit extends to energy ranges beyond what we expect from intuition, we should emphasize that, for all systems in Fig. 2A, the amorphous phase can be made, indicating that any crystalline polymorph below that limit can be accessed downhill on the free energy scale; in other words, it can possibly be synthesized if proper kinetics and pathways are available. For example, even in the GaN system with an amorphous limit exceeding half an electron volt in Fig. 2A, two high-energy polymorphs close to the limit were observed in the laboratory near ambient conditions (35, 36). Moreover, stishovite, a high-pressure SiO2 polymorph, is known to exhibit spontaneous amorphization under decompression (24, 25) and is consistently found to be above the amorphous limit of SiO2. Although we focused on bulk materials here, in materials dominated by surface effects (for example, in nanomaterials), the stability of the amorphous phase may increase relative to the crystalline (26), implying that the “simpler-to-calculate” bulk classification presented may hold in most cases. In general, however, one needs to ensure that the thermodynamic conditions under which the polymorph exists and the calculation of the amorphous limit are consistent. For example, the present analysis pertains to the stability of bulk phases at low/near-ambient pressure; however, for stability under high pressure or under conditions purely dominated by surface/interface effects, one needs to compute the energies and amorphous limits under the corresponding thermodynamic conditions.
The accuracy in classifying synthesizability with the amorphous limit depends on the error due to limited sampling of the PEL (27) and how accurately DFT describes polymorph energetics. In Fig. 4, we show that the sampling error of the amorphous limit decreases with increasing sample size (also see fig. S84) and estimate the error to be typically between ~15 and ~30 meV/atom for sample sizes as small as five to eight configurations, consistent across different chemistries. Despite certain limitations, modern DFT is known to consistently map inorganic energy landscapes, especially within the same chemistry, with errors comparable to experiments (14, 37, 38). In general, the DFT accuracy in the computed relative energies of polymorphs is expected to be within ~24 meV/atom (Supplementary Text) (38). To further analyze the possible effect of DFT errors on our methodology, we first identify Gaussian distributions for random DFT errors that would still identify the observed, correct ground-state structures for each polymorphic system in Fig. 4 with 90% probability (fig. S86 and Supplementary Text). Even when the condition is relaxed to finding the observed ground state to within 5 to 10 meV/atom, we estimate the maximum permissible levels of error for relative energies in such polymorphic systems to be ~12 meV/atom or less, that is, smaller than the ~24 meV/atom Hautier et al. (38) found for reactions. Second, we applied a statistical test to approximate the probability that at least one experimentally verified material is misclassified as unsynthesizable on the basis of abovementioned uncertainties in the amorphous energies and the DFT-calculated crystalline polymorphs, and we confirm that the presented methodology provides statistically significant and sufficiently accurate results for the classification of synthesizability for a wide range of chemistries (fig. S87 and Supplementary Text).
Fig. 4. Amorphous limit sampling error as a function of sample size estimated for four different systems.
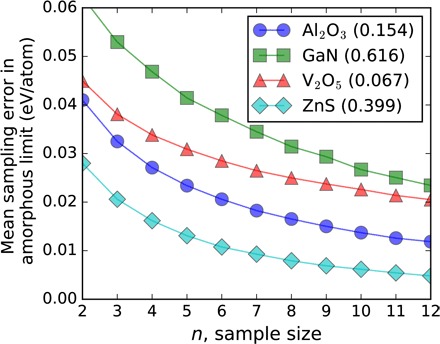
The values of amorphous limits are also given in parentheses in electron volts per atom for comparison with the errors. The error in the amorphous limit due to limited sampling is mostly independent of the value of the amorphous limit. The error is “fail-safe” for materials discovery applications because it is guaranteed to be in only one direction, that is, the actual amorphous limit can only be lower than the limit found by a sample size of n, which prevents excluding potentially revolutionary functionality that is still synthesizable. See Supplementary Text and fig. S84 for further details.
Discovery and synthesis of functional metastable materials for future innovation is an imperative but daunting task. For any polymorph, being within the “amorphous limit” is shown to be a necessary condition for synthesizability because the pathways to polymorphs above it are thermodynamically blocked on the G-T domain, but it is not a sufficient condition because whether a realizable pathway would exist in laboratory is currently not possible to foresee. In the event of successful synthesis, the lifetime of the resulting metastable polymorph will be controlled by the kinetics. The amorphous limit completes a part of this puzzle by identifying the subset of suggested (by experimentalists or theorists) novel materials, which are potentially amenable to synthesis and, most importantly, by ruling out those that are absolutely not. We envision this approach to provide an important first step toward bridging the gap between novel materials prediction and successful synthesis, and toward accelerated materials discovery.
MATERIALS AND METHODS
Study design
Finding the amorphous limit requires exploring the PEL of the amorphous system to locate the low-lying basins. These atomic models of amorphous structures can be generated using a variety of procedures such as melt-quench routes and energy minimization techniques (39, 40). We adopted a common PEL exploration strategy (28, 30) where a certain number of independent configurations from the high-temperature parent liquid were selected and relaxed to their nearby local minima. Our ab initio workflow for generating these amorphous structures starts with constrained random packing of N atoms of the given material in a cubic box using packmol (41) at a molar volume 20% larger than that of the ground-state crystal structure at the same composition in the Materials Project database. N was chosen as the smallest integer larger than 100 that could represent the composition exactly. Starting with this configuration, we performed ab initio molecular dynamics (AIMD) simulations with 5000 steps of equilibration followed by a 5000-step production run for each material in an NVT ensemble with a 2-fs time step. For each material, the AIMD temperature was selected to be in the range of 3000 to 5000 K, at least ~500 K above the melting point to ensure rapid equilibration of the liquid. Around five independent isochronal “snapshots” were selected from the production stage of these AIMD runs and effectively quenched to 0 K by further conjugate gradient optimization of all geometrical degrees of freedom (that is, resembling an extremely fast quench that locks the structure in its basin) to relax the configurations to the local minimum of the corresponding basin in the PEL and obtain the energies of representative amorphous configurations. Generated atomic structures were further investigated in sections below and in the Supplementary Materials to ensure that an amorphous configuration was achieved for each case. This workflow to generate amorphous materials with DFT used pymatgen (42), custodian (42), Fireworks (43), and atomate (42, 43) and can be found at https://github.com/materialsproject/mpmorph.
Verification of amorphous structures
Development of a clearly defined short-range order and lack of crystallization in amorphous structures were confirmed by calculating the partial radial distribution and bond-angle distribution functions (figs. S1 to S42 and S43 to S83, respectively, with corresponding details in Supplementary Text) and can also be observed in their sample ball-stick models provided (fig. S85). For Al2O3, we further compared the radial distribution functions and bond lengths to the experimental data (44) and verified that our procedure accurately captured the local structure of the amorphous phases (Supplementary Text and fig. S1).
DFT calculations
All first-principles calculations (including AIMD and structure optimizations) were performed using Vienna Ab initio Simulation Package (VASP) (45, 46) and the Perdew-Burke-Ernzerhof (47) formulation of generalized gradient approximation with projector-augmented wave potentials (48, 49). AIMD simulations were done using the Γ-point only at the largest VASP-recommended default kinetic energy cutoff of constituent elements. Structure optimization and static calculations of snapshots from trajectories were done using the higher-accuracy DFT settings of the Materials Project (9) for consistency with crystalline materials therein. Structures are visualized using VESTA (50).
Statistical analysis
The sampling error in the amorphous limit as shown in Fig. 4 was estimated by the expected value of the statistical distributions of energies of independent amorphous configurations, obtained from randomly choosing a subset of n configurations from larger populations of amorphous configurations for each system. The population size (N) of independent, ab initio generated amorphous structures is 50 for Al2O3, 35 for GaN, 46 for V2O5, and 44 for ZnS. The statistical distributions, which are shown in fig. S84, were obtained by repeating the random sampling 103 times for each system. Statistical evaluation of permissible random DFT errors in polymorphic systems and their combined effect with the variance in amorphous energies on classification accuracy of amorphous limit is available in the Supplementary Materials.
Continuous PDFs of energies of crystalline polymorphs and amorphous structures in Fig. 2B (solid lines) were represented using a kernel density estimation (KDE) with a bandwidth of 0.035 eV/atom. KDEs were performed using the scikit-learn python package (51).
Supplementary Material
Acknowledgments
Funding: This work was intellectually led by the Center for Next Generation of Materials Design, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Basic Energy Science (award no. DE-AC36-08GO28308). M.A. was also supported as part of the Computational Materials Sciences Program funded by the DOE, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division (award no. DE-SC0014607). This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the DOE under contract no. DE-AC02-05CH1123. Author contributions: M.A. and K.A.P. proposed the concept and conceived this project. M.A. performed the calculations and analysis, with input from S.S.D and W.S. M.A. wrote the manuscript with input from S.S.D., W.S., and K.A.P. All authors extensively contributed to the discussion of the results. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials and/or can be accessed at www.materialsproject.org. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaaq0148/DC1
Supplementary Text
figs. S1 to S42. Radial distribution functions of amorphous configurations.
figs. S43 to S83. Bond-angle distribution functions of amorphous configurations.
fig. S84. Amorphous limit sampling probability.
fig. S85. Snapshots of atomic structures of amorphous materials.
fig. S86. Probability of finding the correct, observed ground states.
fig. S87. PDFs from aggregated uncertainties in the amorphous limit classification of crystalline polymorphs.
table S1. The amorphous limits for the synthesizability of polymorphs.
database S1. Energies of amorphous configurations.
REFERENCES AND NOTES
- 1.Turnbull D., Metastable structures in metallurgy. Metall. Trans. B 12, 217–230 (1981). [Google Scholar]
- 2.Koinuma H., Takeuchi I., Combinatorial solid-state chemistry of inorganic materials. Nat. Mater. 3, 429–438 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Gopalakrishnan J., Chimie douce approaches to the synthesis of metastable oxide materials. Chem. Mater. 7, 1265–1275 (1995). [Google Scholar]
- 4.Prinz G. A., Stabilization of bcc Co via epitaxial growth on GaAs. Phys. Rev. Lett. 54, 1051–1054 (1985). [DOI] [PubMed] [Google Scholar]
- 5.Jones H., Splat cooling and metastable phases. Rep. Prog. Phys. 36, 1425–1497 (1973). [Google Scholar]
- 6.Helmersson U., Lattemann M., Bohlmark J., Ehiasarian A. P., Gudmundsson J. T., Ionized physical vapor deposition (IPVD): A review of technology and applications. Thin Solid Films 513, 1–24 (2006). [Google Scholar]
- 7.Badding J. V., Parker L. J., Nesting D. C., High pressure synthesis of metastable materials. J. Solid State Chem. 117, 229–235 (1995). [Google Scholar]
- 8.Bakker H., Zhou G. F., Yang H., Mechanically driven disorder and phase transformations in alloys. Prog. Mater. Sci. 39, 159–241 (1995). [Google Scholar]
- 9.Jain A., Ong S. P., Hautier G., Chen W., Richards W. D., Dacek S., Cholia S., Gunter D., Skinner D., Ceder G., Persson K. A., Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 11002 (2013). [Google Scholar]
- 10.Hautier G., Fischer C. C., Jain A., Mueller T., Ceder G., Finding nature’s missing ternary oxide compounds using machine learning and density functional theory. Chem. Mater. 22, 3762–3767 (2010). [Google Scholar]
- 11.Hautier G., Jain A., Ping Ong S., Kang B., Moore C., Doe R., Ceder G., Phosphates as lithium-ion battery cathodes: An evaluation based on high-throughput ab initio calculations. Chem. Mater. 23, 3495–3508 (2011). [Google Scholar]
- 12.Jain A., Persson K. A., Ceder G., Research Update: The materials genome initiative: Data sharing and the impact of collaborative ab initio databases. APL Mater. 4, 53102 (2016). [Google Scholar]
- 13.Saal J. E., Kirklin S., Aykol M., Meredig B., Wolverton C., Materials design and discovery with high-throughput density functional theory: The Open Quantum Materials Database (OQMD). JOM 65, 1501–1509 (2013). [Google Scholar]
- 14.Kirklin S., Saal J. E., Meredig B., Thompson A., Doak J. W., Aykol M., Rühl S., Wolverton C., The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. npj Comput. Mater. 1, 15010 (2015). [Google Scholar]
- 15.Price S. L., Why don’t we find more polymorphs? Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 69, 313–328 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Sun W., Dacek S. T., Ong S. P., Hautier G., Jain A., Richards W. D., Gamst A. C., Persson K. A., Ceder G., The thermodynamic scale of inorganic crystalline metastability. Sci. Adv. 2, e1600225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirklin S., Saal J. E., Hegde V. I., Wolverton C., High-throughput computational search for strengthening precipitates in alloys. Acta Mater. 102, 125–135 (2016). [Google Scholar]
- 18.Mueller T., Hautier G., Jain A., Ceder G., Evaluation of tavorite-structured cathode materials for lithium-ion batteries using high-throughput computing. Chem. Mater. 23, 3854–3862 (2011). [Google Scholar]
- 19.Jäckle J., On the glass transition and the residual entropy of glasses. Philos. Mag. B 44, 533–545 (1981). [Google Scholar]
- 20.R. Zallen, The Physics of Amorphous Solids (Wiley-VCH, 2004). [Google Scholar]
- 21.Johari G. P., On the excess entropy of disordered solids. Philos. Mag. B 41, 41–47 (1980). [Google Scholar]
- 22.Angell C. A., Thermodynamic aspects of the glass transition in liquids and plastic crystals. Pure Appl. Chem. 63, 1387–1392 (1991). [Google Scholar]
- 23.Johnson W. L., Crystal-to-glass transformation in metallic materials. Mater. Sci. Eng. 97, 1–13 (1988). [Google Scholar]
- 24.Dachille F., Zeto R. J., Roy R., Coesite and stishovite: Stepwise reversal transformations. Science 140, 991–993 (1963). [DOI] [PubMed] [Google Scholar]
- 25.Sciortino F., Essmann U., Stanley H. E., Hemmati M., Shao J., Wolf G. H., Angell C. A., Crystal stability limits at positive and negative pressures, and crystal-to-glass transitions. Phys. Rev. E 52, 6484–6491 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Reichel F., Jeurgens L. P. H., Mittemeijer E. J., The thermodynamic stability of amorphous oxide overgrowths on metals. Acta Mater. 56, 659–674 (2008). [Google Scholar]
- 27.Sciortino F., Potential energy landscape description of supercooled liquids and glasses. J. Stat. Mech. 2005, P05015 (2005). [Google Scholar]
- 28.Sciortino F., Kob W., Tartaglia P., Inherent structure entropy of supercooled liquids. Phys. Rev. Lett. 83, 3214–3217 (1999). [Google Scholar]
- 29.J. C. Mauro, R. J. Loucks, A. K. Varshneya, P. K. Gupta, in Scientific Modeling and Simulations, S. Yip, T. D. de la Rubia, Eds. (Springer, 2009), pp. 241–281. [Google Scholar]
- 30.Sastry S., Debenedetti P. G., Stillinger F. H., Signatures of distinct dynamical regimes in the energy landscape of a glass-forming liquid. Nature 393, 554–557 (1998). [Google Scholar]
- 31.Belsky A., Hellenbrandt M., Karen V. L., Luksch P., New developments in the Inorganic Crystal Structure Database (ICSD): Accessibility in support of materials research and design. Acta Crystallogr. B 58, 364–369 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Ferlat G., Seitsonen A. P., Lazzeri M., Mauri F., Hidden polymorphs drive vitrification in B2O3. Nat. Mater. 11, 925–929 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Diogo H. P., Minas da Piedade M. E., Dennis T. J. S., Hare J. P., Kroto H. W., Taylor R., Walton D. R. M., Enthalpies of formation of Buckminsterfullerene C60 and of the parent ions and . J. Chem. Soc. Faraday Trans. 89, 3541–3544 (1993). [Google Scholar]
- 34.Revesz A. G., Fehlner F. P., The role of noncrystalline films in the oxidation and corrosion of metals. Oxid. Met. 15, 297–321 (1981). [Google Scholar]
- 35.Xie Y., Quan Y., Wang W., Zhang S., Zhang Y., A benzene-thermal synthetic route to nanocrystalline GaN. Science 272, 1926–1927 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Gajbhiye N. S., Bhattacharyya S., Shivaprasad S. M., Synthesis and characterization of ɛ-Fe3N/GaN, 54/46-composite nanowires. Mater. Res. Bull. 43, 272–283 (2008). [Google Scholar]
- 37.Lejaeghere K., Bihlmayer G., Björkman T., Blaha P., Blügel S., Blum V., Caliste D., Castelli I. E., Clark S. J., Dal Corso A., de Gironcoli S., Deutsch T., Dewhurst J. K., Di Marco I., Draxl C., Dułak M., Eriksson O., Flores-Livas J. A., Garrity K. F., Genovese L., Giannozzi P., Giantomassi M., Goedecker S., Gonze X., Grånäs O., Gross E. K., Gulans A., Gygi F., Hamann D. R., Hasnip P. J., Holzwarth N. A., Iuşan D., Jochym D. B., Jollet F., Jones D., Kresse G., Koepernik K., Küçükbenli E., Kvashnin Y. O., Locht I. L., Lubeck S., Marsman M., Marzari N., Nitzsche U., Nordström L., Ozaki T., Paulatto L., Pickard C. J., Poelmans W., Probert M. I., Refson K., Richter M., Rignanese G.-M., Saha S., Scheffler M., Schlipf M., Schwarz K., Sharma S., Tavazza F., Thunström P., Tkatchenko A., Torrent M., Vanderbilt D., van Setten M. J., Van Speybroeck V., Wills J. M., Yates J. R., Zhang G.-X., Cottenier S., Reproducibility in density functional theory calculations of solids. Science 351, aad3000. (2016). [DOI] [PubMed] [Google Scholar]
- 38.Hautier G., Ong S., Jain A., Moore C., Ceder G., Accuracy of density functional theory in predicting formation energies of ternary oxides from binary oxides and its implication on phase stability. Phys. Rev. B 85, 155208 (2012). [Google Scholar]
- 39.Rosen J., Warschkow O., Electronic structure of amorphous indium oxide transparent conductors. Phys. Rev. B 80, 115215 (2009). [Google Scholar]
- 40.Zawadzki P. P., Perkins J., Lany S., Modeling amorphous thin films: Kinetically limited minimization. Phys. Rev. B 90, 094203 (2014). [Google Scholar]
- 41.Martínez H., Andrade R., Birdgin E. G., Martínez J. M., Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Ong S. P., Richards W. D., Jain A., Hautier G., Kocher M., Cholia S., Gunter D., Chevrier V. L., Persson K. A., Ceder G., Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013). [Google Scholar]
- 43.Jain A., Ong S. P., Chen W., Medasani B., Qu X., Kocher M., Brafman M., Petretto G., Rignanese G.-M., Hautier G., Gunter D., Persson K. A., FireWorks: A dynamic workflow system designed for high-throughput applications. Concur. Comput. Pract. Exp. 27, 5037–5059 (2015). [Google Scholar]
- 44.Lamparter P., Kniep R., Structure of amorphous Al2O3. Phys. B Condens. Matter 234–236, 405–406 (1997). [Google Scholar]
- 45.Kresse G., Furthmüller J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 47.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Blöchl P. E., Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- 49.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- 50.Momma K., Izumi F., VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011). [Google Scholar]
- 51.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Brucher M., Perrot M., Duchesnay É., Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 52.Grunenberg A., Henck J.-O., Siesler H. W., Theoretical derivation and practical application of energy/temperature diagrams as an instrument in preformulation studies of polymorphic drug substances. Int. J. Pharm. 129, 147–158 (1996). [Google Scholar]
- 53.Wu Y.-N., Li L., Cheng H.-P., First-principles studies of Ta2O5 polymorphs. Phys. Rev. B 83, 144105 (2011). [Google Scholar]
- 54.Ghedira M., Vincent H., Marezio M., Launay J. C., Structural aspects of the metal-insulator transitions in V0.985Al0.015O2. J. Solid State Chem. 22, 423–438 (1977). [Google Scholar]
- 55.Jain A., Hautier G., Ong S. P., Moore C. J., Fischer C. C., Persson K. A., Ceder G., Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 84, 045115 (2011). [Google Scholar]
- 56.Aykol M., Wolverton C., Local environment dependent GGA+ U method for accurate thermochemistry of transition metal compounds. Phys. Rev. B 90, 115105 (2014). [Google Scholar]
- 57.Durandurdu M., Hexagonal nanosheets in amorphous BN : A first principles study. J. Non Cryst. Solids 427, 41–45 (2015). [Google Scholar]
- 58.Nord J., Nordlund K., Keinonen J., Molecular dynamics study of damage accumulation in GaN during ion beam irradiation. Phys. Rev. B 68, 184104 (2003). [Google Scholar]
- 59.Cai B., Drabold D. A., Properties of amorphous GaN from first-principles simulations. Phys. Rev. B 84, 075216 (2011). [Google Scholar]
- 60.Aliano A., Catellani A., Cicero G., Characterization of amorphous In2O3: An ab initio molecular dynamics study. Appl. Phys. Lett. 99, 211913 (2011). [Google Scholar]
- 61.Soper A. K., Boroxol rings from diffraction data on vitreous boron trioxide. J. Phys. Condens. Matter 23, 365402 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Benoit M., Ispas S., Jund P., Jullien R., Model of silica glass from combined classical and ab initio molecular-dynamics simulations. Eur. Phys. J. B 13, 631–636 (2000). [Google Scholar]
- 63.Tucker M. G., Keen D. A., Dove M. T., Trachenko K., Refinement of the Si–O–Si bond angle distribution in vitreous silica. J. Phys. Condens. Matter 17, S67–S75 (2005). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaaq0148/DC1
Supplementary Text
figs. S1 to S42. Radial distribution functions of amorphous configurations.
figs. S43 to S83. Bond-angle distribution functions of amorphous configurations.
fig. S84. Amorphous limit sampling probability.
fig. S85. Snapshots of atomic structures of amorphous materials.
fig. S86. Probability of finding the correct, observed ground states.
fig. S87. PDFs from aggregated uncertainties in the amorphous limit classification of crystalline polymorphs.
table S1. The amorphous limits for the synthesizability of polymorphs.
database S1. Energies of amorphous configurations.