

Abstract
The lumen of the endoplasmic reticulum (ER) provides an oxidizing environment to aid in the formation of disulfide bonds, which is tightly regulated by both antioxidant proteins and small molecules. On the cytoplasmic side of the ER, cytochrome P450 (P450) proteins have been identified as a superfamily of enzymes that are important in the formation of endogenous chemicals as well as in the detoxication of xenobiotics. Our previous report described oxidative inhibition of P450 Family 4 enzymes via oxidation of the heme-thiolate cysteine to a sulfenic acid (-SOH) (Albertolle, M. E. et al. (2017) J. Biol. Chem. 292, 11230–11242). Further proteomic analyses of murine kidney and liver microsomes led to the finding that a number of other drug-metabolizing enzymes located in the ER are also redox-regulated in this manner. We expanded our analysis of sulfenylated enzymes to human liver and kidney microsomes. Evaluation of the sulfenylation, catalytic activity, and spectral properties of P450s 1A2, 2C8, 2D6, and 3A4 led to the identification of two classes of redox sensitivity in P450 enzymes: heme-thiolate-sensitive and thiol-insensitive. These findings provide evidence for a mammalian P450 regulatory mechanism, which may also be relevant to other drug-metabolizing enzymes. (Data are available via ProteomeXchange with identifier PXD007913.)
The endoplasmic reticulum (ER)1 is a site of protein translation, posttranslational processing, and small molecule metabolism (1). Posttranslational processing includes glycosylation of proteins for sorting, disulfide bond formation, and specific proteolytic cleavages. The ER lumen is an oxidizing environment that aids in the production of disulfide bonds, which is maintained at a homeostatic level by both small molecules (ascorbate) and proteins (protein disulfide isomerases) (2, 3). The cytosolic side of the ER remains a reducing environment to preserve the normal function of integral ER proteins.
Cytochromes P450 (CYP, P450) are found in the cytoplasmic side of the ER and are most well-known for the ability to metabolize xenobiotics and important endogenous substrates (e.g. steroids and vitamins) (4). These proteins have been of interest in the pharmaceutical industry since their initial discovery, and enzymes in P450 Subfamilies 1A, 2C, 2D, and 3A are involved in the metabolism and clearance of a large majority of small molecule drugs currently approved for clinical use in humans (4). P450 regulation involves genetic and epigenetic aspects, as well as transcriptional regulation by both endogenous and exogenous factors (5). Posttranscriptional mRNA processing is also known and has also been found to be highly controlled (5).
Previous work from our laboratory described the oxidative inhibition of P450 Family 4 enzymes via H2O2 (6). This oxidation occurred through the sulfenylation (-SOH) of the heme-thiolate cysteine ligand, thus reversibly inactivating the enzyme.
Cysteine-sulfenic acid oxidative modifications are important for regulating many cellular processes, including cell metabolism, growth factor receptor signaling, and stress signaling (7). Challenges in the thiol-redox field include identifying sites of oxidation and enzymes capable of reversing the modification and the roles that sulfenylated proteins play in biology (8). Current theories for the production of sulfenic acids include redox relays using peroxiredoxins, local sites of high oxidant production (e.g. NADPH oxidase, mitochondria), and direct diffusion across plasma membranes (9). Sulfenylation can lead to disulfide bond formation with glutathione or other free thiols or can be reversed by either glutaredoxin or protein disulfide isomerase (7). Recently, new evidence has emerged that ER-localized tyrosine-protein phosphatase nonreceptor type 1 (PTP1B) sulfenyl groups can be reduced via thioredoxin reductase (10).
Here we describe the importance of cysteine oxidation in the context of other P450s and identify thiol sensitive and insensitive enzymes. Heme-thiol sensitivity is adopted as a term used here to describe enzymes that experience direct oxidation of the heme-thiolate center, thus altering heme-iron coordination and catalytic activity. Conversely, thiol insensitivity is used to describe enzymes that do not exhibit sensitivity to thiol oxidation up to 1 mm concentrations of H2O2.
Further findings from a proteomic analysis of sulfenylated proteins in both murine kidney and liver microsomes indicated that other P450 enzymes are also sulfenylated, including enzymes in the P450 2a, 2c, 2d, and 3a subfamilies, identified here. A subsequent screen in human liver and kidney microsomes yielded similar results. These analyses of human and murine microsomes from livers and kidneys revealed sulfenylation of many other important drug-metabolizing enzymes including UDP-glucuronyltransferases, epoxide hydrolase, flavin-containing monoxygenases (FMOs), monoamine oxidases, and carboxylesterases. Using enzymatic activity assays, isotope-coded dimedone/iododimedone (ICDID) labeling (Fig. 1), and spectral studies, we further investigated the effect of H2O2 on human recombinant P450s 1A2, 2C8, 2D6, and 3A4. We observed that P450s 2C8, 2D6, and 3A4 experienced heme-thiol sensitivity (with the modification to P450s 2C8, and 2D6 being reversible) and that P450 1A2 is thiol-insensitive to H2O2. This redox phenomenon may represent a new regulatory mechanism for many P450s.
Fig. 1.
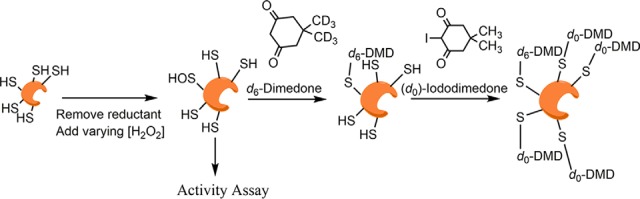
General workflow for isotope-coded dimedone/iododimedone labeling of proteins. In the case of recombinant protein assays, prereduced protein was removed of reductant, exposed to varying concentrations of H2O2, and coincubated with d6-dimedone to label sulfenic acids. Additionally, microsomes were incubated with d6-dimedone. After incubation, samples were then reduced with TCEP and free thiols were counter-alkylated with (d0)-iododimedone. Samples were then subjected to LC-MS/MS analysis.
EXPERIMENTAL PROCEDURES
Chemicals
TCEP, H2O2 (30% w/v), paclitaxel, 6-hydroxypaclitaxel, dextromethorphan, dextrorphan, testosterone, 6β-hydroxytestosterone, phenacetin, and acetaminophen were obtained from Sigma-Aldrich. Iododimedone (melting point 145–146 °C; high-resolution mass spectrometry calculated for C8H12O2I m/z 266.9882 (MH+), observed 266.9881 (Δ 0.4 ppm)) and d6-dimedone (melting point 147–148 °C; high-resolution mass spectrometry calculated for C8H7D6O2 m/z 147.1292 (MH+), observed 147.1284 (Δ 5.4 ppm)) were prepared as described previously (6).
Enzymes
Human P450s 1A2 (11), 2C8 (12), 2D6 (13), and 3A4 (14) (all with C-terminal (His)6 tags) were expressed and purified as described previously. Escherichia coli recombinant rat NADPH-P450 reductase and human liver cytochrome b5 (b5) were prepared as described by Hanna et al. (15) and Guengerich (16), respectively.
Murine Tissue Samples
All experiments using mice were conducted with approved protocols by the Institutional Animal Care and Use Committee of Vanderbilt University and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. 129/Sv mice carrying one copy of the human cytochrome P450 4A11 gene (CYP4A11) (under control of its native promoter were generated as previously described (17)) were provided normal chow diet (Purina Laboratory Rodent 5001; Purina, St. Louis, MO) with free access to water and were housed in an Association for the Assessment and Accreditation of Laboratory Animal Care-accredited, temperature-controlled facility with a 12-h light-dark cycle. All studies were conducted in mice aged 6–28 weeks of age. CYP4A11 transgenic mice were crossed with pure Sv129 wild-type mice and offspring were genotyped for the presence of a single copy of the human CYP411 gene as previously described (17). Organs were collected from male transgenic mice immediately after sacrifice and used for microsomal preparations as described below.
Human Tissue Samples
Tissues were collected and stored by the Vanderbilt University Medical Center Tissue Repository using Cooperative Human Tissue Network approved standard operating procedures under a waiver of consent and anonymized. After collection, areas of necrosis or cauterized areas were removed and then sectioned into normal and tumor tissue. A representative section of each tissue type and/or disease type was fixed in formalin and processed, and a hematoxylin and eosin stained section was obtained and reviewed by a board-certified pathologist to ensure sample integrity and usefulness in research. Kidney and liver samples (five each, collected within one year of analysis, decoded) used for this study were snap-frozen in liquid nitrogen and stored at −80 °C.
Enzymatic Assays
Assays with P450s were performed as described previously with P450 2C8-paclitaxel as substrate (12, 18), P450 2D6—dextromethorphan as substrate (13, 19), P450 3A4—testosterone as substrate (20), and P450 1A2—phenacetin as substrate (21). b5 was only used in the P450 3A4 incubations.
Protein Oxidation
Purified recombinant P450 (5 nmol stock solution, stored at −80 °C in 50 mm potassium phosphate buffer (pH 7.4) containing 20% glycerol (v/v), 1 mm DTT, and 0.1 mm EDTA) was thawed on ice and reduced for 30 min by the addition of 1 mm TCEP at 4 °C. A Zeba Spin Desalting Column (Thermo) pre-equilibrated with oxidation buffer (100 mm potassium phosphate buffer (pH 7.4) containing 0.1 mm EDTA and sparged with Ar) was used to remove reducing agents. Reduced protein was then diluted to a concentration of 500 nm using oxidation buffer. Aliquots were treated with varying amounts of H2O2 or TCEP. Aliquots for the activity assay were incubated with H2O2 or TCEP for 15 min at 37 °C. Human erythrocyte catalase (10 units, Sigma-Aldrich catalogue #C3556) was added and incubations proceeded at 23 °C for 5 min to remove H2O2. These samples were used to measure enzymatic activity as described above.
ICDID Labeling of Recombinant P450s
Additional aliquots of oxidized protein (from above, 100 pmol) were incubated with 5 mm d6-dimedone (from a 50 mm stock suspended in 100 mm sodium 3-[4-(2-hydroxyethyl)-1-piperazinyl]propanesulfonate (HEPPS) buffer (pH 8.0) containing 5% NaCl (w/v)) for 2 h at 37 °C. Trichloroacetic acid was then added to a final concentration of 10% (w/v), and the samples were incubated on ice for 15 min. The enzymes in the samples were pelleted by centrifugation (12,000 × g, 15 min, 4 °C). The supernatant was removed from each sample, and the pellet was washed with ice-cold CH3CN. The pellet was resuspended in 20 μl of 100 mm HEPPS buffer (pH 8.0) containing 2% (w/v) SDS and 1 mm TCEP, and reduction proceeded for 30 min at 37 °C. (d0)-Iododimedone (100 mm, in DMSO) was added to a final concentration of 10 mm, and incubation was done at 23 °C in the dark for 30 min. Samples were subjected to SDS- polyacrylamide gel electrophoresis (10% Bis-Tris, NuPAGE, Invitrogen) separation and stained with SimplyBlue SafeStain (Invitrogen). The Mr region corresponding to each P450 was excised, digested with trypsin (8 ng/μl) for 16 h in 25 mm NH4HCO3 (pH 7.8) at 37 °C, and subjected to LC-MS/MS analysis.
Preparation of Microsomes
Microsomes from human and murine tissues were prepared with slight modifications of published methods (22). For ICDID labeling studies, buffer A (0.10 m Tris acetate buffer (pH 7.4) containing 0.10 m KCl, 1.0 mm EDTA, and 20 μm butylated hydroxytoluene) was sparged with Ar before use. Tissue samples were homogenized in buffer A using a Teflon®-glass Potter-Elvehjem device and centrifuged at 104 × g for 20 min. at 4 °C. Postmitochondrial supernatants were then treated as described (22), and the final microsomal pellets were resuspended in 100 mm HEPPS buffer (pH 8.0) containing 1.0 mm EDTA. Aliquots of these samples were incubated with 10 mm d6-dimedone for 1 h at 37 °C. Aliquots were reduced with 1.0 mm TCEP at 37 °C for 30 min, and then 10 mm (d0)-iododimedone was added. The protein concentrations of microsomes were estimated using a bicinchoninic acid assay (Pierce). Alkylated microsomes (60 μg protein) were subjected to SDS-polyacrylamide gel electrophoresis (10% acrylamide gel, vide supra), and the P450 region (Mr 40–60 kDa) was excised, digested with trypsin (8 ng/μl) for 16 h in 25 mm NH4HCO3 buffer (pH 8.0) at 37 °C, and subjected to LC/MS/MS analysis as described below.
LC-MS/MS Analysis
Extracted peptides from murine microsomes or recombinant P450s were analyzed on a nanoLC Ultra system (Eksigent Technologies, Dublin, CA) interfaced with an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, San Jose, CA). Approximately 2.5 μg (microsomes) or 10 pmol (recombinant P450) of peptides were reconstituted in 0.1% HCO2H (v/v) and pressure-loaded (1.5 μl min−1) onto a 360 μm outer diameter × 100 μm inner diameter microcapillary analytical column packed with Jupiter octadecylsilane (C18) (3 μm, 300 Å, Phenomenex) and equipped with an integrated electrospray emitter tip. Peptides were then separated with a linear gradient formed with 0.1% HCO2H in H2O (solvent A) and 0.1% HCO2H in CH3CN (solvent B) (all v/v) by increasing from 2–45% B (v/v) over a period of 0–45 min at a flow rate of 500 nl min−1. The spray voltage was set to 2.3 kV and the heated capillary temperature to 200 °C. Higher energy collisional dissociation (HCD) and collision-induced dissociation MS/MS spectra were recorded in the data-dependent mode using a Top 2 method for each fragmentation mode. MS1 spectra were measured with a resolution of 70,000, an AGC target of 1e6, and a mass range from m/z 300 to 1,500. HCD MS/MS spectra were acquired with a resolution of 7,500, an AGC target of 1e5, and a normalized collision energy of 35. Collision-induced dissociation MS/MS spectra were acquired with normalized collision energy of 35 with a 50 ms max injection time. Peptide m/z values that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 20 s, with a repeat count of 1.
For samples produced with human microsomes, an analytical column was packed with 20 cm of C18 reverse phase material (Jupiter, 3 μm beads, 300 Å, Phenomenex) directly into a laser-pulled emitter tip. Peptides (500 ng) were loaded on the capillary reverse phase analytical column (360 μm outer diameter × 100 μm inner diameter) using a Dionex Ultimate 3000 nanoLC and autosampler. The mobile phase solvents consisted of 0.1% HCO2H, 99.9% H2O (solvent A) and 0.1% HCO2H, 99.9% CH3CN (solvent B v/v). Peptides were eluted at a flow rate of 400 nl/min. The 90-min gradient consisted of the following: 1–3 min, 2% B (sample loading from autosampler); 3–70 min, 2–40% B; 70–78 min, 40–95% B; 78–79 min, 95% B; 79–80 min, 95–2% B; 80–90 min (column re-equilibration), 2% B (all v/v). A Q-Exactive mass spectrometer (Thermo Scientific), equipped with a nanoelectrospray ionization source, was used to mass analyze the eluting peptides. The instrument method consisted of MS1 using an MS AGC target value of 1e6, followed by up to 20 MS/MS scans of the most abundant ions detected in the preceding MS scan. A maximum MS/MS ion time of 100 ms was used with a MS2 AGC target of 1e5 and an intensity threshold of 5e4. Dynamic exclusion was set to 15 s, HCD collision energy was set to 27 NCE, and peptide match and isotope exclusion were enabled.
Peptide Data Analysis
Raw data files were analyzed using MyriMatch software (Version 2.2.140) (23) and, in the case of the murine microsomes also MS-GF+ (v2016.12.12) (24), against a decoy protein database consisting of a forward and reversed human Uniprot/Swissprot database with only reviewed proteins included (Version 20170202—20,165 entries). For murine microsomes, a murine Uniprot/Swissprot database with only reviewed proteins included was used (Version 20150825—16,718 entries). Trypsin (human microsomes and recombinant P450) or chymotrypsin (murine microsomes) with fully specific digestion was used as the enzyme search parameter. The number of missed cleavages permitted was two. Precursor ion mass tolerance was set at 10 ppm, and the fragmentation tolerance was 20 ppm for the database search. Methionine oxidation (15.9949 Da, dynamic) and cysteine modifications by d6-and d0-dimedone (i.e. derived from iododimedone) (144.1057 and 138.0681 Da, respectively, dynamic) were included as variable search modifications. For the murine microsomes and recombinant P450 assays, an additional low-resolution search was performed with the same databases where precursor ion mass tolerance was set at 10 ppm and fragmentation tolerance set at 0.5 m/z. The maximum Q values of peptide spectrum matches were adjusted to achieve a peptide false discovery rate (FDR) ≤5%, using IDPicker software (Version 3.1.642.0) (25). A spectral library of peptides was then created with IDPicker and loaded into Skyline Software for confident identification and quantitation of precursors pertaining to cysteine-containing peptides. MS1 precursor quantitation was performed as described previously (26, 27). The retention time of the identified peptide was used to position a retention time window (± 2.0 min) across the run lacking the same peptide identification. Second, the resolution for extracting the MS1 filtering chromatogram of the target precursor ions with both light and heavy labeled peptides was set to 60,000 at 400 Th. Then extracted ion chromatograms for the top three isotopic peaks were manually inspected for proper peak picking of MS1 filtered peptides, and those with isotopic dot product scores lower than 0.8 were rejected. Additional criteria were used to further ensure the high accuracy and precision of quantification: S/N> 3.0 and baseline separation was required between the isotopic peaks of a quantifiable peptide and unknown isobaric interference. The ratios of peptide areas of light peptides to their heavy isotopes (RL:H) were calculated automatically. Quantification results were obtained from five biological replicates for human microsomes, four biological replicates for murine microsomes, and two biological replicates for recombinant protein.
Spectroscopy
P450s were oxidized with H2O2 as above, but the procedure was adapted slightly for spectroscopic assays. The enzyme was diluted with oxidation buffer to 1.0 μm prior to introduction of 1.0 mm TCEP or 500 μm H2O2. After oxidation, 30 units of catalase were added, and each sample was incubated at 23 °C for 5 min. To this solution, final concentrations of 1.0 μm NADPH-P450 reductase, 150 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, and 0.1 unit/ml protocatechuate dioxygenase (Sigma) were added to an anaerobic cuvette, with NADPH (300 nmol, in aqueous solution) in a sidearm of the cuvette. Samples were degassed, 20 μm 3,4-dihydroxybenzoate (Sigma, substrate for protocatechuate dioxygenase) was added to remove oxygen (28), and samples were further degassed using a manifold attached to both vacuum and purified Ar (29, 30) and placed under an anaerobic CO atmosphere. The valves of the cuvettes were sealed and multiple UV-visible absorbance spectra were recorded using an OLIS/Hewlett Packard 8452 diode array spectrophotometer (On-Line Instrument Systems, Bogart, GA). Spectra were collected from 380 to 600 nm before and after the addition of NADPH and then sodium dithionite (6).
Experimental Design and Statistical Rationale
All chemoproteomic studies using tissue were performed with at least four biological replicates to evaluate biological variability. Enzymatic activity and labeling assays using recombinant-purified enzymes were performed in biological duplicate and showed consistent results. Values of means with standard deviation are presented.
RESULTS
Identification of Sulfenylated P450s in Murine Microsomes
Following our report that identified oxidation of P450 Subfamily 4 enzymes in murine kidney and liver microsomes (6), further analysis led to the identification of other sulfenylated cysteines from other P450s in Subfamilies 2, 3, and 26 (Table I, Supplemental Table S1). Of these, the murine P450 3a, 2c, 2d, and 2e proteins are known drug-metabolizing enzymes in mice (31). These results indicate that many P450s may be inhibited by oxidative posttranslational modification of cysteines.
Table I. Sulfenylated murine microsomal proteins. Proteins with respective gene names identified in microsomes of murine livers and kidneys at 5% peptide FDR. Modifications are presented as cysteine (C) and sequence location. Tissue location is denoted as liver (L) or kidney (K) by the modification.
| Description | Gene Name | Modification |
|---|---|---|
| Carboxylesterase 3B | Ces3b | C434 (L) |
| Cytochrome P450 1a2 | Cyp1a2 | C404 (L) |
| Cytochrome P450 2c29 | Cyp2c29 | C435 (L)1, C216 (L), C226 (L), C372 (L) |
| Cytochrome P450 2c50 | Cyp2c50 | C435 (L)1 |
| Cytochrome P450 2c54 | Cyp2c54 | C435 (L)1 |
| Cytochrome P450 2d10 | Cyp2d10 | C462 (L), C496 (L) |
| Cytochrome P450 2d26 | Cyp2d26 | C462 (L/K)2, C496 (L) |
| Cytochrome P450 2d9 | Cyp2d9 | C462 (L/K)2, C496 (L) |
| Cytochrome P450 2e1 | Cyp2e1 | C488 (L/K) |
| Cytochrome P450 2f2 | Cyp2f2 | C487 (L) |
| Cytochrome P450 3a11 | Cyp3a11 | C64 (L), C443(L), C98(L), C239(L) |
| Cytochrome P450 4a12A | Cyp4a12a | C373 (L/K) |
| Cytochrome P450 4b1 | Cyp4b1 | C453 (K), C369 (K), C373 (K) |
| Leukotriene-B4 ω-hydroxylase 2 | Cyp4f3 | C384 (L) |
| Dimethylaniline monooxygenase, N-oxide-forming 2 | Fmo2 | C146 (K) |
| Dimethylaniline monooxygenase, N-oxide-forming 5 | Fmo5 | C248 (L), C468 (L) |
| Epoxide hydrolase 1 | Ephx1 | C312 (L) |
Shared peptide sequences: 1STGKRICAGEGLARMELF, 2LFFTCLLQRF.
Identification of Sulfenylated Drug-Metabolizing Enzymes in Human Kidney and Liver Microsomes
The murine data (Table I, Supplemental Table S1) led to the expansion of the proteomic study to human microsomes. Frozen human kidney and liver tissues from five individuals were rapidly fractionated into microsomal fractions in deaerated buffer and labeled using an isotope-coded ICDID strategy for relative quantitation (Fig. 1) (32). After excising the 40–60 kDa P450 region and digesting the proteins with trypsin, we identified 347 modified proteins in the kidney microsomes at a 5% peptide FDR (Table II, Supplemental Table S2, Fig. S1). In addition, 380 modified proteins were identified in the liver microsomal fractions (5% peptide FDR, Table II, Supplemental Table S3, Fig. S1). Of these proteins, 11 P450s were sulfenylated in the kidney microsomes and 24 P450s in liver microsomes. The identified P450s included enzymes that are important in the metabolism of both endogenous and xenobiotic substrates.
Table II. Sulfenylated human microsomal proteins. Proteins with respective gene names identified in microsomes prepared from human livers and kidneys, at 5% peptide FDR. Modifications are presented as cysteine (C) and sequence location, with the percentage sulfenylation (±SD, n = 5 different tissue samples) indicated in parentheses.
| Description | Gene Name | Modifications Found in Liver | Modifications Found in Kidney | ||||
|---|---|---|---|---|---|---|---|
| Arylacetamide deacetylase | AADAC | C340 (28 ± 5%) | – | ||||
| Alcohol dehydrogenase 1A | ADH1A | C1331 (32 ± 2%) | C328 (31 ± 6%) | C1714 | C212 | – | |
| C1122 (35 ± 4%) | C98 (29 ± 8%) | C1754 | C47 | ||||
| C2413 (37 ± 3%) | C196 | ||||||
| Alcohol dehydrogenase 1B | ADH1B | C47 (27 ± 3%) | C1122 | C1754 | – | ||
| C985 (34 ± 2%) | C2413 | C1966 | |||||
| C1331 | C1714 | C2126 | |||||
| Alcohol dehydrogenase 1C | ADH1C | C1122 | C985 | C1967 | C2413 | – | |
| Alcohol dehydrogenase 4 | ADH4 | C246 (28 ± 2%) | C23 (37 ± 4%) | C287 | – | ||
| C274 (39 ± 5%) | C99 (32 ± 4%) | C2016 | |||||
| C47 (35 ± 3%) | C105 | C2176 | |||||
| Alcohol dehydrogenase 6 | ADH6 | C1967 | – | ||||
| Aldehyde dehydrogenase family 16 member A1 | ALDH16A1 | C350 | C496 | ||||
| Aldehyde dehydrogenase family 1 member A3 | ALDH1A3 | – | C196 | C197 | |||
| Aldehyde dehydrogenase, mitochondrial | ALDH2 | C386 (33 ± 3%) | C66 | C472 | C66 | C472 | |
| Aldehyde oxidase | AOX1 | – | C386 (23 ± 13%) | C179 | |||
| Liver carboxylesterase 1 | CES1 | C285 (40 ± 3%) | C390 (37 ± 1%) | C116 (37 ± 3%) | C390 | ||
| C274 (37 ± 5%) | |||||||
| Steroid 17 α-hydroxylase/17,20 lyase | CYP17A1 | – | C442 | ||||
| P450 1A2 | CYP1A2 | C504 (42 ± 5%) | C159 (44 ± 5%) | – | |||
| P450 26A1 | CYP26A1 | – | C170 | ||||
| Sterol 26-hydroxylase, mitochondrial | CYP27A1 | C228 (36 ± 6%) | C427 (43 ± 1%) | C228 | |||
| P450 2A6 | CYP2A6 | C82 (34 ± 4%) | C439 (36 ± 3%) | – | |||
| P450 2B6 | CYP2B6 | C436 (32 ± 3%) | C180 (27 ± 7%) | C152 (46 ± 2%) | C79 | – | |
| P450 2C18 | CYP2C18 | C3388 (42 ± 2%) | C216 (38 ± 4%) | C486 | C26610 | – | |
| C1519 (38 ± 2%) | |||||||
| P450 2C19 | CYP2C19 | C3388 | C372 (40 ± 4%) | C216 | C1519 | – | |
| C435 | C26610 | C179 | |||||
| P450 2C8 | CYP2C8 | C435 (37 ± 3%) | C26610 (39 ± 3%) | C216 | C172 | C26610 | |
| C1519 (41 ± 3%) | C486 (37 ± 4%) | C225 | C175 | ||||
| C51 (34 ± 2%) | C3388 | C179 | C164 | ||||
| P450 2C9 | CYP2C9 | C372 (38 ± 1%) | C216 (38 ± 3%) | C164 | C3388 | C1519 | C216 |
| C486 (38 ± 2%) | C172 | C179 | C1519 | ||||
| C266 (41 ± 5%) | |||||||
| P450 2D6 | CYP2D6 | C443 (45 ± 7%) | C191 (39 ± 4%) | C57 (27 ± 7%) | – | ||
| P450 2E1 | CYP2E1 | C261 (39 ± 6%) | C488 (34 ± 2%) | C480 (39 ± 4%) | – | ||
| C437 (37 ± 3%) | C268 (42 ± 2%) | ||||||
| P450 3A4 | CYP3A4 | C9811 (33 ± 1%) | C46812 (40 ± 2%) | C58 | – | ||
| P450 3A5/3A7 | CYP3A5/A7 | C9811 | C46712 | – | |||
| P450 4A11 | CYP4A11 | C25613 (40 ± 2%) | C200 (33 ± 4%) | C51314 | C375 | C25613 (41 ± 5%) | C86 |
| C53 (36 ± 5%) | C86 (36 ± 4%) | C200 (38 ± 6%) | C51314 | ||||
| P450 4A22 | CYP4A22 | C25613 | C51314 | C25613 | C51314 | ||
| P450 4F11 | CYP4F11 | C354 (36 ± 2%) | C46817 (43 ± 6%) | C38418* | C260 | C38418* | |
| C5016 | C102 (84 ± 5%) | C276 | |||||
| P450 4F12 | CYP4F12 | C35415 | C46817 | C384 | C5016 | – | |
| P450 4F2 | CYP4F2 | C102 (37 ± 2%) | C5019 (36 ± 6%) | C35415 | C468 | C35415 (43 ± 5%) | C102 |
| C384 (43 ± 13%) | |||||||
| P450 4F22 | CYP4F22 | C5819 | – | ||||
| P450 4F3 | CYP4F3 | C35415 | C402 | C38418* | C46817 | C35415 | C38418* |
| C50 (38 ± 2%) | |||||||
| P450 4F8 | CYP4F8 | – | C35415 | C38418* | |||
| P450 4V2 | CYP4V2 | C282 (37 ± 5%) | C383 (37 ± 5%) | C483 (40 ± 4%) | C406 | – | |
| P45051A1 | CYP51A1 | C337 | C345 | C366 (31 ± 6%) | C449 | – | |
| Epoxide hydrolase 1 | EPHX1 | C80 (39 ± 1%) | C304 (38 ± 7%) | C232 (39 ± 2%) | C80 (43 ± 4%) | C304 (46 ± 14%) | |
| C182 (38 ± 1%) | C182 (41 ± 3%) | C232 | |||||
| Dimethylaniline monooxygenase | FMO1 | – | C63 (43 ± 4%) | C111 (33 ± 3%) | |||
| [N-oxide-forming] 1 | C423 | ||||||
| Dimethylaniline monooxygenase | FMO3 | C170 (38 ± 1%) | C466 (43 ± 4%) | C68 (37 ± 3%) | C21 | C294 | |
| [N-oxide-forming] 3 | C397 (36 ± 1%) | C197 (41 ± 2%) | C294 (36 ± 9%) | C30 | |||
| C146 (36 ± 4%) | |||||||
| Dimethylaniline monooxygenase | FMO5 | C112 (35 ± 4%) | C468 (29 ± 10%) | C147 | – | ||
| [N-oxide-forming] 5 | C248 (28 ± 0%) | C31 | C22 | ||||
| Amine oxidase [flavin-containing] A | MAOA | C37421* (39 ± 2%) | C165 (32 ± 1%) | C306 (31 ± 2%) | C266 | C20120 (40 ± 4%) | C165 |
| C20120 (35 ± 1%) | C37421*(42 ± 4%) | C266 | |||||
| Amine oxidase [flavin-containing] B | MAOB | C156 (36 ± 1%) | C297 (37 ± 1%) | C36521* | C19220 | C297 (37 ± 1%) | C19220 |
| C156 (41 ± 3%) | C36521* | ||||||
| Serum paraoxonase/arylesterase 1 | PON1 | C42 (29 ± 2%) | C284 | – | |||
| Serum paraoxonase/lactonase 3 | PON3 | C42 (38 ± 7%) | – | ||||
| UDP-glucuronosyltransferase 1–4 | UGT1A4 | C187 (37 ± 4%) | – | ||||
| UDP-glucuronyltransferase 1–6 | UGT1A6 | C176 | C176 | ||||
| UDP-glucuronyltransferase 1–7 | UGT1A7 | – | C174 | ||||
| UDP-glucuronyltransferase 1–9 | UGT1A9 | – | C153 | ||||
| UDP-glucuronyltransferase 2A3 | UGT2A3 | – | C31 | ||||
| UDP-glucuronyltransferase 2B10 | UGT2B10 | C28122 (40 ± 4%) | – | ||||
| UDP-glucuronyltransferase 2B15 | UGT2B15 | C28322 | – | ||||
| UDP-glucuronyltransferase 2B17 | UGT2B17 | C28322 | – | ||||
| UDP-glucuronyltransferase 2B4 | UGT2B4 | C127 (28 ± 6%) | C514 | – | |||
| UDP-glucuronyltransferase 2B7 | UGT2B7 | C282 (35 ± 5%) | – | ||||
Shared peptide sequences: 1RFTCR, 2NPESNYCLK, 3ELGATECINPQDYK, 4VCLIGCGFSTGYGSAVNVAK, 5VIPLFTPQCGK, 6VTPGSTCAVFGLGGVGLSAVMGCK, 7VTPGSTCAVFGLGGVGLSVVMGCK, 8SPCMQDR, 9CLVEELRK, 10DFIDCFLIK, 11ECYSVFTNR, 12VLQNFSFKPCK, 13ACQLAHQHTDQVIQLR, 14RLPNPCEDKDQL, 15HPEYQEQCR, 16LQCFPQPPK, 17NCIGQAFAMAEMK, 18EIEWDDLAQLPFLTMCI(/L)K, 19LRCFPQPPR, 20QCGGTTR, 21I(/L)CELYAK, 22PFLPNVDFVGGLHCK.
* Single amino acid variation (leucine/isoleucine) in peptide sequences that cannot be determined from MS/MS fragmentation.
In addition to the P450 enzymes, we identified other modified proteins important in drug metabolism, including UDP-glucuronyl transferases, epoxide hydrolase, FMOs, monoamine oxidases, and carboxylesterases. Other previously known sulfenylated proteins were also identified, including protein disulfide isomerases (33) and aldehyde dehydrogenases (34). MS1 precursor intensities for peptides were quantified using Skyline software (26).
Spectral Analysis of Recombinant P450s for Thiol Sensitivity to H2O2
P450s 1A2, 2C8, 2D6, and 3A4 were selected for further analysis. To confirm that these P450 enzymes were affected by sulfenylation, we utilized their spectral properties to determine if heme coordination or interaction with NADPH-P450 reductase was disrupted. In the presence of CO, disruption of the heme-thiol ligation either prevents reduction of the heme or yields the inactive form, cytochrome P420 (the five-coordinate ferrous-CO complex, which has a maximal absorbance at P420 nm) so that a characteristic 450 nm spectrum is not observed (35). tris-(Carboxyethyl)phosphine (TCEP)-pretreated P450s were treated with 500 μm H2O2 and (after the removal of residual H2O2 by catalase) reconstituted with NADPH-P450 reductase, deaerated, and placed under an anaerobic atmosphere of CO. Following the addition of NADPH, spectra were recorded for the TCEP-reduced (Fig. 2, left column) and H2O2-oxidized (Fig. 2, right column) samples. Sodium dithionite, which reduces both sulfenic acids and the heme iron, was subsequently added to both samples (Fig. 3). P450 1A2 was very insensitive to H2O2, with only a slight increase in 450 nm absorbance after dithionite addition (Fig. 2A). P450s 2D6 (Fig. 2B) and 2C8 (Fig. 2C) both exhibited sensitivity to H2O2-dependent oxidation, with a noted decrease in 420 nm absorbance (indicative of the inactive form, cytochrome P420) and an increase in 450 nm absorbance indicative of reestablishment of the heme-thiol ligand (Fig. 3). P450 3A4 (Fig. 2D) exhibited a complete loss of 450 nm absorbance in the oxidized sample, which was not reversible upon addition of dithionite and increased absorbance at 420 nm.
Fig. 2.

Spectral analysis of oxidized P450s. TCEP-pretreated P450s were treated with 500 μm H2O2 and (after the removal of residual H2O2 by catalase) reconstituted with NADPH-P450 reductase, deaerated, and placed under an anaerobic atmosphere of CO. Following the addition of NADPH, spectra were recorded for the TCEP-reduced (Fig. 2A) and H2O2-oxidized (Fig. 2B) samples (NADPH-P450 reductase (POR), blue traces). Following NADPH addition, sodium dithionite (Na2S2O4) was added as a stronger reducing agent (dithionite, orange traces). Analysis was performed for P450s 1A2 (A), 2C8 (B), 2D6 (C), and 3A4 (D).
Fig. 3.

Mechanism of loss of activity due to sulfenylation. In the presence of H2O2, the heme-thiolate ligand becomes oxidized and loses its coordination with the heme iron. The sulfenic acid is not reduced by NADPH-P450 reductase but can be reduced by dithionite, re-forming the heme coordination.
Oxidative Inhibition of P450 1A2, 2C8, 2D6, and 3A4 Catalytic Activities
The enzymatic activity of P450 1A2 was largely uninhibited with preincubation of up to 1 mm H2O2, using phenacetin as a substrate, consistent with the spectral results (Fig. 4A). P450 2C8 showed sensitivity to H2O2, with an approximate IC50 of 150 μm and a loss of 87% activity at 1 mm H2O2, using taxol as a substrate (Fig. 4B). P450 2D6 was also inhibited by H2O2, with an estimated IC50 of ∼300 μm (Fig. 4C) and loss of 70% activity at 1 mm H2O2, using dextromethorphan as a substrate. An IC50 of ∼300 μm H2O2 was determined for P450 3A4, using testosterone as a substrate (Fig. 4D), with a loss of 95% of activity at 1 mm H2O2.
Fig. 4.
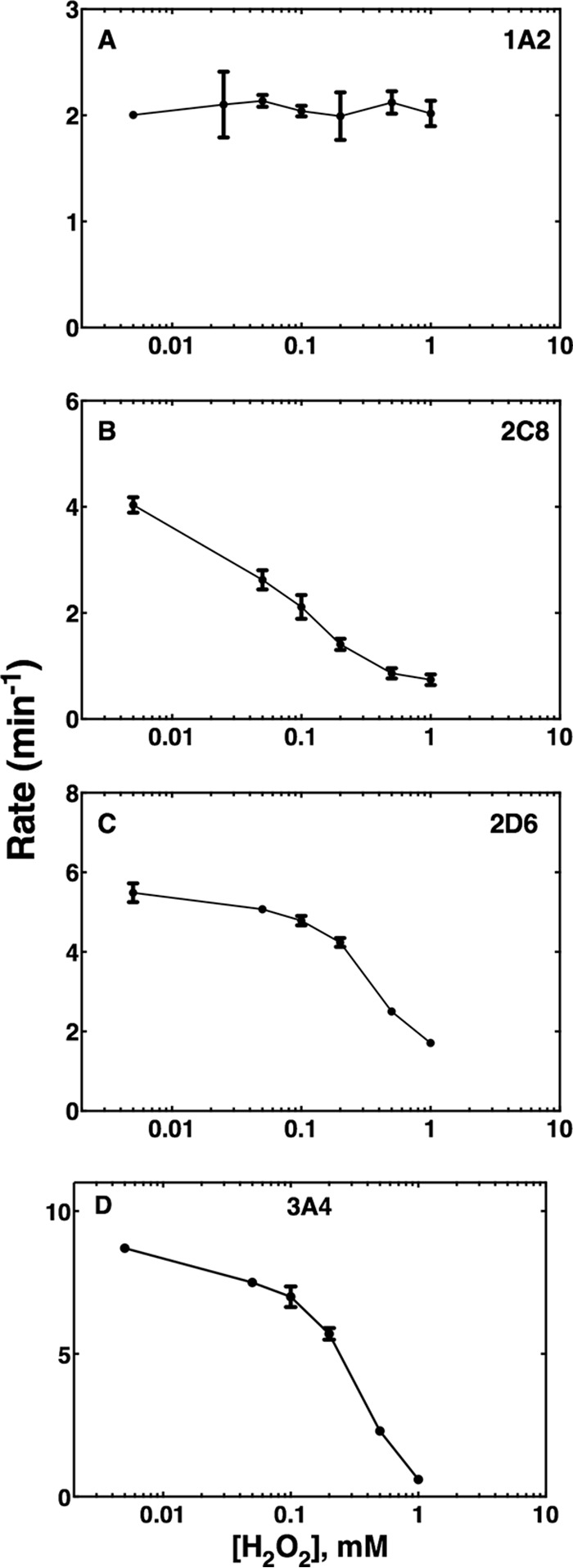
Oxidative inhibition of P450 catalytic activity. Prereduced P450 was incubated with varying concentrations of H2O2, or TCEP as a reduced control. Enzymes were then reconstituted and enzymatic activity assays were performed as in the experimental procedures. (A) P450 1A2 was incubated with the substrate phenacetin, and acetaminophen was monitored as the product. The reduced control enzymatic rate was 1.85 ± 0.01 min−1. (B) P450 2C8 was incubated with taxol, and 6α-hydroxytaxol was monitored as the product. The reduced control enzymatic rate was calculated as 5.5 ± 0.4 min−1. (C) P450 2D6 was incubated with dextromethorphan and dextrorphan was monitored as the product. The reduced control enzymatic rate was 5.6 ± 0.1 min−1. (D) P450 3A4 was incubated with testosterone and 6β-hydroxytestosterone was monitored as the product. The reduced control enzymatic rate was 11.3 ± 0.3 min−1. Samples were acquired in biological duplicate and presented as means ± S.D. (range).
Sulfenylation of P450s 1A2, 2C8, 2D6, 3A4
P450s 1A2, 2C8, 2D6, and 3A4 were subjected to ICDID labeling followed by LC-MS/MS analysis of tryptic peptides (Fig. 1). For P450 1A2, this assay allowed three of the seven cysteines to be quantified (Fig. 5A, supplemental Fig. S2). Cys-458 (heme-thiol ligand) and Cys-504 showed low levels of sulfenylation up to 1 mm H2O2. Additionally, Cys-159 showed very high levels of oxidation, but this did not impair the enzymatic activity of P450 1A2 (Figs. 4A and 5A), indicating that P450 1A2 is insensitive to thiol oxidation.
Fig. 5.

Sulfenylation of P450s 1A2, 2C8, 2D6, and 3A4. Prereduced enzyme was coincubated with d6-dimedone and varying concentrations of H2O2 or TCEP. Proteins were then reduced and counter alkylated with d0-iododimedone. Labeled samples were then subjected to LC-MS/MS analysis and heavy/light ratios were quantified as described in the experimental procedures. Peptides for P450 1A2 (A), 2C8 (B), 2D6 (C), and 3A4 (D) were acquired in biological duplicate and presented as means ± S.D.
Five thiol-containing peptides were quantified for P450 2C8 (Fig. 5B, Supplemental Fig. S3). Cys-338, Cys-51, Cys-151, and Cys-435 (heme-thiol ligand) were sulfenylated in an H2O2-dependent manner, while Cys-266 was oxidation insensitive.
Two peptides of P450 2D6 could be quantified (Fig. 5C, Supplemental Fig. S4). Cys-443 (heme-thiol ligand) showed an H2O2-dependent increase in sulfenylation compared with the reduced control.
P450 3A4 ICDID labeling allowed for the quantitation of four of the seven cysteines (Fig. 5D, Supplemental Fig. S5). The heme-thiol cysteine (Cys-442) showed a significant H2O2-dependent increase in sulfenylation. Cys-468 also showed an H2O2-dependent increase in sulfenylation, but peptides containing the di- and trioxidized cysteine sulfinic (SO2−) and sulfonic (SO3−) acids (respectively) were also found (Supplemental Fig. S6), suggesting that Cys-468 is very sensitive to oxidation.
DISCUSSION
ICDID labeling of murine liver and kidney microsomes provided the interesting finding that P450s other than the Subfamily 4 enzymes previously described (6) also contained oxidatively modified cysteines. This result may seem inconsistent with previous activation studies involving other P450s and dithiothreitol, which found no significant differences in enzymatic rates (6). However, this is probably because it is a regular practice to dialyze against dithiothreitol when removing imidazole after His6-nitrilotriacetic acid/nickel (NTA-Ni2+) purification and storage. P450 4A11 is unusual in its ability to readily oxidize in the presence of air, at least under these conditions. This labeling strategy was then expanded to human liver and kidney microsomes. Many other P450s were found to be sulfenylated in these samples (Table II, Supplemental Tables S2 and S3). While our knowledge of transcriptional regulation, sequence variation, and inhibition is extensive for many P450s (4), knowledge of posttranslational regulation of P450s is limited, with research mostly focused on glycosylation, phosphorylation, ubiquitination, and nitration (24, 36). Cysteine oxidation may play a role in physiological posttranslational regulation as well.
Cysteine sulfenylation was found in a total of 57 drug-metabolizing enzymes. The group includes UDP-glucuronyl transferases, FMOs, and carboxylesterases, suggesting that many microsomal enzymes are modified by oxidation. These results seem reasonable, in that many enzymes have been found to be regulated by oxidation to form cysteine sulfenic acids (34, 37). Additionally, a comparison of the results presented here and a recent study on sulfenylated proteins (38) revealed that 21% (kidney microsomes) and 14% (liver microsomes) of proteins were identified in both datasets. This comparison may indicate that the RKO adenocarcinoma cell line studied by Gupta et al. (38) likely has similar basal proteins that are oxidized. More in-depth studies of the non-P450 enzymes will be required to further verify sensitivity of catalytic activity to oxidation and effects that oxidation may have on activity and regulation.
Relative quantitation of the modified cysteines (Table II, Supplemental Fig. S1) yielded similar levels of d6-dimedone labeling, which may be due in part to the one hour incubation time with microsomes. While dimedone has been shown to be selective, the low reactivity of dimedone with sulfenic acids requires extended incubation times to achieve sufficient labeling (rate of 0.8 min−1 under the reported conditions) (39, 40). The modified cysteines identified are likely sensitive to oxidation, but the amount of labeling observed may be representative of the equilibrium of a saturated system. Overall, 35 and 33% of all cysteine-containing peptides identified were modified with d6-dimedone in kidney and liver microsomes, respectively. Since more than half of the peptides identified were not sulfenylated, the likelihood that this observation is an artifact would seem to be low. Furthermore, a control experiment was performed in which kidney and liver tissues were homogenized in 5 mm TCEP, fractionated into microsomes, labeled, and subjected to LC-MS/MS analysis as described under “Experimental Procedures”. Approximately 6% of all cysteine-containing peptides from both the kidney and liver samples were modified with d6-dimedone, suggesting that some sulfenylated cysteines may have been inaccessible to the TCEP reducing agent (Supplemental Fig. S7). Also, the fractionation and labeling protocol may have introduced a slightly oxidative environment that modified highly susceptible cysteines. Nevertheless, this experiment serves as a control for our positive results.
Four drug-metabolizing P450s were chosen for more in-depth oxidative inhibition studies.
Interestingly, P450 1A2 was resistant to oxidation, as established by spectral, inhibition, and dimedone labeling studies (Figs. 2A, 4A, and 5A, respectively). Of note was the hyperoxidation of Cys-159, which did not affect the catalytic activity of P450 1A2 (Fig. 5A). This ancillary cysteine is positioned away from the active site, and its modification has a negligible effect on function (Fig. 6A). The resistance to oxidation is proposed to be related to access of oxidants or to stabilization of the heme-thiol system due to either the surrounding amino acid residues or the overall structure of the protein.
Fig. 6.

Crystal structure of P450s highlighting positions of cysteine. Crystal structures for 1A2 (A), 2C8 (B), 2D6 (C), and 3A4 (D) were adapted from the RCSB Protein Data Bank (rcsb.org). The accession numbers used were 2HI4, 2NNJ, 5TFT, and 5G5J, respectively.
P450 2C8 showed an 87% loss of catalytic activity at 1 mm H2O2, compared with the reduced control (Fig. 3B). This inhibition may seem surprising in light of the large number of cysteines contained in P450 2C8 (Fig. 7), but the heme thiol peptide was still modified. Despite the gradual loss of activity, the spectral and ICDID labeling studies (Figs. 2B and 5B) showed a high loss and recovery of the heme-thiol ligand spectrally and significant sulfenic acid labeling (Cys-435). Only four of the 16 thiols were quantified because of the clustering of thiols in the sequence; i.e. some tryptic peptides contained up to four cysteines and spanned >25 amino acids.
Fig. 7.
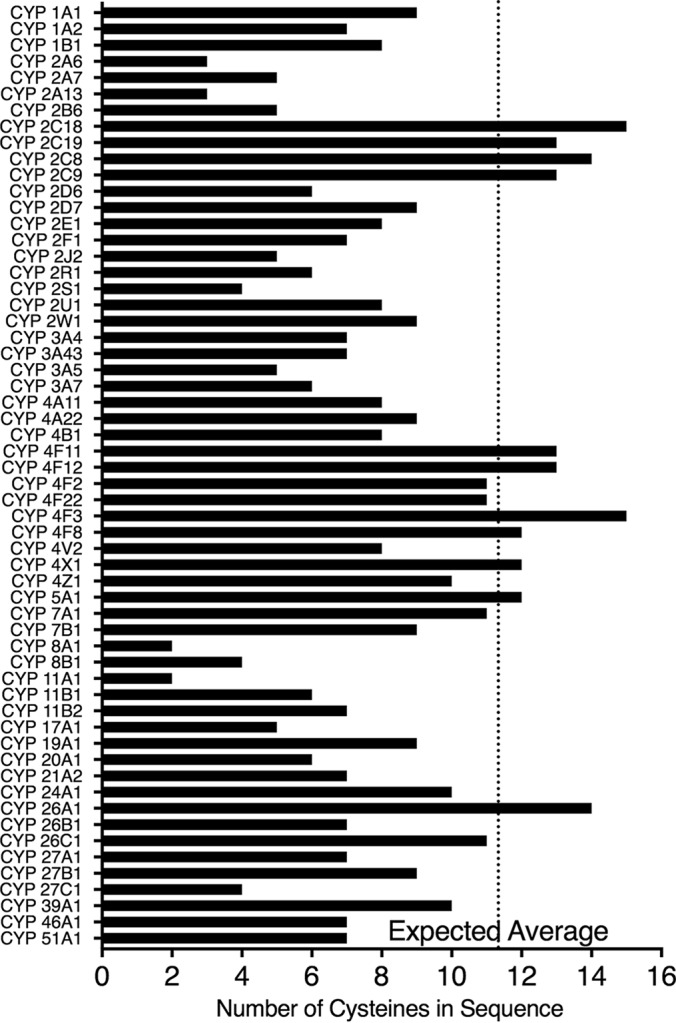
Number of cysteines found in human P450 enzymes. Cysteines of P450 sequences obtained from Uniprot (uniport.org) were counted. The average number of cysteines in the human proteome (49) was applied to the average length of human P450s (501 amino acids) to determine the expected number of cysteines to be 11.3 (dotted vertical line).
P450 2D6 showed less loss of activity (70%) than P450 2C8 but still exhibited inhibition after treatment with H2O2 (Fig. 4C) and the ability of the heme-thiol to re-ligand the heme iron in the spectral analysis (Fig. 2C). This diminished response was reflected in ICDID analysis, which showed lower amounts of sulfenylation of the heme-thiol Cys-443 and also Cys-191 (Fig. 5C).
Oxidized P450 3A4 showed an inability of the heme-thiol to re-ligand to the heme-iron (after dithionite reduction), as judged by the spectra (Fig. 2D). This irreversible inactivation is proposed to be related to stabilization of the oxidized thiol by surrounding residues and is a unique feature among P450s tested. ICDID labeling showed high amounts of sulfenylation on several cysteine thiols, including the heme-thiol Cys-442, indicating a high susceptibility to oxidation. This sensitivity of the non-heme thiols can be considered in light of the report that a cysteine-depleted variant of P450 3A4 has increased activity compared with the wild-type enzyme (41). We concur that the cysteine residues are not essential but that the presence of the (non-heme peptide) sulfenic acids appears to be inhibitory to catalytic activity, through an unknown mechanism.
There has been difficulty designing probes that can directly measure local bursts of H2O2 and not just a cumulative or average amount over time, especially in prominent production areas such as the plasma membrane, endoplasmic reticulum, and peroxisomes (42). When looking at redox issues, one mainly focuses on a concentration of H2O2 for which the effect can be reversed. This is the case for three of the four P450s tested spectrally, suggesting that this treatment does not irreversibly destroy the protein.
These spectral studies have been challenging to interpret because the proper conditions have not yet been identified to produce a homogenous sample of stable sulfenylated heme-thiol cysteine P450 protein in large quantities. The spectral studies in conjunction, with the observation that sulfenylated cysteines cause enzymatic inhibition, lead us to believe this phenomenon is blocking steps in the P450 catalytic cycle. This blocked step may be reduction of the heme iron. This could be occurring directly (most likely) or through a peripheral oxidation that limits reductase binding. It may also reduce the ability for the heme iron to bind oxygen (or carbon monoxide in the case of the inhibitory or spectral studies, respectively). The spectral studies also require physical changes (i.e. vacuum and slight bubbling) to remove the existing oxygen in the sample. These manipulation conditions likely affect some protein irreversibly, accounting at least in part for the cytochrome P420 seen in the pre-reduced samples. What is most relevant is the change in the amount of P450 seen between the reductase- and dithionite -reduced spectra (Fig. 2).
It has been known that some P450s can catalyze reactions with the use of H2O2 alone for quite some time (43–46). Also, mutating residues around the proximal heme ligand allows for alterations in both the heme redox potential and reactivity (47). P450s are also known to produce H2O2 as a byproduct of catalysis (48). CYP4A11 H2O2 production was measured to be 5 μm min−1, which would be 25 nmol H2O2 min−1 under our experimental conditions (6). Typical incubation times would likely not produce enough H2O2 to produce an inhibitory effect. These data point to the significance of H2O2 in the catalytic cycle of P450s and of the residues surrounding the heme-thiol ligand. The presence of a biological mechanism that limits potentially harmful H2O2 shunting in certain mammalian P450s, but is not present in others (e.g. P450 1A2), seems reasonable but more studies are needed to evaluate the significance of this phenomenon.
Our finding of both thiol-sensitive and insensitive P450s expands the knowledge of potential post-translational modifications found in drug-metabolizing enzymes. In general, cysteines are reactive, underrepresented in the proteome, and conserved among proteomes (49). In an accounting of all human P450s, the number of cysteines varies from two to 15 (Fig. 7). Using the average length of all human P450s (501 amino acids) and the overall percent of cysteines found in the human proteome (2.26% (49)), the expected number of cysteines in human P450s would be 11. Due to the deviation from this number, these cysteines may play important roles other than heme coordination and further investigation in cellular systems may be of interest.
The drug-metabolizing enzymes found to be sulfenylated in our proteomic screen and the subsequent validation of the inhibitory nature of this oxidative modification in P450s pose many new questions and potentially provide insight into the biological regulation of P450s. In further studies, a major goal is to determine the biological role of heme-thiolate sulfenylation and its relevance to oxidative stress.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD007913.
Supplementary Material
Acknowledgments
We thank K. Trisler for assistance in preparation of the manuscript.
Footnotes
* This work was supported by National Institutes of Health Grants R01 GM118122 (to F.P.G.) and F31 F31HL136133 (M.E.A.) and American Heart Association Predoctoral Fellowship PRE33410007 (M.E.A.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This paper constitutes part of the requirements to fulfill a Ph.D. thesis (M.E.A.) at Vanderbilt University.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ER
- endoplasmic reticulum
- AGC
- automatic gain control
- CID
- collision-induced dissociation
- DTT
- dithiothreitol
- FDR
- false discovery rate
- FMO
- microsomal flavin-containing monooxygenase
- HCD
- higher-energy collisional dissociation
- HEPPS
- 3-[4-(2-hydroxyethyl)-1-piperazinyl]propanesulfonate
- ICDID
- isotope-coded dimedone/iododimedone (labeling)
- MH+
- exact mass of protonated parent ion
- NCE
- normalized collision energy
- P450 or CYP
- cytochrome P450
- TCEP
- tris(carbethoxymethyl)phosphine.
REFERENCES
- 1. Fewell S. W., Travers K. J., Weissman J. S., and Brodsky J. L. (2001) The action of molecular chaperones in the early secretory pathway. Annu. Rev. Genet. 35, 149–191 [DOI] [PubMed] [Google Scholar]
- 2. Saaranen M. J., Karala A. R., Lappi A. K., and Ruddock L. W. (2010) The role of dehydroascorbate in disulfide bond formation. Antioxid. Redox Signal. 12, 15–25 [DOI] [PubMed] [Google Scholar]
- 3. Tavender T. J., Springate J. J., and Bulleid N. J. (2010) Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 29, 4185–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guengerich F. P. (2015) Human cytochrome P450 enzymes, in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R. ed.), 4th Ed., Springer, New York, pp 523–785 [Google Scholar]
- 5. Gotoh S., Ohno M., Yoshinari K., Negishi M., and Kawajiri K. (2015) Nuclear receptor-mediated regulation of cytochrome P450 genes, in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R. ed.), 4th Ed., Springer, New York, pp 787–811 [Google Scholar]
- 6. Albertolle M. E., Kim D., Nagy L. D., Yun C. H., Pozzi A., Savas Ü., Johnson E. F., and Guengerich F. P. (2017) Heme-thiolate sulfenylation of human cytochrome P450 4A11 functions as a redox switch for catalytic inhibition. J. Biol. Chem. 292, 11230–11242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Devarie-Baez N. O., Silva Lopez E. I., and Furdui C. M. (2016) Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radical Res. 50, 172–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forman H. J., Davies M. J., Krämer A. C., Miotto G., Zaccarin M., Zhang H., and Ursini F. (2017) Protein cysteine oxidation in redox signaling: Caveats on sulfenic acid detection and quantification. Arch. Biochem. Biophys. 617, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Travasso R. D. M., Sampaio Dos Aidos F., Bayani A., Abranches P., and Salvador A. (2017) Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 12, 233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dagnell M., Pace P. E., Cheng Q., Frijhoff J., Östman A., Arner E. S. J., Hampton M. B., and Winterbourn C. C. (2017) Thioredoxin reductase 1 and NADPH directly protect protein tyrosine phosphatase 1B from inactivation during H2O2 exposure. J. Biol. Chem. 292, 14371–14380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sandhu P., Guo Z., Baba T., Martin M. V., Tukey R. H., and Guengerich F. P. (1994) Expression of modified human cytochrome P450 1A2 in Escherichia coli: Stabilization, purification, spectral characterization, and catalytic activities of the enzyme. Arch. Biochem. Biophys. 309, 168–177 [DOI] [PubMed] [Google Scholar]
- 12. Bajpai P., Srinivasan S., Ghosh J., Nagy L. D., Wei S., Guengerich F. P., and Avadhani N. G. (2014) Targeting of splice variants of human cytochrome P450 2C8 (CYP2C8) to mitochondria and their role in arachidonic acid metabolism and respiratory dysfunction. J. Biol. Chem. 289, 29614–29630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanna I. H., Kim M.-S., and Guengerich F. P. (2001) Heterologous expression of cytochrome P450 2D6 mutants, electron transfer, and catalysis of bufuralol hydroxylation: the role of aspartate 301 in structural integrity. Arch. Biochem. Biophys. 393, 255–261 [DOI] [PubMed] [Google Scholar]
- 14. Hosea N. A., Miller G. P., and Guengerich F. P. (2000) Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry 39, 5929–5939 [DOI] [PubMed] [Google Scholar]
- 15. Hanna I. H., Teiber J. F., Kokones K. L., and Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 [DOI] [PubMed] [Google Scholar]
- 16. Guengerich F. P. (2005) Reduction of cytochrome b5 by NADPH-cytochrome P450 reductase. Arch. Biochem. Biophys. 440, 204–211 [DOI] [PubMed] [Google Scholar]
- 17. Savas Ü., Wei S., Hsu M. H., Falck J. R., Guengerich F. P., Capdevila J. H., and Johnson E. F. (2016) 20-Hydroxyeicosatetraenoic acid (HETE)-dependent hypertension in human cytochrome P450 (CYP) 4A11 transgenic mice: Normalization of blood pressure by sodium restriction, hydrochlorothiazide, or blockade of the type 1 angiotensin II receptor. J. Biol. Chem. 291, 16904–16919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaclavikova R., Soucek P., Svobodova L., Anzenbacher P., Simek P., Guengerich F. P., and Gut I. (2004) Different in vitro metabolism of paclitaxel and docetaxel in humans, rats, pigs, and minipigs. Drug Metab. Dispos. 32, 666–674 [DOI] [PubMed] [Google Scholar]
- 19. Yu A., Dong H., Lang D., and Haining R. L. (2001) Characterization of dextromethorphan O- and N-demethylation catalyzed by highly purified recombinant human CYP2D6. Drug Metab. Dispos. 29, 1362–1365 [PubMed] [Google Scholar]
- 20. Yamazaki H., Nakano M., Imai Y., Ueng Y. F., Guengerich F. P., and Shimada T. (1996) Roles of cytochrome b5 in the oxidation of testosterone and nifedipine by recombinant cytochrome P450 3A4 and by human liver microsomes. Arch. Biochem. Biophys. 325, 174–182 [DOI] [PubMed] [Google Scholar]
- 21. Shimada T., and Guengerich F. P. (2006) Inhibition of human cytochrome P450 1A1-, 1A2-, and 1B1-mediated activation of procarcinogens to genotoxic metabolites by polycyclic aromatic hydrocarbons. Chem. Res. Toxicol. 19, 288–294 [DOI] [PubMed] [Google Scholar]
- 22. Guengerich F. P. (2014) Analysis and characterization of enzymes and nucleic acids relevant to toxicology, in Hayes' Principles and Methods of Toxicology (Hayes A. W., and Kruger C. L., eds.), 6th Ed., CRC Press-Taylor & Francis, Boca Raton, FL, pp 1905–1964 [Google Scholar]
- 23. Tabb D. L., Fernando C. G., and Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim S. M., Wang Y., Nabavi N., Liu Y., and Correia M. A. (2016) Hepatic cytochromes P450: Structural degrons and barcodes, posttranslational modifications and cellular adapters in the ERAD-endgame. Drug Metab. Rev. 48, 405–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holman J. D., Ma Z. Q., and Tabb D. L. (2012) Identifying proteomic LC-MS/MS data sets with Bumbershoot and IDPicker. Curr. Prot. Bioinform. Chapter 13, Unit 13.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schilling B., Rardin M. J., MacLean B. X., Zawadzka A. M., Frewen B. E., Cusack M. P., Sorensen D. J., Bereman M. S., Jing E., Wu C. C., Verdin E., Kahn C. R., Maccoss M. J., and Gibson B. W. (2012) Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: Application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun R., Fu L., Liu K. K., Tian C. P., Yang Y., Tallman K. A., Porter N. A., Liebler D. C., and Yang J. (2017) Chemoproteomics reveals chemical diversity and dynamics of 4-oxo-2-nonenal modifications in cells. Mol. Cell. Proteomics 16, 815–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patil P. V., and Ballou D. P. (2000) The use of protocatechuate dioxygenase for maintaining anaerobic conditions in biochemical experiments. Anal. Biochem. 286, 187–192 [DOI] [PubMed] [Google Scholar]
- 29. Burleigh B. D. Jr, Foust G. P., and Williams C. H. Jr. (1969) A method for titrating oxygen-sensitive organic redox systems with reducing agents in solution. Anal. Biochem. 27, 536–544 [DOI] [PubMed] [Google Scholar]
- 30. Guengerich F. P., Krauser J. A., and Johnson W. W. (2004) Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry 43, 10775–10788 [DOI] [PubMed] [Google Scholar]
- 31. Jiang X. L., Gonzalez F. J., and Yu A. M. (2011) Drug-metabolizing enzyme, transporter, and nuclear receptor genetically modified mouse models. Drug Metab. Rev. 43, 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seo Y. H., and Carroll K. S. (2011) Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew. Chem., Int. Ed. 50, 1342–1345 [DOI] [PubMed] [Google Scholar]
- 33. Hatahet F., and Ruddock L. W. (2009) Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850 [DOI] [PubMed] [Google Scholar]
- 34. Muñoz-Clares R. A., González-Segura L., Murillo-Melo D. S., and Riveros-Rosas H. (2017) Mechanisms of protection against irreversible oxidation of the catalytic cysteine of ALDH enzymes: Possible role of vicinal cysteines. Chem. Biol. Interact. 276, 52–64 [DOI] [PubMed] [Google Scholar]
- 35. Omura T., and Sato R. (1962) A new cytochrome in liver microsomes. J. Biol. Chem. 237, 1375–1376 [PubMed] [Google Scholar]
- 36. Aguiar M., Masse R., and Gibbs B. F. (2005) Regulation of cytochrome P450 by posttranslational modification. Drug Metab. Rev. 37, 379–404 [DOI] [PubMed] [Google Scholar]
- 37. Peralta D., Bronowska A. K., Morgan B., Dóka É., Van Laer K., Nagy P., Gräter F., and Dick T. P. (2015) A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 11, 156–163 [DOI] [PubMed] [Google Scholar]
- 38. Gupta V., Yang J., Liebler D. C., and Carroll K. S. (2017) Diverse redoxome reactivity profiles of carbon nucleophiles. J. Am. Chem. Soc. 139, 5588–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gupta V., and Carroll K. S. (2016) Profiling the reactivity of cyclic C-nucleophiles towards electrophilic sulfur in cysteine sulfenic acid. Chem. Sci. 7, 400–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gupta V., Paritala H., and Carroll K. S. (2016) Reactivity, selectivity, and stability in sulfenic acid detection: A comparative study of nucleophilic and electrophilic probes. Bioconjug. Chem. 27, 1411–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sevrioukova I. F. (2017) High-level production and properties of the cysteine-depleted cytochrome P450 3A4. Biochemistry 56, 3058–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pundir C. S., Deswal R., and Narwal V. (2018) Quantitative analysis of hydrogen peroxide with special emphasis on biosensors. Bioprocess Biosyst. Eng. 41, 313–329 [DOI] [PubMed] [Google Scholar]
- 43. Nordblom G. D., White R. E., and Coon M. J. (1976) Studies on hydroperoxide-dependent substrate hydroxylation by purified liver microsomal cytochrome P-450. Arch. Biochem. Biophys. 175, 524–533 [DOI] [PubMed] [Google Scholar]
- 44. Joo H., Lin Z., and Arnold F. H. (1999) Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature 399, 670–673 [DOI] [PubMed] [Google Scholar]
- 45. Shoji O., and Watanabe Y. (2014) Peroxygenase reactions catalyzed by cytochromes P450. J. Biol. Inorg. Chem. 19, 529–539 [DOI] [PubMed] [Google Scholar]
- 46. Matthews S., Belcher J. D., Tee K. L., Girvan H. M., McLean K. J., Rigby S. E., Levy C. W., Leys D., Parker D. A., Blankley R. T., and Munro A. W. (2017) Catalytic determinants of alkene production by the cytochrome P450 peroxygenase OleTJE. J. Biol. Chem. 292, 5128–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matsumura H., Wakatabi M., Omi S., Ohtaki A., Nakamura N., Yohda M., and Ohno H. (2008) Modulation of redox potential and alteration in reactivity via the peroxide shunt pathway by mutation of cytochrome P450 around the proximal heme ligand. Biochemistry 47, 4834–4842 [DOI] [PubMed] [Google Scholar]
- 48. Nordblom G. D., and Coon M. J. (1977) Hydrogen peroxide formation and stoichiometry of hydroxylation reactions catalyzed by highly purified liver microsomal cytochrome P-450. Arch. Biochem. Biophys. 180, 343–347 [DOI] [PubMed] [Google Scholar]
- 49. Miseta A., and Csutora P. (2000) Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 17, 1232–1239 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD007913.