

Abstract
Helicobacter pylori is a common pathogen that is estimated to infect half of the human population, causing several diseases such as duodenal ulcer. Despite one of the first pathogens to be sequenced, its proteome remains poorly characterized as about one-third of its proteins have no functional annotation. Here, we integrate and analyze known protein interactions with proteomic and genomic data from different sources. We find that proteins with similar abundances tend to interact. Such an observation is accompanied by a trend of interactions to appear between proteins of similar functions, although some show marked cross-talk to others. Protein function prediction with protein interactions is significantly improved when interactions from other bacteria are included in our network, allowing us to obtain putative functions of more than 300 poorly or previously uncharacterized proteins. Proteins that are critical for the topological controllability of the underlying network are significantly enriched with genes that are up-regulated in the spiral compared with the coccoid form of H. pylori. Determining their evolutionary conservation, we present evidence that 80 protein complexes are identical in composition with their counterparts in Escherichia coli, while 85 are partially conserved and 120 complexes are completely absent. Furthermore, we determine network clusters that coincide with related functions, gene essentiality, genetic context, cellular localization, and gene expression in different cellular states.
Helicobacter pylori (H. pylori) 1 is a pervasive pathogen that is uniquely adapted to life in the acidic environment of the human stomach and associated with gastric inflammation and duodenal ulcer (1, 2). Persisting in such an environment by tightly associating with epithelial cells, H. pylori affects an estimated half of the human population. As a consequence, H. pylori is notorious for causing low-level inflammation and duodenal ulcer as well as stomach carcinoma and MALT (mucosa-associated lymphoid tissue) lymphoma (1–3), causing 700,000 deaths annually worldwide (4).
The genome of H. pylori reference strain 26695 was completely sequenced in 1997 (5) and encodes ∼1,587 proteins with about 34% remaining uncharacterized (6). Given its impact on world health, a concerted effort is required to understand this significant number of proteins and their role in infection and disease.
Interactions between proteins are needed for almost all biological processes, helping to understand pathways as well as linking poorly or uncharacterized proteins. Only a few comprehensive bacterial interactome studies have been published to date, such as Escherichia coli (7), Campylobacter jejuni (8) and Mycobacterium tuberculosis (9). In particular, protein interactions of H. pylori were among the first to be determined in bacteria (10), an interactome that has been recently expanded (11), capturing roughly 70% of the proteome. While such interactomes have been detected using yeast two-hybrid methods, a few studies also identified bacterial protein complexes (12–15).
Several studies have attempted to characterize the proteome of H. pylori. Bumann et al. (16) found more than 1,800 protein spots on 2-dimensional gels, of which 200 were identified. Similarly, Jungblut et al. (17) found up to 1,800 protein spots on 2-dimensional gels. 152 were identified, including 27 proteins that corresponded to hitherto hypothetical proteins (17). Govorun et al. (18) analyzed the proteomes of four H. pylori clinical isolates and identified 126 proteins. More recently, Jungblut et al. (19) used intensive prefractionation to identify a total of 567 proteins (36.6% of the proteome). Recently, we have identified 1,190 and 1,143 proteins by 2D-LC-MS and GeLC-MS, respectively (20, 21), representing roughly 72% of the H. pylori proteome.
As proteomes and interactomes have been determined independently, their relationship remains unclear. Here, we integrate proteomic quantitative measurements in a network of roughly 3,000 protein–protein interactions (10, 11). Our analyses of diverse datasets allow us to explore the role of abundance in both the proteome and interactome as well as the structure and functionality of networked patterns. Investigating the proteomes of spiral and coccoid forms of H. pylori, we find that proteins that are critical for the control of the underlying interactome are significantly enriched with genes that are differentially expressed in the spiral form. Such observations potentially point to single proteins that play a role in the adaptability of the pathogen to different physiological conditions. Furthermore, we predict the function of more than 300 previously poorly annotated genes as well as protein complexes and functional network clusters in H. pylori. As a consequence, our integration and analysis of various large-scale datasets provide new insights into the proteome, interactome, and physiology to significantly improve our knowledge of this important pathogen.
EXPERIMENTAL PROCEDURES
Essential Genes
We collected essentiality data from several comprehensive genetic studies in H. pylori (22, 23). Furthermore, we added genes that were essential for H. pylori colonization (24, 25).
Relative and Absolute Protein Quantification
Data on relative changes in protein abundance between coccoid and spiral cells were extracted from our previous study (21). Briefly, four biological replicates of coccoid and spiral H. pylori cells were measured by LC-MS using a stable isotope labeling in cell culture based approach, capturing 1,143 proteins.
As for proteomic abundances, we collected data from a previous study as well (20). Nine replicates were measured by LC-MS/MS analysis without sample fractionation, capturing the abundance values of 1,190 proteins in H. pylori.
Protein–protein Interactions in H. pylori
We collected a total of 3,002 protein interactions from two high-throughput studies. In particular, we considered a set called PIM1 from (10) and PIM2 from (11) that both were determined by yeast two-hybrid approaches. We also identified 1,466 interactions that were classified as core data (i.e. high confidence) as they represent the overlap of PIM1 and PIM2 (10, 11).
Clustering Analysis
We used the Markov Clustering (26) algorithm (MCL) to identify clusters of interactions in the combined core H. pylori network. Applying different combinations of parameters, we automatically assessed each cluster's ability to significantly enrich coherent proteins. In particular, we utilized functional annotations from the Comprehensive Microbial Resource (27), gene ontology (GO) (28) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database (29). Furthermore, we utilized gene essentiality from (22–25). For microarray analyses, we utilized 16 sets of gene expression analyses of H. pylori (27) from the Comprehensive Microbial Resource and considered three cases: genes up-regulated (+), genes down-regulated (−), and genes differently regulated (+ or −). Each experiment is identified by a number, a title, and the author (Table S4). For GO term enrichment analysis, we used the TopGo python library (30). For other annotations, we used Fisher's exact test and considered clusters if they enriched genes with p < 0.05.
Functional Classes of Proteins
H. pylori proteins were grouped according to broad functional classes that were defined by clusters of orthologous groups (COGs) (31, 32) since COGs provide a consistent classification of bacterial genes based on orthologous groups.
Enrichment Analysis
Binning proteins with a certain characteristic d (e.g. with a given number of interactions), we calculated the fraction of proteins that had a feature i in each group d, fi(d). As a null model, we randomly sampled protein sets with feature i of the same size 10,000 times and calculated the corresponding random fraction, fi,r(d). The enrichment/depletion of proteins with feature i in a group d is then defined as
Interactions Between Functional Classes
Proteins of H. pylori were grouped according to their protein abundance. Focusing on a set of protein interactions, we counted the occurrence of different abundance group combinations (33). For each combination of abundance groups i, j, we determined its probability po(i, j) = , where N is the total number of interactions between the underlying abundance groups. As a null model, we determined an expected probability of interactions between classes i, j: pe(i, j) = . Specifically, vi is the number of viable proteins in group i (i.e. proteins of group i that are involved in at least one interaction in the underlying set), and Ji,j is the number of genes that are involved in both groups. Combining these probabilities, we determined a log-odds ratio: r = . For large samples, we estimated the variance of the odds distribution σ2 = nij−1 + (N − nij)−1 + a−1 + (b − a)−1 where a = (vivj) − and b = . In particular, we calculated a p value for the significance of a link between two groups by a Z-test, where z = and considered each link with p < 0.05 (33).
Bacterial Meta-protein Interaction Data
We used 2,231 binary interactions between E. coli proteins that we have previously determined through yeast two-hybrid screens (7). As for other yeast two-hybrid screen sets, we utilized 12,012 interactions in Campylobacter jejuni (8), 3,121 interactions in Mesorhizobium loti (34), 3,236 interactions in Synechocystis sp. PCC6803 (35), 2,519 interactions in Streptococcus pneumoniae (36), 3,684 interactions in Treponema pallidum (33), 783 interactions in Bacillus subtilis (37), and 8,042 interactions in M. tuberculosis (9).
Utilizing all-versus-all BLASTP searches with the InParanoid script (38) in protein sets of two species, sequence pairs with mutually best scores were selected as central orthologous pairs. Proteins of both species that showed such an elevated degree of homology were clustered around these central pairs, forming orthologous groups. The quality of the clustering was further assessed by a standard bootstrap procedure. We only considered the central orthologous sequence pair with a confidence level of 100% as the real orthologous relationship. Protein sequence information of bacterial organisms was retrieved from Uniprot (39).
Functional Prediction of Unknown Proteins in H. pylori
We modeled the prediction of a functional class σ of a protein i as a Potts model (40). In particular, we considered functional annotation of proteins in H. pylori using COG classes as of the EggNOG database (24). All proteins without a functional annotation as well as proteins that were either classified as unknown or had a general function (such as membrane protein or ABC transporter) were randomly assigned a function out of the remaining 23 classes. In particular, we minimized the following global function E = −Σi,jJijδ(σi,σj) − Σihi(σi), where Jij is the adjacency matrix of the interaction network that accounts for unclassified proteins. In particular, Jij = 1 if unclassified proteins i and j interact and vice versa. δ(i,j) is the discrete δ function, where δ = 1 if unclassified proteins i and j have the same function (i.e. σi = σj) and vice versa. As a consequence, the first term allows us to optimize the number of interactions between unclassified proteins if they are predicted to have the same function. Depending on the assigned function to an otherwise unclassified protein, the second term aims to optimize support for the assigned function of protein i. In particular, we determine the number of classified proteins hi(σi) that interact with unclassified protein i with the same function σ that was assigned to unclassified protein i. To minimize E, we applied a simulated annealing approach that features an effective temperature T. After initially assigning random functions to all unclassified proteins, we randomly selected a protein, changed its function to a different class, and determined the energy of the new configuration. If the difference of energies ΔE ≤ 0, the new configuration was accepted. If ΔE > 0, the new configuration was accepted with probability p = e−ΔE/T. To obtain stabilized functional configurations, we repeated such a Monte Carlo step 10,000 times (40). Subsequently, we increased the inverse of T by 0.01 in each step and repeated such Monte Carlo steps. Since minimum energy solutions are not unique, we repeated such runs of simulated annealing 100 times and considered the fraction of times an unclassified protein i was observed in a certain functional state σ as an estimate of the probability that protein i belongs to class σ.
Heterogeneity of Functional Prediction
The Simpson s-index considers the fractions with which a given protein was assigned to a functional class. In particular, we calculated its heterogeneity of functional fractions as a Simpson diversity (41) index defined as s = Σi=1Npi2, where pi is the fraction with which a given protein was assigned to functional class i. Such a measure tends to 1 if one function dominates the distribution of fractions and vice versa.
Three-dimensional Modeling of Protein Structures
To model the structures of proteins (Fig. 6), we used Protein Data Bank (PDB) (42) structures 1A50 (TrpAB), 1PII (TrpCF), 1KGZ, (TrpD), 1I1Q (TrpFE), 2EEY (MoaC), 2FUW (Mog), 3RPF (MoaDE), and 2BZ0 (RibA). We created images with PyMOL v1.5.0.1.
Fig. 6.

Examples of PPI enrichments in and between genomic operons. Interacting proteins are symbolized by connected gene symbols (heteromers) and colored gene symbols (homomers). In the right panel, protein structures of Trp and Moa orthologs highlight detected (solid arrows) and undetected (dashed arrows) intermolecular interactions that are known from the enzymes′ tertiary structures.
Determination of Critical, Intermediate, and Redundant Proteins
We defined a set S ⊆ V of nodes in a network G = (V, E) as a minimum dominating set (MDSet) if every node v ∈ V is either an element of S or adjacent to an element of S. In a binary integer linear programming problem (ILP) we assigned a binary variable xv = 1 when a protein v ∈ V that participates in interactions E in a protein interaction network G is an element of the MDSet, and xv = 0 otherwise. The smallest set of MDSet nodes is obtained by minΣv ∈ Vxv, subject to the constraint xv + Σw ∈ Γ(v)xw ≥ 1 where Γ(v) was the set of interaction partners of protein v. However, many optimal solutions exist that provide MDSets of the same size. Such characteristics suggest the existence of subset of nodes that always (critical nodes), never (redundant nodes), and sporadically appear in MDSets (intermediate nodes). To find such subsets, our objective is to determine if v ∈ MDSet always appear in the MDSet of any solution. For each v ∈ MDSet, we create an ILP as before and assume that xv = 0 (i.e. not participating in the MDSet). After solving the ILP, we determine the size of the corresponding MDSet Nv that we obtained with xv = 0. If Nv > N, v is a critical node and intermediate otherwise. For all nodes that did not participate in the original MDSet, v ∉ MDSet, we need to check if they always appear outside MDSets. For v ∉ MDSet, we create an ILP as before and assume that xv = 1 (i.e. participating in the MDSet). After solving the ILP, we determine the size of the corresponding MDSet Nv that we obtained with xv = 1. If Nv > N, v is a redundant node and intermediate otherwise (43, 44). To solve these ILP problems, we utilized a branch-and-bound algorithm (45) as implemented by the lpSolve library.
Betweeness Centrality
As a global measure of its centrality, we calculated a node's betweenness, indicating a node's appearance in shortest paths through the whole network. In particular, we defined betweenness centrality cB of a node v as cB(v) = , where sst was the number of shortest paths between proteins s and t, while sst (v) was the number of shortest paths running through v.
RESULTS
Proteome Versus Interactome
We combine interaction datasets that have been determined by yeast two-hybrid approaches and obtain an interactome of H. pylori that connects 1,060 proteins (∼70% of the proteome) through roughly 3,000 interactions (10, 11). Furthermore, a “core” interactome, capturing high-confidence interactions, connects 759 proteins (49% of the proteome) through 1,466 interactions. In Fig. 1A, we label each protein with its functional class, essentiality, and abundance in this high-quality core network of protein interactions. As for estimating absolute quantities, we utilize data from our previous study (20) where we measured the abundance of proteins in H. pylori without sample fractionation with a LC-MS approach. Only accounting for proteins with at least three unique peptides, we obtain abundance values of 1,130 proteins that correspond to 831 interacting proteins in the combined interactions network of H. pylori. In Fig. 1B, we determine the functions of the most abundant proteins by binning proteins according to their abundance in three groups. Utilizing functional annotations from the EggNOG database (46), we find that most abundant proteins are involved in metabolite biosynthesis, transport and catabolism, protein turnover, translation and energy production. Fig. 2A indicates that essential proteins are significantly overrepresented among highly abundant proteins (Student's t test, p < 10−2). To account for interactions between proteins, we determine the proteins' propensity to interact with proteins of certain abundance levels. In particular, we calculate each protein's abundance specific Z-score and group proteins in bins of certain Z-scores. In Fig. 2B, we determine the enrichment of interactions between proteins that appear in a given Z-score bin, utilizing the combined network of all protein interactions. Generally, we observe that proteins predominately interact with proteins of similar abundance. Still, our results further indicate that proteins in low-abundance bins appear to interact with proteins in intermediate-abundance bins.
Fig. 1.

The interactome and proteome of H. pylori. (A) We map all high-quality interactions between proteins in the core protein interaction network and their corresponding abundances in H. pylori. Furthermore, we label all proteins with their protein functions and essentiality. (B) We bin protein abundances in three groups (high: top 20%, low: bottom 20%, intermediate: remainder) and determine the enrichments of functions in each bin. We find that highly abundant proteins preferably are enriched with metabolic functions.
Fig. 2.
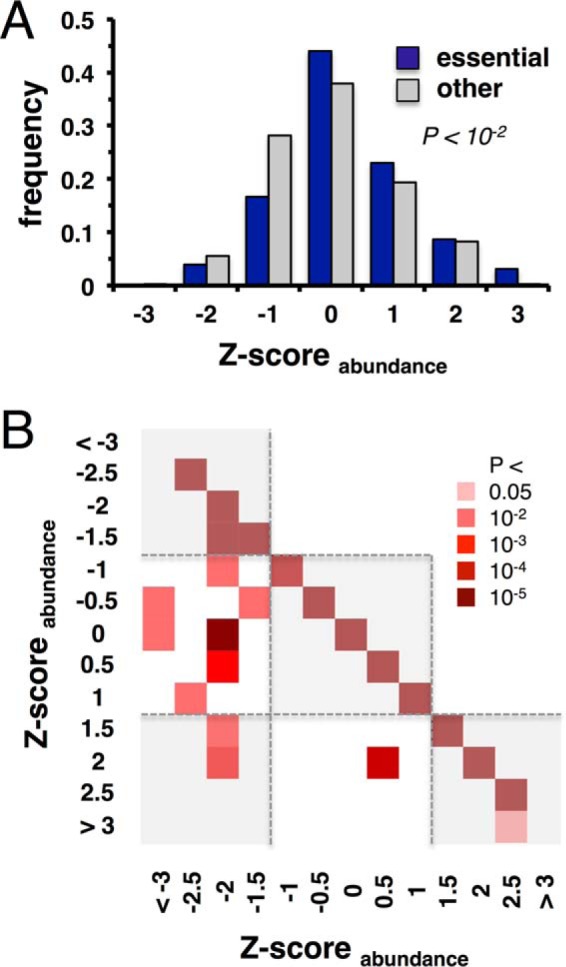
Essentiality and abundances. (A) Grouping proteins into bins of abundances, we observe that essential proteins are more abundant than their nonessential counterparts (p < 10−2, Student's t test). (B) Using the combined interaction network in H. pylori, proteins appear to predominately interact with proteins of similar abundance. In turn, interactions that involve proteins in low-abundance bins tend to interact with proteins in intermediate-abundance bins.
The Functional Cross-talk of the Interactome
Resembling other interactome hairballs (Fig. 1A) the H. pylori interactome does not show clear functional clustering. However, the interaction data are well supported by interactions between proteins of the same functional group (Fig. 3). This observation can be used to validate the reliability (or at least plausibility) of an interaction dataset since random interactions would provide no significant enrichment. Interestingly, we additionally observe some unexpected functional cross-talk. For example, ribosomal proteins and proteins involved in translation interact with proteins involved in motility more often than expected by chance (groups I and K in Fig. 3). Similarly, motility proteins also interact with proteins involved in amino acid metabolism.
Fig. 3.

Functional crosstalk in the interactome of H. pylori. Determining the prevalence of interactions between functional groups, we observe that the majority of interactions appear between proteins of the same functional class.
Predicting Functions of Proteins Using a Bacterial Meta-interactome
To investigate the functional predictive power of our initial network of experimentally determined interactions in H. pylori, we randomly pick 80% of all functionally annotated proteins 1,000 times to predict the functions of the remaining 20% in each random run. Using a stochastic model (40), we represent every protein by a profile that reflects the probability of having a certain function. Applying different probability thresholds for the presence of a functional annotation, we determine receiver operating characteristic curves and consider the corresponding area under the curve as a measure of prediction quality (47) (Fig. 4A). To increase the predictive power of the underlying protein interaction network, we augment our network in H. pylori with protein interactions from other bacteria (36, 48). Specifically, we consider interactions that have at least one interacting protein with a functionally annotated ortholog in H. pylori, while its interacting counterpart is at least functionally annotated in the corresponding organism. Focusing on the same previously sampled sets of proteins, we predict the functions of the corresponding 20% by utilizing the augmented network. Notably, we observe a significant shift toward increased values of the area under the receiver operating characteristic curve (p < 10−50, Student's t test), suggesting that the augmentation of the original network with interactions from other bacteria significantly improves the quality of functional predictions (Fig. 4A). Since each protein is represented by a profile of function-specific probabilities, we calculate the Simpson s-index (41) as a measure of heterogeneity of predicted functions. Such a measure tends to be 1 if one function dominates the distribution of fractions (i.e. has a high probability). In turn, the s-index approaches 0 if probabilities are equally distributed. Since our sampling approach randomly picks a subset of proteins and predicts functions based on the remaining proteins in both the original interaction network of H. pylori and the augmented network, we directly compare the impact of the augmented network on the homogeneity of functional prediction. In Fig. 4B, we calculate the mean s-indices of each protein, suggesting that functional predictions of the majority of proteins benefit from the addition of the bacterial meta-interactome. Based on our observations that interactions from other bacteria have a considerable benefit on our ability to predict functions, we apply our approach to the functional prediction of 337 poorly characterized or previously unknown H. pylori proteins. While we determine the probability that a given protein has a particular function, we assess the significance of our predictions by randomly sampling known functions 100 times. Applying a Z-test, we determine a corrected p value for each score (49) that we consider significant if FDR<0.05. The heatmap in Fig. 4C shows the range of functions predicted for these proteins, including a sizeable fraction to be involved in transcriptional and translational activities. In Supplemental Table S1, we present the functional profiles of all proteins in the order in which they appear in Fig. 4C.
Fig. 4.

Functional prediction of unknown proteins in H. pylori using a bacterial meta-interactome. (A) To assess the quality of our classification procedure, we randomly sample 20% of all functionally annotated proteins in H. pylori and utilize the remainder to predict their functions. To measure prediction quality we calculate the area under the receiver operating characteristic curve, suggesting that the addition of the bacterial meta-interactome allows for better functional prediction (p < 10−50, Student's t test). (B) We consider all randomized samples and calculate the mean s-indices of each gene of unknown function (circles) in both the original network of H. pylori and the augmented network. In the scatter-plot the homogeneity of the functional prediction of the majority of genes (78.6%) benefit from including the bacterial meta-interactome. (C) Combining the network of protein interactions of H. pylori and the bacterial meta-interactome, we predict the functions of 337 proteins with unknown or poorly characterized functions (FDR < 0.05).
Control of the H. pylori Protein Interaction Network
Considering the network of protein–protein interactions in H. pylori, we aim at the elucidation of proteins that are important for the topological controllability of the underlying network (43, 44, 50). In particular, networks are dominated by minimum dominating sets (MDSet) that can be determined by an ILP. Such a method allows us to find the smallest set of nodes where each non-MDSet node is adjacent to a node in the MDSet. However, many different configurations of MDSets exist that have the same number of critical proteins. As such, an assumption implies sets of nodes that always, partially, or never participate in MDSets. Therefore, we define proteins as critical if they always participated in the MDSet of a given configuration (Fig. 5A). Furthermore, we consider redundant nodes that never appeared in MDSets while intermediate nodes sporadically occur in MDSets. Applying an algorithm that allows us to determine such sets of nodes (43, 44), we observe that the percentage of critical nodes is roughly <10%, while intermediate nodes constitute <30% of all proteins (Fig. 5B). The mean degree of critical proteins far exceeds the corresponding values of intermediate and redundant proteins that are close to the mean degree of all proteins in the underlying interaction networks (Fig. 5B).
Fig. 5.

Controlling the H. pylori protein interaction network. (A) In a toy network we illustrate the concept of critical, intermediate, and redundant nodes. (B) In the table, we present statistics of the protein interaction network of H. pylori and of its corresponding critical, intermediate, and redundant proteins. Notably, critical proteins are highly connected, while degrees of intermediate and redundant nodes revolve around the mean degree of all proteins (dashed line). (C) We define the top 20% of proteins with the highest node betweeness as a set of bottleneck proteins. Randomly sampling sets of critical, intermediate, and redundant proteins 10,000 times, we find that critical nodes are strongly enriched with bottlenecks. While intermediate nodes are moderately enriched, we also find a significant depletion of redundant nodes in the underlying set of bottleneck proteins. (D) In the Volcano plot of the fold change of proteins that compares their abundance levels in the coccoid and spiral form, we label all proteins with their critical, intermediate, and redundant role in the underlying network of protein interactions of H. pylori. We define proteins with a fold change of >0.5 and <−0.5 (p < 0.05) as regulated proteins (shaded areas), suggesting that critical, regulated proteins predominantly appear as being present in the spiral form. (E) As a corollary, we randomly sample sets of regulated proteins 10,000 times. We observe that critical proteins are significantly enriched with regulated genes. (F) We determine the enrichment of functions in the set of critical proteins by randomly sampling their functions. We observe that critical proteins predominately appear in transcriptional and posttranslational modification functions.
As for other topological characteristics, we calculate the betweeness centrality of all nodes in the underlying network. Defining the top 20% of proteins with highest betweeness centrality as a set of bottleneck nodes, we calculate the enrichment of such proteins in sets of critical, intermediate, and redundant proteins. Given all proteins in the underlying interaction network, we sample sets of proteins by randomly shuffling their labels, generating nonoverlapping, random sets of critical, intermediate, and redundant proteins. We observe that critical proteins in all organisms are strongly enriched with bottlenecks (p < 10−4). Albeit insignificantly, intermediate proteins are enriched with bottleneck nodes as well, while critical proteins hardly are bottlenecks (p < 10−4, Fig. 5C).
Additionally, we compare protein levels in the spiral and coccoid cells of H. pylori based on previously published proteomic data (21) to link the generated interaction network with cell physiology. These cellular forms were analyzed by LC-MS, allowing the comparison of relative changes between the two states with high accuracy. Determining the fold change and the corresponding p value using a Student's t test of proteins comparing the spiral and coccoid expression levels, we generate a Volcano plot where we label each protein as critical, intermediate or redundant (Fig. 5D). Qualitatively, we observe that critical proteins seem to have a higher abundance in the spiral form of H. pylori. As a corollary, we consider a set of regulated genes defined as proteins with −0.5 ≤ fold change ≥ 0.5 and p < 0.05 (21). Randomly sampling sets of regulated genes 10,000 times (Fig. 5E), we observe that critical proteins are significantly enriched with regulated genes (p < 0.05) while redundant proteins are found diluted (p < 0.05). Assuming that critical proteins play a role in the transition between the spiral and coccoid forms, we perform an analysis of their functions. Fig. 5F indicates that functions of critical proteins mostly revolve around transcriptional and posttranslational modification functions. In Supplemental Table S2, we annotate each protein with its role, fold change comparing coccoid to spiral form, and functional annotation.
Genomic Organization and the Interactome
Bacterial genomes are typically organized through functional gene clusters such as operons that encode functional units such as protein complexes. Interdependence of genomic loci and protein interaction maps has been demonstrated for the T. pallidum interactome (33) as well as for phage interactomes (51, 52). To reveal such genomic links we map protein interactions onto operons in the H. pylori genome (53) (Fig. 6). For instance, the well characterized urease gene cluster (operons 4721–4722) reveals interactions among the enzyme's core components (UreA-UreB), between the urease accessory factors UreE-UreG and UreF-UreH, and finally between UreA and UreH (see Fig. 6 in (11)). Another example is observed in operon 4,987, a gene cluster that encodes enzymes involved in tryptophan biosynthesis. A comparison with experimental protein structures from orthologs shows that our protein-protein interaction (PPI) studies (11) capture most PPI interactions that are important to assemble the enzyme complexes (Fig. 6, right panel): TrpA-TrpB and TrpE-TrpG are organized as heterotetramers with two subunits of each protein. TrpD is a homodimer, and TrpCF is a single protein that functions as a monomer. Likewise, operon 4888 encodes for enzymes involved in molybdopterin biosynthesis while the protein interaction map accurately reflects the organization of the enzyme complex.
An example for enriched crosstalk between different gene clusters is found between two operons (4960 and 5013), encoding ribosomal proteins or products that are related to protein translation and tRNA modification, respectively. The hypothetical protein HP1414, the first gene in the miaA operon, binds the ribosomal proteins L19 and S16 as well as the hypothetical protein HP1150 (which belongs to COG1837, a family of putative RNA-binding proteins). In fact, HP1414 is the H. pylori homologue of the ribosomal silencing factor RsfS (=RsfA) that we previously showed to bind to ribosomal protein L14, preventing association of the small and large ribosomal subunit (54). L19 is located in the direct neighborhood of L14 in the ribosome-forming bridges (B8 and B6) to the small ribosomal subunit (55), potentially representing a novel or additional hotspot for RsfS action. Both operons are functionally associated since both encode for products that are involved in protein translation.
One more example of interconnected operons is found between operon 4815 and 5035 that encode several uncharacterized (HP0469-HP0465) and flagellar rod proteins (FliE, FlgC, FlgB), respectively, suggesting that the 4815 operon may be involved in motility. Involvement of HP0466 in flagellar biosynthesis has already been suggested by others based on its interaction with FlgB and homology comparisons of the operon member HP0465 with motility accessory factors of C. jejuni (56). Moreover, transposon insertion into the HP0466 locus causes a colonization defect (24) whereas for HP0468 no functional information is available.
Protein Complexes in H. pylori
Protein functions are often mediated by protein complexes that are defined as stable assemblies of multiple proteins. Since complexes have not been studied systematically in H. pylori, we utilize extensive experimental data on protein complexes from E. coli (57) and M. pneumoniae (14) to predict homologous complexes in H. pylori. Utilizing orthologous protein information from the COG database (31, 32), we find that H. pylori shares 786 orthologous proteins with E. coli but only 260 with M. pneumoniae (Fig. 7A). As an example for different levels of complex conservation, we observe that the murein tripeptide transporter, a hetero-pentamer, is well conserved in H. pylori, while only three out of five subunits in the periplasmic nitrate reductase are present. In contrast to E. coli, the cascade complex is completely missing from H. pylori (Fig. 7B). In Fig. 7C, we count the number of complexes with different degrees of conservation in H. pylori using reference sets of E. coli complexes from the EcoCyc database (58) and the dataset of (57). Furthermore, we use protein complex information from M. pneumoniae (14). The degree of conservation (Fig. 7A) prompts us to focus on E. coli, indicating that E. coli may be a good model for some processes in epsilon-proteobacteria but not for others. Using a reference set of 285 well-studied E. coli complexes from EcoCyc (58), we predict 80 H. pylori complexes to be identical (in composition) with their counterparts in E. coli. Another 85 complexes are partially conserved while 120 are completely absent. All predicted complexes in H. pylori are available in Supplemental Table S3.
Fig. 7.

Predicted protein complexes in H. pylori. (A) Proteomes of E. coli, M. pneumoniae, and H. pylori overlap substantially using orthologous proteins. Proteins not belonging to COGs were excluded. (B) We show selected protein complexes indicating different degrees of complex conservation. Dashed circles indicate proteins in E. coli complexes that are absent in H. pylori. Stoichiometry of protein complexes is indicated if they diverge from one subunit. (C) We count the number of complexes with different degrees of conservation in H. pylori.
Functional Integration of Gene Expression
To identify functionally relevant network clusters, we systematically analyze the H. pylori high-quality core protein interaction network to identify subnetworks that overrepresent certain functional groups, using functional terms from the Comprehensive Microbial Resource (27), GO (28), KEGG (29), gene essentiality, genetic context, cellular localization, and gene expression data. Some of these clusters are illustrated in Fig. 8 while detailed results can be found in Supplemental Table S4. For instance, cluster 16 consists of nine proteins that are highly interconnected by interactions. Involved genes are co-expressed under different conditions: Six genes of the cluster are up-regulated when the growth conditions are shifted to low pH values while three members are up-regulated under limited iron accessibility in the stationary phase. Finally, three proteins have an increased expression level when H. pylori is grown in contact with liver cells versus medium alone. While cluster members belong to very different pathways (e.g. Cag17 and Cag20 belong to the type IV secretion system, IspA is a geranyltranstransferase, CeuE a periplasmic iron-binding protein, and Ggt is a gamma-glutamyltranspeptidase), they are connected by interactions and gene expression. While such discordant expression patterns are found in other clusters as well (e.g. clusters 17, 26, and 36), our results suggest the presence of conditions under which these genes are co-expressed, allowing proteins to interact. Clusters 26 and 36 are enriched for proteins related to chemotaxis and motility. Notably, our screens detect all three CheV paralogs in the H. pylori genome (HP0019, HP0393, and HP0616) to bind to the hemolysin secretion protein precursor HylB (cluster 36). Moreover, we find that HP0019 and HP0393 interact with the methyl-accepting chemotaxis transducer (TlpC) but not HP0616. Cluster 6 shows that this combination strategy unearthed additional interesting aspects that cannot be detected when one parameter is analyzed in isolation. While seven members that belong to three different operons are connected, GO assignments of HP0164 (signal-transducing protein, histidine kinase) and OmpR (response regulator HP0166) suggest an involvement of the cluster members in two-component signaling.
Fig. 8.

Selected interaction clusters with various enriched functional terms, genomic context, co-expression, co-localization, and phenotypes. We depict interaction clusters that we derive from the combined H. pylori core protein interaction map with significant enrichments (p < 0.05). All results can be found in Supplemental Table S5. Node labels represent protein names (if available) or the locus number of the corresponding protein. Co-expressed genes based on operons are given in the legends by the transcription unit number as used in (53) and gene expression data (Supplemental Table S5). For each cluster, we provide a separate legend highlighting the enriched properties. In particular, clusters 16 and 17 are enriched for differentially, co-expressed proteins when growth is shifted to low pH values conditions. Clusters 13 and 65 are enriched for essential genes, while cluster 23 is enriched with ribosomal proteins. Clusters 26 and 36 include flagellar/chemotaxis proteins, and cluster 6 is enriched for intra- and interconnected operons.
DISCUSSION
Given that H. pylori is a major human pathogen causing millions of ulcers and other health problems each year surprisingly little is known about its molecular biology. To fill this gap, we investigate the proteome and interactome in a more systematic way.
Investigating the abundance of proteins in H. pylori, we find that highly abundant proteins revolve around translational, posttranslational modification, protein turnover, metabolite biosynthesis, transport, catabolism, and energy production functions. Furthermore, we find that abundant proteins are typically encoded by essential genes.
Combining protein interactions with protein abundances, we observe that proteins of similar abundance preferably interact with each other. While we find some interactions between proteins of different abundances, our results clearly confirm assumptions that interacting pairs of proteins are usually present in roughly stoichiometric ratios.
Notably, H. pylori still encodes a large number (∼500) of uncharacterized proteins. Among proteins of known function, we find that interactions usually connect proteins of similar activity. Based on such characteristics, we utilize a bacterial meta-interactome of closely related bacteria to predict the functions of unknown proteins. In particular, we account for interactions that are conserved in other closely related bacteria. Such an augmentation of our initial network of protein interactions allows us to increase the accuracy of our classification method significantly and to predict the function of more than 300 proteins with previously poorly annotated or unknown function. Resembling the spectrum of functions of abundant proteins, we find that the majority of proteins thus obtained mostly revolve around translational and posttranslational modification functions.
Utilizing our network of protein interactions in H. pylori, we determine sets of proteins that topologically control the underlying network. In particular, we find sets of critical, intermediate, and redundant proteins that always, partially, or never appear in different control configurations of the underlying network. In particular, each control configuration features a minimum dominating set (MDSet) so that every node is either an element of the MDSet or adjacent to a protein of the MDSet. Notably, critical proteins appear to be enriched with regulated genes that are significantly present in the spiral form of H. pylori. The spiral form of H. pylori is mostly dividing while the coccoid is a nonculturable but viable form. The observation that genes that are overexpressed in the spiral compared with the coccoid form are enriched with critical proteins suggests that the underlying topology network plays a role in the switch of the two bacterial forms. Notably, critical proteins are also enriched with proteins of high betweeness, representing central topological proteins with a propensity to connect different, disparate parts of the network. Therefore, we surmise that critical proteins may assume the role of levers that allow the bacteria to activate certain functions to change between forms as well as integrate different parts of the network to carry out the transformation from coccoid to spiral form. As a corollary, we hypothesize that such proteins carry functions that contribute to the spiral form. Indeed, we find that critical proteins are mostly enriched with transcriptional and posttranslational modification functions.
As for protein complexes, we integrate protein complex information of E. coli and M. pneumoniae and infer potential complexes in H. pylori by determining evolutionarily conserved complex components. As expected, we find a higher rate of conserved complexes when we consider E. coli protein complexes. Such a result may be rooted in the fact that E. coli has almost six times as many proteins than M. pneumoniae. Furthermore, its protein complexes are better investigated than their counterparts in Mycoplasma, suggesting that E. coli is a better model for protein complexes as H. pylori shares significantly more orthologs with E. coli than M. pneumoniae. Moreover, we integrate the interactome with functional and expression profiles of genes in H. pylori, allowing us to find significant protein clusters. Our analysis reveals an abundance of different network clusters that combine certain functions that integrate the placement of operons of cluster members as well. Such an observation clearly suggests that expression, function, and operon regulation are driving forces of the observed network clusters.
Supplementary Material
Footnotes
* This work was supported by National Institutes of Health grant NIH R01GM109895.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- H. pylori
- Helicobacter pylori
- LC-MS/MS
- Liquid chromatography-mass spectrometry
- PPI
- protein-protein interactions
- MDSet
- minimum dominating set
- PIM
- protein interaction map
- GO
- gene ontology
- KEGG
- Kyoto Encyclopedia of genes and genomes
- COG
- Clusters of orthologous genes
- MCL
- Markov clustering
- PDB
- Protein Data Bank
- MALT
- mucosa-associated lymphoid tissue.
REFERENCES
- 1. Warren J. R., and Marshall B. (1983) Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1, 1273–1275 [PubMed] [Google Scholar]
- 2. Marshall B. J., and Warren J. R. (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315 [DOI] [PubMed] [Google Scholar]
- 3. Kusters J. G., van Vliet A. H., and Kuipers E. J. (2006) Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bauer B., and Meyer T. F. (2011) The Human gastric pathogen Helicobacter pylori and its association with gastric cancer and ulcer disease. Ulcers 2011, 340157 [Google Scholar]
- 5. Tomb J. F., White O., Kerlavage A. R., Clayton R. A., Sutton G. G., Fleischmann R. D., Ketchum K. A., Klenk H. P., Gill S., Dougherty B. A., Nelson K., Quackenbush J., Zhou L., Kirkness E. F., Peterson S., Loftus B., Richardson D., Dodson R., Khalak H. G., Glodek A., McKenney K., Fitzegerald L. M., Lee N., Adams M. D., Hickey E. K., Berg D. E., Gocayne J. D., Utterback T. R., Peterson J. D., Kelley J. M., Cotton M. D., Weidman J. M., Fujii C., Bowman C., Watthey L., Wallin E., Hayes W. S., Borodovsky M., Karp P. D., Smith H. O., Fraser C. M., and Venter J. C. (1997) The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547 [DOI] [PubMed] [Google Scholar]
- 6. The UniProt, C. (2017) UniProt: The universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rajagopala S. V., Sikorski P., Kumar A., Mosca R., Vlasblom J., Arnold R., Franca-Koh J., Pakala S. B., Phanse S., Ceol A., Häuser R., Siszler G., Wuchty S., Emili A., Babu M., Aloy P., Pieper R., and Uetz P. (2014) The binary protein–protein interaction landscape of Escherichia coli. Nat. Biotechnol. 32, 285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parrish J. R., Yu J., Liu G., Hines J. A., Chan J. E., Mangiola B. A., Zhang H., Pacifico S., Fotouhi F., DiRita V. J., Ideker T., Andrews P., and Finley R. L. Jr. (2007) A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 8, R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y., Cui T., Zhang C., Yang M., Huang Y., Li W., Zhang L., Gao C., He Y., Li Y., Huang F., Zeng J., Huang C., Yang Q., Tian Y., Zhao C., Chen H., Zhang H., and He Z. G. (2010) Global protein–protein interaction network in the human pathogen Mycobacterium tuberculosis H37Rv. J. Proteome Res. 9, 6665–6677 [DOI] [PubMed] [Google Scholar]
- 10. Rain J. C., Selig L., De Reuse H., Battaglia V., Reverdy C., Simon S., Lenzen G., Petel F., Wojcik J., Schächter V., Chemama Y., Labigne A., and Legrain P. (2001) The protein–protein interaction map of Helicobacter pylori. Nature 409, 211–215 [DOI] [PubMed] [Google Scholar]
- 11. Häuser R., Ceol A., Rajagopala S. V., Mosca R., Siszler G., Wermke N., Sikorski P., Schwarz F., Schick M., Wuchty S., Aloy P., and Uetz P. (2014) A second-generation protein–protein interaction network of Helicobacter pylori. Mol. Cell. Proteomics 13, 1318–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Butland G., Peregrín-Alvarez J. M., Li J., Yang W., Yang X., Canadien V., Starostine A., Richards D., Beattie B., Krogan N., Davey M., Parkinson J., Greenblatt J., and Emili A. (2005) Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537 [DOI] [PubMed] [Google Scholar]
- 13. Arifuzzaman M., Maeda M., Itoh A., Nishikata K., Takita C., Saito R., Ara T., Nakahigashi K., Huang H. C., Hirai A., Tsuzuki K., Nakamura S., Altaf-Ul-Amin M., Oshima T., Baba T., Yamamoto N., Kawamura T., Ioka-Nakamichi T., Kitagawa M., Tomita M., Kanaya S., Wada C., and Mori H. (2006) Large-scale identification of protein–protein interaction of Escherichia coli K-12. Genome Res. 16, 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kühner S., van Noort V., Betts M. J., Leo-Macias A., Batisse C., Rode M., Yamada T., Maier T., Bader S., Beltran-Alvarez P., Castaño-Diez D., Chen W. H., Devos D., Güell M., Norambuena T., Racke I., Rybin V., Schmidt A., Yus E., Aebersold R., Herrmann R., Bottcher B., Frangakis A. S., Russell R. B., Serrano L., Bork P., and Gavin A. C. (2009) Proteome organization in a genome-reduced bacterium. Science 326, 1235–1240 [DOI] [PubMed] [Google Scholar]
- 15. Hu P., Janga S. C., Babu M., Diaz-Mejía J. J., Butland G., Yang W., Pogoutse O., Guo X., Phanse S., Wong P., Chandran S., Christopoulos C., Nazarians-Armavil A., Nasseri N. K., Musso G., Ali M., Nazemof N., Eroukova V., Golshani A., Paccanaro A., Greenblatt J. F., Moreno-Hagelsieb G., and Emili A. (2009) Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins. PLoS Biol. 7, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bumann D., Meyer T. F., and Jungblut P. R. (2001) Proteome analysis of the common human pathogen Helicobacter pylori. Proteomics 1, 473–479 [DOI] [PubMed] [Google Scholar]
- 17. Jungblut P. R., Bumann D., Haas G., Zimny-Arndt U., Holland P., Lamer S., Siejak F., Aebischer A., and Meyer T. F. (2000) Comparative proteome analysis of Helicobacter pylori. Mol. Microbiol. 36, 710–725 [DOI] [PubMed] [Google Scholar]
- 18. Govorun V. M., Moshkovskii S. A., Tikhonova O. V., Goufman E. I., Serebryakova M. V., Momynaliev K. T., Lokhov P. G., Khryapova E. V., Kudryavtseva L. V., Smirnova O. V., Toropyguine I. Y., Maksimov B. I., and Archakov A. I. (2003) Comparative analysis of proteome maps of Helicobacter pylori clinical isolates. Biochemistry 68, 42–49 [DOI] [PubMed] [Google Scholar]
- 19. Jungblut P. R., Schiele F., Zimny-Arndt U., Ackermann R., Schmid M., Lange S., Stein R., and Pleissner K. P. (2010) Helicobacter pylori proteomics by 2-DE/MS, 1-DE-LC/MS and functional data mining. Proteomics 10, 182–193 [DOI] [PubMed] [Google Scholar]
- 20. Müller S. A., Findeiβ S., Pernitzsch S. R., Wissenbach D. K., Stadler P. F., Hofacker I. L., von Bergen M., and Kalkhof S. (2013) Identification of new protein coding sequences and signal peptidase cleavage sites of Helicobacter pylori strain 26695 by proteogenomics. J. Proteomics 86, 27–42 [DOI] [PubMed] [Google Scholar]
- 21. Müller S. A., Pernitzsch S. R., Haange S. B., Uetz P., von Bergen M., Sharma C. M., and Kalkhof S. (2015) Stable isotope labeling by amino acids in cell culture based proteomics reveals differences in protein abundances between spiral and coccoid forms of the gastric pathogen Helicobacter pylori. J. Proteomics 126, 34–45 [DOI] [PubMed] [Google Scholar]
- 22. Salama N. R., Shepherd B., and Falkow S. (2004) Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J. Bacteriol. 186, 7926–7935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chalker A. F., Minehart H. W., Hughes N. J., Koretke K. K., Lonetto M. A., Brinkman K. K., Warren P. V., Lupas A., Stanhope M. J., Brown J. R., and Hoffman P. S. (2001) Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 183, 1259–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baldwin D. N., Shepherd B., Kraemer P., Hall M. K., Sycuro L. K., Pinto-Santini D. M., and Salama N. R. (2007) Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect. Immun. 75, 1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kavermann H., Burns B. P., Angermuller K., Odenbreit S., Fischer W., Melchers K., and Haas R. (2003) Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exper. Med. 197, 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Enright A. J., Van Dongen S., and Ouzounis C. A. (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peterson J. D., Umayam L. A., Dickinson T., Hickey E. K., and White O. (2001) The Comprehensive Microbial Resource. Nucleic Acids Res. 29, 123–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., and Sherlock G. (2000) Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanehisa M., and Goto S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexa A., Rahnenführer J., and Lengauer T. (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 [DOI] [PubMed] [Google Scholar]
- 31. Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., Krylov D. M., Mazumder R., Mekhedov S. L., Nikolskaya A. N., Rao B. S., Smirnov S., Sverdlov A. V., Vasudevan S., Wolf Y. I., Yin J. J., and Natale D. A. (2003) The COG database: An updated version includes eukaryotes. BMC Bioinformatics 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., and Jensen L. J. (2013) STRING v9.1: Protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Titz B., Rajagopala S. V., Goll J., Häuser R., McKevitt M. T., Palzkill T., and Uetz P. (2008) The binary protein interactome of Treponema pallidum—The syphilis spirochete. PloS One 3, e2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shimoda Y., Shinpo S., Kohara M., Nakamura Y., Tabata S., and Sato S. (2008) A large scale analysis of protein–protein interactions in the nitrogen-fixing bacterium Mesorhizobium loti. DNA Res. 15, 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sato S., Shimoda Y., Muraki A., Kohara M., Nakamura Y., and Tabata S. (2007) A large-scale protein–protein interaction analysis in Synechocystis sp. PCC6803. DNA Res. 14, 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wuchty S., Rajagopala S. V., Blazie S. M., Parrish J. R., Khuri S., Finley R. L. Jr., and Uetz P. (2017) The protein interactome of Streptococcus pneumoniae and bacterial meta-interactomes improve function predictions. mSystems 2, e00019–e00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marchadier E., Carballido-López R., Brinster S., Fabret C., Mervelet P., Bessieres P., Noirot-Gros M. F., Fromion V., and Noirot P. (2011) An expanded protein–protein interaction network in Bacillus subtilis reveals a group of hubs: Exploration by an integrative approach. Proteomics 11, 2981–2991 [DOI] [PubMed] [Google Scholar]
- 38. Remm M., Storm C. E., and Sonnhammer E. L. (2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 314, 1041–1052 [DOI] [PubMed] [Google Scholar]
- 39. UniProt. (2015) UniProt: A hub for protein information. Nucleic Acids Res. 43, D204–D212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vazquez A., Flammini A., Maritan A., and Vespignani A. (2003) Global protein function prediction from protein–protein interaction networks. Nat. Biotechnol. 21, 697–700 [DOI] [PubMed] [Google Scholar]
- 41. Simpson E. H. (1949) Measurement of diversity. Nature 163, 688 [Google Scholar]
- 42. Rose P. W., Prlić A., Altunkaya A., Bi C., Bradley A. R., Christie C. H., Costanzo L. D., Duarte J. M., Dutta S., Feng Z., Green R. K., Goodsell D. S., Hudson B., Kalro T., Lowe R., Peisach E., Randle C., Rose A. S., Shao C., Tao Y. P., Valasatava Y., Voigt M., Westbrook J. D., Woo J., Yang H., Young J. Y., Zardecki C., Berman H. M., and Burley S. K. (2017) The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 45, D271–D281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishitsuka M., Akutsu T., and Nacher J. C. (2016) Critical controllability in proteome-wide protein interaction network integrating transcriptome. Sci. Rep. 6, 23541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nacher J. C., and Akutsu T. (2014) Analysis of critical and redundant nodes in controlling directed and undirected complex networks using dominating sets. J. Compl. Networks 2, 394–412 [Google Scholar]
- 45. Land A. H., and Doig A. G. (1960) An automatic method of solving discrete programming-problems. Econometrica 28, 497–520 [Google Scholar]
- 46. Huerta-Cepas J., Szklarczyk D., Forslund K., Cook H., Heller D., Walter M. C., Rattei T., Mende D. R., Sunagawa S., Kuhn M., Jensen L. J., von Mering C., and Bork P. (2016) EggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fawcett T. (2006) An introduction to ROC analysis. Pattern Recogn. Lett. 27, 861–874 [Google Scholar]
- 48. Caufield J. H., Wimble C., Shary S., Wuchty S., and Uetz P. (2017) Bacterial protein meta-interactomes predict cross-species interactions and protein function. BMC Bioinformatics 18, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. Roy Stat. Soc. B Met. 57, 289–300 [Google Scholar]
- 50. Wuchty S. (2014) Controllability in protein interaction networks. Proc. Natl. Acad. Sci. U.S.A. 111, 7156–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sabri M., Häuser R., Ouellette M., Liu J., Dehbi M., Moeck G., García E., Titz B., Uetz P., and Moineau S. (2011) Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp-1. J. Bacteriol. 193, 551–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Häuser R., Sabri M., Moineau S., and Uetz P. (2011) The proteome and interactome of Streptococcus pneumoniae phage Cp-1. J. Bacteriol. 193, 3135–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sharma C. M., Hoffmann S., Darfeuille F., Reignier J., Findeiss S., Sittka A., Chabas S., Reiche K., Hackermüller J., Reinhardt R., Stadler P. F., and Vogel J. (2010) The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255 [DOI] [PubMed] [Google Scholar]
- 54. Häuser R., Pech M., Kijek J., Yamamoto H., Titz B., Naeve F., Tovchigrechko A., Yamamoto K., Szaflarski W., Takeuchi N., Stellberger T., Diefenbacher M. E., Nierhaus K. H., and Uetz P. (2012) RsfA (YbeB) proteins are conserved ribosomal silencing factors. PLoS Genet. 8, e1002815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gao H., Sengupta J., Valle M., Korostelev A., Eswar N., Stagg S. M., Van Roey P., Agrawal R. K., Harvey S. C., Sali A., Chapman M. S., and Frank J. (2003) Study of the structural dynamics of the E. coli 70S ribosome using real-space refinement. Cell 113, 789–801 [DOI] [PubMed] [Google Scholar]
- 56. Karlyshev A. V., Linton D., Gregson N. A., and Wren B. W. (2002) A novel paralogous gene family involved in phase-variable flagella-mediated motility in Campylobacter jejuni. Microbiology 148, 473–480 [DOI] [PubMed] [Google Scholar]
- 57. Caufield J. H., Abreu M., Wimble C., and Uetz P. (2015) Protein complexes in bacteria. PLoS Comput. Biol. 11, e1004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keseler I. M., Mackie A., Santos-Zavaleta A., Billington R., Bonavides-Martinez C., Caspi R., Fulcher C., Gama-Castro S., Kothari A., Krummenacker M., Latendresse M., Muñiz-Rascado L., Ong Q., Paley S., Peralta-Gil M., Subhraveti P., Velázquez-Ramírez D. A., Weaver D., Collado-Vides J., Paulsen I., and Karp P. D. (2017) The EcoCyc database: Reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res. 45, D543–D550 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.