

Abstract
Tumor: tumor microenvironment (TME) interactions are critical for tumor progression and the composition and structure of the local extracellular matrix (ECM) are key determinants of tumor metastasis. We recently reported that activation of Wnt/beta-catenin signaling in Ewing sarcoma cells induces widespread transcriptional changes that are associated with acquisition of a metastatic tumor phenotype. Significantly, ECM protein-encoding genes were found to be enriched among Wnt/beta-catenin induced transcripts, leading us to hypothesize that activation of canonical Wnt signaling might induce changes in the Ewing sarcoma secretome. To address this hypothesis, conditioned media from Ewing sarcoma cell lines cultured in the presence or absence of Wnt3a was collected for proteomic analysis. Label-free mass spectrometry was used to identify and quantify differentially secreted proteins. We then used in silico databases to identify only proteins annotated as secreted. Comparison of the secretomes of two Ewing sarcoma cell lines revealed numerous shared proteins, as well as a degree of heterogeneity, in both basal and Wnt-stimulated conditions. Gene set enrichment analysis of secreted proteins revealed that Wnt stimulation reproducibly resulted in increased secretion of proteins involved in ECM organization, ECM receptor interactions, and collagen formation. In particular, Wnt-stimulated Ewing sarcoma cells up-regulated secretion of structural collagens, as well as matricellular proteins, such as the metastasis-associated protein, tenascin C (TNC). Interrogation of published databases confirmed reproducible correlations between Wnt/beta-catenin activation and TNC and COL1A1 expression in patient tumors. In summary, this first study of the Ewing sarcoma secretome reveals that Wnt/beta-catenin activated tumor cells upregulate secretion of ECM proteins. Such Wnt/beta-catenin mediated changes are likely to impact on tumor: TME interactions that contribute to metastatic progression.
The local and metastatic progression of solid tumors is critically dependent on interactions and crosstalk between tumor cells and their local tumor microenvironment (TME)1. Both cellular and noncellular components of the TME can bind and activate surface receptors on tumor cells to impact on cell signaling and cell behavior (1). The proteinaceous extracellular matrix (ECM) is a key player in tumor: TME crosstalk and changes in the composition and structure of the ECM can profoundly alter cell signaling (2). The main source of ECM proteins is secretion from fibroblasts (3), however, ECM secretion does occur in other normal physiological processes, such as secretion of collagens from osteoblasts during bone formation (4). In cancer, aberrant secretion of proteins from nontumor stromal cells, as well as tumor cells themselves, can disturb homeostatic signaling and promote disease progression. Indeed, each of the major hallmarks of cancer (5) is impacted by secreted proteins in the TME - i.e. VEGF and it's role in angiogenesis, MMPs and their role in matrix degradation, and cytokines and their recruitment of immune cells (6).
Ewing sarcoma is an aggressive tumor of bone and soft tissue that has a peak incidence in adolescents and young adults (7). Although much has been learned about the genetic basis of Ewing sarcoma, and the specific role of EWS/ETS fusion genes in tumorigenesis, relatively little is known about the cellular mechanisms that underlie metastasis and even less is known about the contribution of the local TME (8). Previous reports have shown that the bone TME specifically promotes metastatic progression of Ewing sarcoma (9) and that its ability to grow in bone is dependent on the osteolytic phenotype (10). Moreover, a hypoxic microenvironment promotes activation of metastasis-associated gene expression in tumor cells and enhanced metastatic progression in xenograft models (11, 12). In addition, gene expression profiling studies of primary localized tumor specimens demonstrated the important contribution of tumor stroma to relapse and patient survival (13). Thus, crosstalk between the Ewing sarcoma cells and their TME plays a key role in tumor progression. However, the Ewing sarcoma secretome is undefined, and its role in dictating ECM composition and tumor progression is yet to be elucidated.
We previously showed that activation of Wnt/beta-catenin in Ewing sarcoma cells induces transition to a more migratory cellular phenotype and enhances metastatic engraftment (14). In the current study, we have investigated whether activation of canonical Wnt signaling in Ewing sarcoma cells impacts on their secretion of ECM proteins. To address this, we combined mass spectrometry, proteomics, and bioinformatics tools to define the Ewing sarcoma secretome. In addition, we determined if the secretome is impacted by exposure to exogenous Wnt3a, a canonical Wnt ligand abundant in the bone microenvironment (15, 16). Our results demonstrate that the Ewing sarcoma secretome is rich in ECM proteins, as well as IGF binding proteins. Activation of Wnt/beta-catenin modulates the secretion of ECM proteins, altering the local TME. These data provide novel insight into the nature of the Ewing sarcoma secretome and how it is altered by Wnt/beta-catenin signaling.
EXPERIMENTAL PROCEDURES
Cell Lines
Ewing sarcoma cell line TC32 was maintained in RPMI 1640 media (Gibco, Gaithersburg, MD) supplemented with 10% FBS (Atlas Biologicals, Fort Collins, CO) and 2mmol/l-glutamine (Life Technologies, Carlsbad, CA). CHLA10 was maintained in IMDM media (Fisher Scientific, Chicago, IL) supplemented with 20% FBS, 2mmol/l-glutamine, and 1× Insulin-Transferrin-Selenium (Gibco). Both cell lines are late passage cell lines and were obtained from the Children's Oncology Group (COG) cell bank (cogcell.org). Identity of all cell lines was confirmed by STR profiling and absence of mycoplasma was confirmed within 6 months of the experiments performed in this manuscript.
Biological Annotation of the Differentially Expressed Transcripts
To understand the biological functions of the transcripts that were differently expressed in Wnt3a treated samples compared with that of the baseline group, we used the Genomatics software suite. The signal transduction pathway enrichment analyses were done using the Genomatix Pathway System (GePS) tool (www.genomatix.de/) embedded in Genomatix software. GePS uses information extracted from public and proprietary databases to display canonical pathways or to create networks based on literature data. Cellular compartment was determined using DAVID 6.8 (updated October 2016). All genes identified to be up-regulated by Wnt3a±RSPO2 in our previously published work ((14) GSE75859) were compared against the human genome. All cellular compartments shown have FDR < 1%.
Experimental Design and Statistical Rationale
Cells were either treated with vehicle (PBS, Life Technologies) or 100 ng/μl of recombinant Wnt3a (Wnt3a; R&D Systems, Minneapolis, MN). Cells were treated once a day for 3 days in media with full serum before changing to either RPMI or IMDM lacking FBS. Cells were cultured in 15 cm dishes for all experiments and media was collected from a single dish for each biologic replicate. Cells were then treated once a day for two additional days before media was collected. Protein in the media was concentrated using three kilodalton cutoff Amicon Ultra Centrifugal Filter Units (Fisher). Protein concentration was measured using the DC Protein Assay (Bio-Rad, Des Plaines, IL). Two cell lines were used for analysis with each condition. For each cell line, three replicates of control (vehicle treated samples) were compared against three replicates of Wnt3a treated samples. All samples for each cell line were processed at the same time to account for batch effect.
Protein Identification by LC-tandem Mass Spectrometry
Equal amounts of proteins (50 μg) were used for processing. Cysteines were reduced with 10 mm DTT at 55 °C for 30 min and alkylated using 50 mm chloroacetamide (RT, 30 min). Digestion with 500 ng of sequencing grade, modified trypsin (Promega, Fitchburg, WI) was carried out overnight at 37 °C. Reaction was terminated by acidification with trifluoroacetic acid (0.1% v/v) and peptides were purified using SepPak C18 cartridge following manufacturer's protocol (Waters Corp, Milford, MA). An aliquot of the resulting peptides (∼2 ug) were resolved on a nano-capillary reverse phase column (Acclaim PepMap C18, 2 micron, 50 cm, ThermoScientific, San Jose, CA) using 0.1% formic acid/acetonitrile gradient at 300 nl/min (2–25% acetonitrile in 105 min; 25–40% acetonitrile in 20 min followed by a 90% acetonitrile wash for 10 min and a further 30 min re-equilibration with 2% acetonitrile) and directly introduced in to Q Exactive HF mass spectrometer (Thermo Scientific). MS1 scans were acquired at 60K resolution (AGC target = 3e6, max IT = 50ms). Data-dependent high-energy C-trap dissociation MS/MS spectra were acquired for the 20 most abundant ions (Top20) following each MS1 scan (15K resolution; AGC target = 1e5; relative CE ∼28%). Data was analyzed using Proteome Discoverer (v2.1, Thermo Scientific). Proteins were identified by searching the data using Sequest HT against Homo sapiens (Swissprot, v2016–11-30, database includes 20,213 proteins) appended with common contaminants (100 total proteins). Search parameters included MS1 mass tolerance of 10 ppm and fragment tolerance of 0.2 Da; the protease used was trypsin; two missed cleavages were allowed; carbamidimethylation of cysteine was considered fixed modification and oxidation of methionine, deamidation of asparagine and glutamine, phosphorylation of serine, threonine and tyrosine were considered as potential modifications. False discovery rate (FDR) was determined using Percolator and proteins/peptides with a FDR of ≤1% were retained for further analysis.
Identification of Secreted Proteins
In order to gather more information about the nature of the identifications, the predicted proteins were initially mapped to the Human Protein Atlas (HPA) Secretome and Proteome (17). HPA maintains a list of genes and proteins annotated as potentially secreted based on several inputs, including results from 3 bioinformatics applications (SignalP (18), Phobius (19), and SPOCTOPUS (20)) and one large-scale study (17). Proteins found in any of these lists were then classified as secreted and subject to further analysis.
Statistical Analysis and Interpretation of Results
Differential protein expression of proteins identified to be secreted in both vehicle and Wnt3a treated samples were analyzed by QPROT as described previously in (21). QPROT is developed specifically for modeling protein abundances in label free proteomics estimated using either MS1 intensity or the number of MS2 peptide-spectral-matches (PSMs). Proteins were statistically significant if they had fold change score >2 or <.5 and local FDR as computed by QPROT of < 10%. The fold change score was calculated by averaging the total PSMs across all replicates in Wnt plus another factor (in this case we used 1) divided by the average PSMs across all replicates in PBS plus the same factor. Any proteins that had <2 unique peptides, and were only identified in 1 of 6 replicates, were not considered differentially secreted. Gene set enrichment analysis (GSEA) was conducted using the GSEA v2.1.0 software (22). Gene ontology of secreted proteins was performed using DAVID 6.8 with the human genome as the background. Pathway analysis for the baseline secretome and Wnt-activated secretome was done using the Reactome pathway knowledgebase (23).
Quantitative Real-time PCR
Total RNA was extracted from cells at the same time as protein collection using Quick-RNA MicroPrep (Zymo Research, Irvine, CA) and cDNA was generated using iScript (Bio-Rad). Quantitative real-time PCR (qRT-PCR) was performed using universal SYBR-Green Supermix (Bio-Rad) for designed primers. Analysis was performed in triplicate using the Light-Cycler 480 System (Roche Applied Science, Branchburg, NJ) and average Cp values were normalized relative to the geometric mean of the two housekeeping genes GAPDH and HPRT. The following primers were used: LEF1 forward - 5′TGGATCTCTTTCTCCACCCA3′ and reverse - 5′CACTGTAAGTGATGAGGGGG3′, NKD1 forward - 5′TCGCCGGGATAGAAAACTACA3′ and reverse - 5′CAGTTCTGACTTCTGGGCCAC3′, AXIN2 forward - 5′AAGTGCAAACTTTCGCCAAC and reverse - 5′ACAGGATCGCTCCTCTTGAA, HPRT forward - 5′TGACACTGGCAAAACAATGCA3′ and reverse - 5′GGTCCTTTTCACCAGCAAGCT3′, GAPDH forward - 5′TGCACCACCAACTGCTTAGC3′v and reverse - 5′GGCATGGACTGTGGTCATGAG3′, TNC forward - 5′GCAGCTCCACACTCCAGGTA3′ and reverse - 5′TTCAGCAGAATTGGGGATTT3′, and COL1A1 forward - 5′CTGGACCTAAAGGTGCTGCT3′ and reverse - 5′GCTCCAGCCTCTCCATCTTT3′.
Flow Cytometry
Stably transduced 7xTcf-eGFP (7TGP) (14) TC32 and CHLA10 cells were stimulated with Wnt3a (100 ng/ml) once a day for 5 days before collecting cells to measure GFP fluorescence. Fluorescence was measured and quantified on an Accuri C6 cytometer (BD Biosciences, Franklin Lakes, NJ).
Western Blots
Western blot analysis was performed using the Bio-Rad Mini-PROTEAN Tetra System. Following transfer, nitrocellulose membranes were blocked in 5% BSA in TBS-T for 1 h. Membranes were washed once with TBS-T and then incubated overnight at 4 °C with either mouse-anti human Tenascin C (1:500, Sigma T2551, St. Louis, MO) or rabbit-anti Collagen 1 (1:1000, Abcam ab34710, Cambridge, MA). Membranes were then washed three times in 1× TBS-T for 5 min each and then incubated with secondary antibody IRDye 800CW goat anti-mouse (1:10,000, LiCor) or IRDye 800CW goat anti-rabbit (1:10,000, LiCor, Lincoln, NE) for 1 h. They were then washed two times with 1X TBS-T and once with 1X TBS for 5 min before scanning the membrane using the LiCor Imaging system.
Gene Expression in Ewing Sarcoma Patient Tumors
Pearson correlation coefficients of gene expression and associated confidence intervals in primary patient tumors were determined using three previously reported, independent data sets: GSE 63157 (13), GSE 34620 (24), and GSE 17679 (25). Only data from patient tumors were extracted from the GSE17679 data set and the average value for duplicate assays from each of 44 unique patient tumors was used.
RESULTS
Wnt-activated Cells Upregulate Pathways Involved in Tumor: TME Crosstalk
To begin to understand the mechanisms through which up-regulation of the Wnt/beta-catenin signaling axis contribute to metastatic phenotypes and poor outcomes in Ewing sarcoma, we performed pathways analysis on our previously reported RNA-sequencing study of Wnt/beta-catenin activated cells (14). In this previous study, 1157 transcripts were identified as being significantly induced by Wnt activation and 1221 were significantly induced by Wnt activation with potentiation by Rspondin2. Mapping of these transcripts against published signal transduction pathways using the Genomatix Pathway System tool revealed significant enrichment of signaling pathways that are known to play key roles in mediating tumor: TME interactions. Signaling pathways that depend on crosstalk between receptors on tumor cells and proteins in the surrounding ECM were identified, including, integrin linked kinase, focal adhesion kinase, cadherin, angiogenesis, TGF beta and matrix metalloproteinase pathways (supplemental Fig. 1A). Additionally, gene ontology analysis of cellular localization revealed significant enrichment of proteins found in focal adhesions, extracellular exosomes and the ECM (supplemental Fig. 1B). Thus, activation of canonical Wnt signaling in Ewing sarcoma cells leads to a significant increase in the transcription of genes that encode ECM proteins as well as other proteins that play key roles in tumor: TME interaction and crosstalk.
Identification of Secreted Proteins in Ewing Sarcoma Using Label Free Mass Spectrometry
ECM proteins in the local TME of solid tumors are most often secreted by tumor-associated fibroblasts and other nonmalignant stromal cells (1). However, unlike many other solid tumors, nontumor stromal cells are infrequently detected in Ewing sarcoma biopsies (13). This led us to speculate that the composition of the ECM in Ewing sarcoma may be largely determined by proteins that are secreted by the tumor cells themselves. Furthermore, considering our observation that activation of canonical Wnt signaling induces transcription of ECM-encoding genes, we hypothesized that Wnt activation would alter the Ewing sarcoma secretome. To address this hypothesis we used tandem mass spectrometry to define the secretomes of control and Wnt-activated Ewing sarcoma cells. TC32 and CHLA10 cells were chosen for these studies given that both are well-established Ewing sarcoma cell lines that grow as adherent monolayers on uncoated tissue culture plates. They were also found to generate less cell debris when exposed to serum free conditions than other Ewing sarcoma cell lines and to robustly activate TCF-dependent transcription in response to Wnt3a. An outline of the experimental approach is summarized in Fig. 1.
Fig. 1.

Identification of secreted proteins in Ewing sarcoma using label free mass spectrometry. Both TC32 and CHLA10 were treated with either vehicle (PBS) or recombinant human Wnt3a (100 ng/uL) once a day for 5 days before collection of media. Cell debris was monitored over time and only replicate cultures with excellent cell viability and minimal debris in the conditioned culture medium were collected for analysis. Media was collected gently to minimize cell rupture. All conditions were collected in triplicate and cell debris removed by centrifugation. Conditioned media was then concentrated and submitted for Tandem mass spectrometry and subsequent protein identification using Swissprot.
Conditioned media from control and experimental cells was analyzed by mass spectrometry, as described above, and the overall distribution of proteins identified across all conditions was first quantified. Because we used unlabeled mass spectrometry, all quantification was done using spectral counting and peptide-spectra-match (PSM) counts for each protein (26). The total number of proteins identified in the conditioned media was 2336 proteins for TC32 and 857 for CHLA10 cells (supplemental Table S1). One caveat of using mass spectrometry for secretome analysis of conditioned media is that many proteins can be detected that are not considered part of the secretome. These proteins include those that are present in the media because of cell rupture, cell shedding, as debris from dying cells, or simply because of their overwhelming abundance in cultured cells. To address this key issue, and to minimize assignment of nonsecreted proteins to the defined secretome, we gently collected conditioned media from the culture dishes without disrupting the cell monolayers and removed any cellular debris by centrifugation before mass spectrometry analysis. Next, we used four independent computational methods to analyze the mass spectrometry data to further ensure that the data represented secreted proteins. Four different analytic tools have been developed to rigorously assign identified proteins to the secretome: Human Protein Atlas, SignalP4.0, Phobius, and SPOCTOPUS (17–20). These tools predict the likelihood of a detected protein being secreted based on presence of a signal peptide sequence and absence of a transmembrane domain. Following application of these computational tools, 543 of 2336 (23%) proteins were identified as secreted proteins in the conditioned media of TC32 cells. For CHLA10 cells, 259 of the 857 (30%) proteins were classified as secreted. This proportion of secreted proteins in conditioned media is consistent with prior reports and, thus, the reproducible protein data are likely to be highly representative of the Ewing sarcoma secretome (27, 28). Alignment of the secreted proteins to all proteins encoded by the human genome confirmed highly significant enrichment for proteins in the “extracellular region” and “extracellular space” compartments, with FDR of 6.03E-72 and 2.47E-68 respectively, validating their designation as secreted proteins. Finally, to ensure that the designation of proteins to the secretome was not biased in favor of either low- or high- abundance proteins, we compared the distribution of spectral counts from the list of secreted proteins to the list of all proteins identified. A similar range of counts was evident across both the total and the secreted protein lists, confirming that we had identified an unbiased list of proteins that belong to the Ewing sarcoma secretome (supplemental Fig. S2). This computationally-validated list of secreted proteins was used for all further analyses and validation studies (supplemental Table S2).
Defining the Baseline Ewing Sarcoma Secretome
Having defined the identities of secreted proteins in the conditioned media of cultured Ewing sarcoma cells we next sought to define the nature of these proteins in the two different cell lines in basal conditions. To our knowledge, the Ewing sarcoma secretome has not previously been characterized. Under basal conditions, 138 and 85 proteins were identified as being secreted into the media by TC32 and CHLA10 cells, respectively (Fig. 2A and supplemental Table S3). Principal component analysis of the data highlights that the secretome signature differs between the two cell lines, and that reproducibility among biologic replicates is high (Fig. 2B). Interestingly, however, over a third of proteins in the TC32 secretome and two-thirds of proteins in the CHLA10 secretome were shared, indicating that, despite some overall heterogeneity, there is evidence of a common secretome. Specifically, 56 proteins were reproducibly secreted by both Ewing sarcoma cell lines under basal, serum-free conditions (supplemental Table S3). In addition, seven of the 20 most abundantly secreted proteins in each cell line, based on total PSM number, were shared (Fig. 2C). Pathway analysis of the commonly secreted 56 proteins revealed enrichment for proteins involved in IGF transport, extracellular matrix organization, and elastic fiber formation (Fig. 2D). This is especially interesting given that many insulin-like growth factor (IGF) family members are targets of the pathognomonic EWS-FLI1 fusion and that alterations in IGF signaling are characteristic of Ewing sarcoma (29, 30). In addition, fibronectin, a major component of the ECM, was robustly secreted by both cell lines demonstrating that a primary source of fibronectin in the local Ewing sarcoma TME is the tumor cells themselves (31, 32). Thus, from these data we conclude that modulators of IGF pathway signaling and ECM composition and organization are reproducibly and abundantly secreted by Ewing sarcoma tumor cells.
Fig. 2.

The baseline Ewing sarcoma secretome. A, Venn diagram showing the intersection among proteins identified at baseline in TC32 and CHLA10. 56 of the proteins were found in both samples. B, Principal component analysis of three TC32 and three CHLA10 replicates with vehicle treatment only. C, Total PSMs were added among all three replicates for each cell line and then ranked in descending order based on total PSM. Seven of the top 20 most abundant proteins were shared among both cell lines and are displayed here. D, Proteins present at baseline in both TC32 and CHLA10 were subject to pathway analysis using the reactome database. Top 10 pathways are shown here and all pathways have FDR < 1%.
Defining the Wnt-dependent Ewing Sarcoma Secretome
Having defined the basal secretomes of both TC32 and CHLA10 cells we next sought to assess the impact of canonical Wnt-activation. We previously observed that the response of Ewing sarcoma cells to exogenous Wnt3a ligand is highly heterogeneous (14). Thus, we used flow cytometry analysis of TCF/LEF-GFP reporter cells, as previously described (14), to validate the extent of canonical Wnt signaling in Ewing sarcoma cells following the defined five-day exposure to Wnt3a. As shown, approximately one-third of cells in both TC32 and CHLA10 cell lines demonstrated activation of Wnt/beta-catenin-dependent transcription under the experimental conditions (Fig. 3A). In addition, parallel analysis of mRNA from the same cultures confirmed robust and reproducible up-regulation of the previously validated Wnt/beta-catenin target genes, LEF1, NKD1, and AXIN2, in Wnt-activated samples (Fig. 3B).
Fig. 3.

The wnt-dependent Ewing sarcoma secretome. A, Cells were transduced with 7TGP plasmid (plasmid #24305 from Addgene). Cells were treated for 5 days with Wnt3a (100 ng/uL) once a day before collection of cells for flow cytometry analysis. Analysis was run in triplicate and percentage of GFP positive cells are shown. Statistical significance was determined using student's t test and *** = p value <.0001. B, qRT-PCR validation of established Wnt targets in Ewing sarcoma. LEF1, NKD1, and AXIN2 were all up-regulated in all replicates submitted for mass spectrometry. C, 170 proteins were identified to be secreted in TC32, 70 were identified in CHLA10, and 50 were identified in both cell lines. D, Principal component analysis of the Wnt-activated secretomes of each cell line. E, The total PSMs were added together for all three replicates of Wnt3a treatment and ranked in descending order based on total PSM. Of the top 20 proteins in each cell line, 7 were secreted by both cell lines. These proteins are shown here. F, Proteins present in both TC32 and CHLA10 were subject to pathway analysis using the reactome database. Top 10 pathways are shown here and all pathways have FDR < 1%.
Similar to the basal secretomes, the Wnt-activated secretome of TC32 contained more proteins than that of CHLA10. Whereas 170 secreted proteins were identified in the conditioned media of TC32 cells, 70 proteins were identified in the CHLA10 secretome. Principal component analysis again confirmed that the Wnt-activated secretomes differed between the two cell lines (Fig. 3D). Nevertheless, 50 proteins were again shared (supplemental Table S4), including IGFBP2, fibronectin, as well as other proteins that were also abundant in basal conditions (Fig. 3E), Notably, two proteins that are integral to ECM composition and bone remodeling, collagen type 1 alpha 2 chain (COL1A2) (33) and secreted protein acidic and cysteine rich (SPARC) (34), were among the most abundant and reproducibly secreted under conditions of canonical Wnt activation (Fig. 3E). Consistent with basal secretomes, analysis of the 50 shared proteins in Wnt-activated secretomes revealed enrichment for pathways involved in IGF signaling as well as ECM composition, organization, and degradation (Fig. 3F).
Wnt Activation in Ewing Sarcoma Cells Promotes Secretion of Proteins That Alter the Composition and Structure of the ECM
Next, we assessed each cell line independently to determine if secretion of individual proteins changed in response to Wnt/beta-catenin activation. Total PSMs were used to determine differential expression between control and Wnt3a-treated samples. Differentially expressed proteins were defined as proteins that had a local FDR < 10% (computed using QPROT), a fold change score of >2 or <0.5, and detection of at least 2 unique peptides (see methods for further details) (21). Using these very stringent criteria, we identified significant changes in the secretomes of both cell lines following activation of canonical Wnt signaling. Specifically, TC32 cells showed altered secretion of 33 proteins, 27 of which were up-regulated (Fig. 4A). In CHLA10 cells, Wnt/beta-catenin activation led to altered secretion of 16 proteins with 14 of these having increased secretion (Fig. 4B). Shared proteins with Wnt-dependent increases in secretion were Wnt3a, TNC, pro-enkephalin (PENK), and members of the proprotein convertase subtilisin/kexins family (PCSK2 and PCSK9). Given that Wnt3a was added to the media for these experiments, it is important to note that it was identified in both lists, confirming the validity of the analysis and serving as an internal quality control.
Fig. 4.
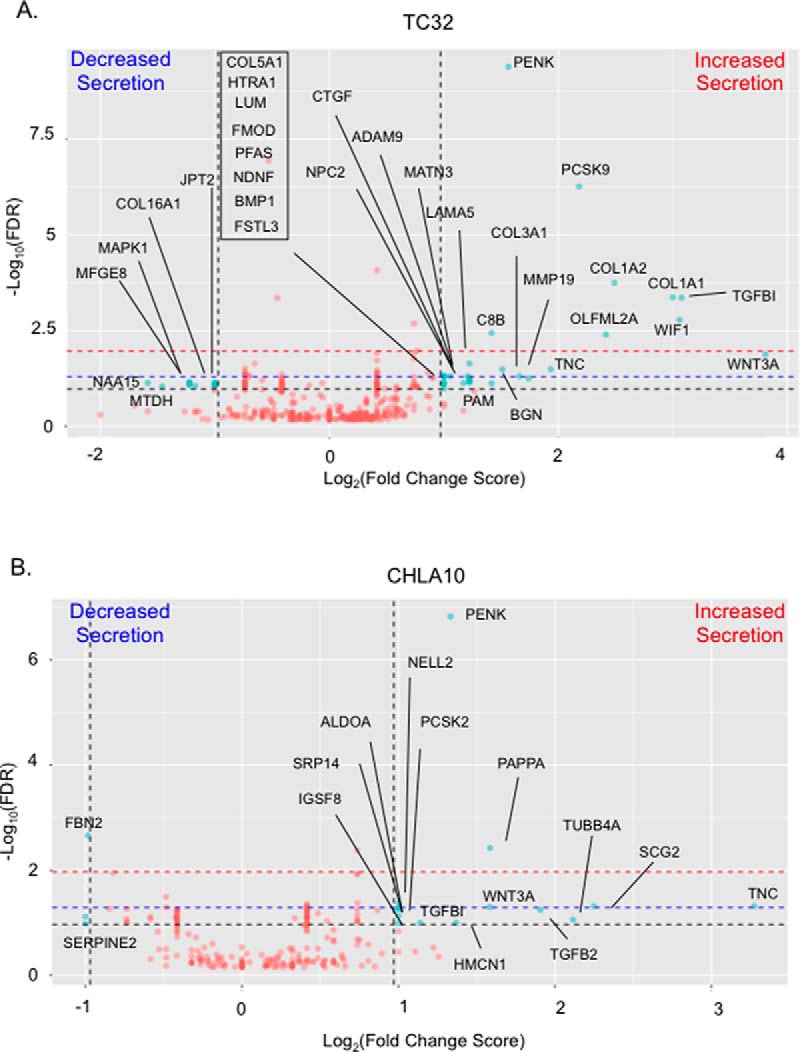
Stimulation with Wnt3a changes the Ewing sarcoma secretome. Volcano plots showing the Log2(Fold Change Score) versus -Log10(localFDR). Red dots indicate proteins that were not differentially secreted and blue dots indicate proteins that were considered to be differentially secreted. Proteins that were considered differentially secreted have Fold Change Score >2 or <.5 and QPROT computed local FDR of differential expression <.1 (indicated by the black dashed lines). We also have noted that FDR <.05 results in 16 differentially secreted proteins in TC32 and 5 in CHLA10 (proteins that are above the blue dashed line). An FDR <.01 results in 8 proteins in TC32 and 2 in CHLA10 to be differentially secreted (proteins that are above the red dashed line).
To better understand the nature of Wnt-dependent secretome, and to remove bias associated with a priori assignment of significance based on fold change, we performed Gene Set Enrichment Analysis (GSEA) on the ranked list of detected proteins and searched against the KEGG and Reactome databases (22). GSEA revealed the Wnt-dependent secretome to be enriched, in both cell lines, for proteins that play key roles in ECM organizational structure and ECM: cell receptor interactions (supplemental Fig. S3). Notably, 5 of 27 proteins with increased secretion in Wnt/beta-catenin activated TC32 cells are structural proteins (COL1A1, COL1A2, COL3A1, COL5A1, and LAMA5), and another 9 have been published to play a role in collagen fibril and extracellular matrix organization (BGN (35), TGFBI (36–38), CTGF (39), MATN3 (40), ADAM9 (41), MMP19 (42), FMOD (33), LUM (33, 43) and TNC (44–46)). Thus, these data show that the secretome of TC32 cells is particularly responsive to canonical Wnt activation, and secretion of structural proteins that contribute to ECM organization and remodeling is significantly increased. Interestingly, the secretome of CHLA10 cells, an inherently highly metastatic cell line (47), contained abundant collagens at baseline and secretion was not further induced by Wnt stimulation (supplemental Table S2).
To validate our findings, we used orthogonal methods to assess the impact of Wnt/beta-catenin activation on expression and secretion of TNC and collagen I. Both ECM proteins are well-established mediators of tumor engraftment and metastasis (46, 48). We previously showed that stimulation of Ewing sarcoma cells with Wnt3a for 48 h leads to a 2-fold increase in expression of TNC mRNA (14). Analysis of cells following the five-day stimulation protocol used for secretome analysis confirmed robust up-regulation of TNC mRNA to greater than 5-fold (Fig. 5A). Expression of the COL1A1 transcript was also induced (Fig. 5B). Next, we sought to validate whether activation of Wnt/beta-catenin in tumors in vivo is also associated with increased expression of these ECM protein-encoding genes. To achieve this we interrogated three independent, publicly available databases (13, 24, 25) and assessed correlations between expression of LEF1 and TNC and COL1A1 in primary patient tumors. We have previously shown that LEF1 is a robust biomarker of Wnt/beta-catenin activation in Ewing sarcoma cells and tumors (14). As shown, expression of TNC was significantly positively correlated with LEF1 in all data sets and COL1A1 correlated with LEF1 in two of the three patient cohorts (Fig. 5C). As a control, we tested correlations of two other ECM protein-encoding genes, FBLN1 and LTBP3, whose protein products were detected in the Ewing sarcoma secretome but were not impacted by Wnt activation (supplemental Table S2). As shown, expression of neither of these genes correlated with LEF1 in any of the three primary Ewing sarcoma data sets (Fig. 5C). Thus, these data support the conclusion that, consistent with their response in vitro, activation of Wnt/beta-catenin in Ewing sarcoma cells in vivo leads to up-regulated expression of the ECM protein-encoding genes TNC and COL1A1.
Fig. 5.

Activation of Wnt/beta-catenin is associated with high expression of TNC and COL1A1 in Ewing sarcoma patients. A, qRT-PCR of TNC expression after 5 days of treatment with Wnt3a. B, qRT-PCR of COL1A1 expression after 5 days of treatment with Wnt3a. C, Confidence intervals for Pearson correlation of LEF1 with TNC, COL1A1, FBLN1, and LTBP3 from microarray analysis of three different patient cohorts, Volchenbaum 2015 (13), Postel Vinay 2012 (24), and Savola 2009 (25). The number of unique patient tumors in each data set is shown (legend on right). D, Validation of increased secretion of TNC by Ewing sarcoma cells post Wnt3a treatment (Western blot of conditioned media). Note that although TNC is expressed by Ewing sarcoma cells, its presence is not robustly detected in the secretome in the absence of Wnt3a stimulation. Statistical significance was determined using student's t test with * = p value < 0.05 and ** = p value <0.001. E, Validation of increased secretion of COL1A1 post Wnt3a treatment in TC32 and high basal levels in CHLA10.
Finally, we sought to directly validate our mass spec findings that TNC and collagen I are secreted by Ewing sarcoma cells. To achieve this, we performed Western blot on concentrated conditioned media collected from independent experiments. As shown, increased secretion of TNC was evident in the conditioned media of both TC32 and CHLA10 cells following Wnt3a stimulation (Fig. 5D). Likewise, an increase in collagen I secretion was detected in the conditioned media of Wnt-stimulated TC32 cells (Fig. 5E). Consistent with our mass spec results, Collagen I was readily detected in the media of Wnt-activated CHLA10 cells but was not increased beyond the high levels that were already secreted under basal conditions (Fig. 5E).
Together these studies confirm that activation of Wnt/beta-catenin signaling in Ewing sarcoma cells leads to increased expression and secretion of ECM proteins that have the potential to impact on tumor progression by altering tumor: TME crosstalk.
DISCUSSION
Secreted proteins play a role in every hallmark of cancer and are critical to tumor progression (6). Our data show that under basal, nonstimulated conditions, Ewing sarcoma cells secrete abundant proteins that play key roles in modulation of IGF signaling and the composition of the local tumor ECM. In addition, we find that, on activation of Wnt/beta-catenin, Ewing sarcoma cells alter their secretomes in a manner that leads to increased secretion of matricellular and structural ECM proteins. We previously reported that activation of Wnt/beta-catenin signaling in Ewing sarcoma cells leads to an increase in expression of TNC, and that this is associated with enhanced engraftment and metastatic potential (14). In the current study we have confirmed that, not only is TNC expression up-regulated, but that its secretion by tumor cells into the local TME is robustly increased. TNC interacts with FN1, another abundant ECM protein detected in our secretome analysis. TNC contains up to 17 FNIII repeats and can bind to the FNIII domain of FN1 through syndecan 4 (49). Thus, the increase in TNC secretion is expected to alter the physical interaction between TNC and FN1, resulting in altered TME structure. TNC has previously been reported to play a role in establishing the pre-metastatic niche in breast cancer, in addition to being highly expressed in the secretome of aggressive colorectal cancers (50, 51). Thus, these studies lend evidence to support the hypothesis that Wnt/beta-catenin dependent activation of TNC contributes to the metastatic phenotype of Ewing sarcoma by altering the local ECM.
Despite intertumor heterogeneity, our studies of two different cell lines revealed substantial overlap in secreted proteins. We particularly noted the presence of members the IGF pathway. The IGF1-receptor is necessary for EWS-FLI1-induced malignant transformation and activation of IGF signaling contributes to tumor initiation and progression (29, 30, 52). We detected abundant secretion of IGF family members, specifically IGFBPs, in the Ewing sarcoma secretome, and the overall level of secretion did not change under conditions of Wnt/beta-catenin activation. Interestingly, however, Wnt/beta-catenin activation did lead to a reproducible increase in secretion of the IGFBP protease, pappalysin-1 (PAPPA) (Fold change scores 2.2 and 3.0 for TC32 and CHLA10 cells, respectively). PAPPA cleaves IGFBPs, thereby releasing IGF ligands, and promoting activation of the IGF signaling axis (53). Thus, a Wnt-dependent increase in PAPPA secretion could increase in the availability of IGF ligands in the local TME and activate IGF signaling in tumor cells. Despite its tremendous potential as a therapeutic target, and clear evidence of efficacy in preclinical models, the efficacy of IGF1-R targeted therapies in clinical trials has been mixed with response rates ranging from 10–14% in three independent clinical trials (54–56). To date, it is not known why some patients are so responsive to anti-IGF1-R strategies, nor how to identify them (57). Intriguingly, in a phase II study of 115 patients with relapsed or refractory Ewing sarcoma, 10 of 11 responses occurred in patients with bone tumors, suggesting that there may be something about bone tumor microenvironment that sensitizes tumors to IGF1 pathway targeted therapy (56). Given the high level of canonical Wnt ligands that are present in the bone, we speculate that activation of Wnt/beta-catenin in bone Ewing sarcoma cells may alter their secretome, resulting in an altered TME, promotion of IGF1 pathway activation, and enhanced sensitivity to anti-IGF approaches.
In addition to IGFBPs, FN1 and other proteins involved in elastic fiber formation were abundantly secreted by Ewing sarcoma cells. This is consistent with earlier reports that Ewing sarcoma cells synthesize fibronectin, collagens, and laminin (31, 32). Notably, in the Wnt-activated secretomes, we detected further abundance of proteins that contribute to the structure and composition of the ECM. In particular, secretion of collagens as well as other glycoproteins involved in mediating collagen fibril formation (i.e. TNC, BGN, TGFBI, LUM, MATN3, ADAM9, MMP19, and FMOD) was significantly increased in TC32 cells, a cell line in which canonical Wnt activation results in enhanced metastatic engraftment (14). Changes in the structural and biochemical properties of the ECM play fundamental roles in cancer progression (2). Not only does changing the biochemical properties lead to aberrant alterations in classical signaling pathways, such as TGFβ and Wnt signaling (58, 59), but changes in synthesis, deposition, and degradation of ECM components, including collagen I, III, and V, are able to further promote tumor progression (60). Hayashi et al., recently reported that inhibition of canonical Wnt signaling in Ewing sarcoma cells resulted in an increase in metastasis-free survival in orthotopic bone tumor models and this was associated with decreased expression of COL1A2 and COL3A1 (61). Moreover, in the current study, COL1A1, COL1A2, COL3A1, and COL5A1 were abundant in CHLA10 secretomes, under both basal and Wnt-activated conditions. CHLA10 is an inherently metastatic cell line (47), irrespective of Wnt-activation, leading us to speculate that secretion of collagens by Ewing sarcoma cells may be a key determinant of their metastatic potential. Indeed, the propensity of undifferentiated soft tissue sarcomas to metastasize is, in part, dependent on modification and organization of collagens in the local TME (62). Further experiments are now needed to understand the contribution of altered collagen secretion and organization to metastatic progression of Ewing sarcoma.
In summary, through an unbiased proteomics approach we have defined the Ewing sarcoma secretome, under both basal and Wnt/beta-catenin activated conditions. Our findings demonstrate that Ewing sarcoma cells secrete numerous proteins that modulate IGF signaling as well as ECM composition and structure, and that activation of the canonical Wnt pathway leads to increased secretion of ECM matricellular and structural components. These findings support the conclusion that activation of canonical Wnt signaling in Ewing sarcoma cells leads to changes in the local TME that have the potential to impact on tumor crosstalk and metastatic progression.
DATA AVAILABILITY
Data are available via ProteomeXchange - https://www.ebi.ac.uk/pride/archive/ with identifier: PXD007909.
Supplementary Material
Acknowledgments
We would like to thank members of the Lawlor lab and Nesvizhskii lab for helpful discussions.
Footnotes
This work was supported by the following grants from the NIH/NCI: SARC Sarcoma SPORE U54 CA168512, R01GM094231, and U24CA210967, U-M Cancer Center support grant P30CA046592, Cancer Biology T32 CA 009676, Advanced Proteome Informatics of Cancer T32 CA140044; and with the support of the Russell G. Adderley endowment from the Department of Pediatrics.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- TME
- tumor microenvironment
- BGN
- biglycan
- COL1A1
- collagen type 1 alpha 1 chain
- COL1A2
- collagen type 1 alpha 2 chain
- COL3A1
- collagen type 3 alpha 1 chain
- COL5A1
- collagen type 5 alpha 1 chain
- ECM
- extracellular matrix
- FMOD
- fibromodulin
- FN1
- Fibronectin
- IGF-1
- Insulin like growth factor 1
- IGFBP2
- Insulin like growth factor binding protein 2
- IGFBP3
- Insulin like growth factor binding protein 3
- LAMA5
- laminin subunit alpha 5
- LEF1
- Lymphoid enhancer factor 1
- LUM
- lumican
- MMP
- Matrix Metalloprotease
- PCSK
- proprotein convertase subtilisin/kexin
- PENK
- proenkephalin
- PSM
- peptide-spectra-match
- TCF/LEF
- T-cell factor/Lymphoid enhancer factor
- TGFBI
- transforming growth factor beta induced protein
- TGF
- transforming growth factor
- TNC
- Tenascin C
- VEGF
- Vascular Endothelial Growth Factor.
REFERENCES
- 1. Quail D. F., and Joyce J. A. (2013) Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu P., Weaver V. M., and Werb Z. (2012) The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turoverova L. V., Khotin M. G., Yudintseva N. M., Magnusson K. E., Blinova M. I., Pinaev G. P., and Tentler D. G. (2009) Analysis of extracellular matrix proteins produced by cultured cells. Cell Tissue Biol. 3, 497–502 [PubMed] [Google Scholar]
- 4. Jensen E. D., Gopalakrishnan R., and Westendorf J. J. (2010) Regulation of gene expression in osteoblasts. Biofactors 36, 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 6. Paltridge J. L., Belle L., and Khew-Goodall Y. (2013) The secretome in cancer progression. Biochim. Biophys. Acta 1834, 2233–2241 [DOI] [PubMed] [Google Scholar]
- 7. Balamuth N. J., and Womer R. B. (2010) Ewing's sarcoma. Lancet Oncol. 11, 184–192 [DOI] [PubMed] [Google Scholar]
- 8. Lawlor E. R., and Sorensen P. H. (2015) Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Critical Rev. Oncogenesis 20, 155–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldstein S. D., Hayashi M., Albert C. M., Jackson K. W., and Loeb D. M. (2015) An orthotopic xenograft model with survival hindlimb amputation allows investigation of the effect of tumor microenvironment on sarcoma metastasis. Clin. Exp. Metastasis 32, 703–715 [DOI] [PubMed] [Google Scholar]
- 10. Picarda G., Matous E., Amiaud J., Charrier C., Lamoureux F., Heymann M. F., Tirode F., Pitard B., Trichet V., Heymann D., and Redini F. (2013) Osteoprotegerin inhibits bone resorption and prevents tumor development in a xenogenic model of Ewing's sarcoma by inhibiting RANKL. J. Bone Oncol. 2, 95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. El-Naggar A. M., Veinotte C. J., Cheng H., Grunewald T. G., Negri G. L., Somasekharan S. P., Corkery D. P., Tirode F., Mathers J., Khan D., Kyle A. H., Baker J. H., LePard N. E., McKinney S., Hajee S., Bosiljcic M., Leprivier G., Tognon C. E., Minchinton A. I., Bennewith K. L., Delattre O., Wang Y., Dellaire G., Berman J. N., and Sorensen P. H. (2015) Translational activation of HIF1alpha by YB-1 promotes sarcoma metastasis. Cancer Cell 27, 682–697 [DOI] [PubMed] [Google Scholar]
- 12. Krook M. A., Nicholls L. A., Scannell C. A., Chugh R., Thomas D. G., and Lawlor E. R. (2014) Stress-induced CXCR4 promotes migration and invasion of ewing sarcoma. Mol. Cancer Res. 12, 953–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Volchenboum S. L., Andrade J., Huang L., Barkauskas D. A., Krailo M., Womer R. B., Ranft A., Potratz J., Dirksen U., Triche T. J., and Lawlor E. R. (2015) Gene Expression Profiling of Ewing Sarcoma Tumors Reveals the Prognostic Importance of Tumor-Stromal Interactions: A Report from the Children's Oncology Group. J. Pathol. Clin. Res. 1, 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pedersen E. A., Menon R., Bailey K. M., Thomas D. G., Van Noord R. A., Tran J., Wang H., Qu P. P., Hoering A., Fearon E. R., Chugh R., and Lawlor E. R. (2016) Activation of Wnt/beta-Catenin in Ewing Sarcoma Cells Antagonizes EWS/ETS Function and Promotes Phenotypic Transition to More Metastatic Cell States. Cancer Res. 76, 5040–5053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parr B. A., Shea M. J., Vassileva G., and McMahon A. P. (1993) Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 119, 247–261 [DOI] [PubMed] [Google Scholar]
- 16. Day T. F., Guo X., Garrett-Beal L., and Yang Y. (2005) Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 [DOI] [PubMed] [Google Scholar]
- 17. Uhlen M., Fagerberg L., Hallstrom B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., and Ponten F. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 18. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 19. Kall L., Krogh A., and Sonnhammer E. L. (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 20. Viklund H., Bernsel A., Skwark M., and Elofsson A. (2008) SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics 24, 2928–2929 [DOI] [PubMed] [Google Scholar]
- 21. Choi H., Kim S., Fermin D., Tsou C. C., and Nesvizhskii A. I. (2015) QPROT: Statistical method for testing differential expression using protein-level intensity data in label-free quantitative proteomics. J. Proteomics 129, 121–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., McKay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., and D'Eustachio P. (2016) The reactome pathway Knowledgebase. Nucleic Acids Res. 44, D481–D487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Postel-Vinay S., Veron A. S., Tirode F., Pierron G., Reynaud S., Kovar H., Oberlin O., Lapouble E., Ballet S., Lucchesi C., Kontny U., Gonzalez-Neira A., Picci P., Alonso J., Patino-Garcia A., de Paillerets B. B., Laud K., Dina C., Froguel P., Clavel-Chapelon F., Doz F., Michon J., Chanock S. J., Thomas G., Cox D. G., and Delattre O. (2012) Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat. Genet. 44, 323–327 [DOI] [PubMed] [Google Scholar]
- 25. Savola S., Klami A., Myllykangas S., Manara C., Scotlandi K., Picci P., Knuutila S., and Vakkila J. (2011) High expression of complement component 5 (C5) at tumor site associates with superior survival in Ewing's sarcoma family of tumour patients. ISRN Oncol. 2011, 168712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nahnsen S., Bielow C., Reinert K., and Kohlbacher O. (2013) Tools for label-free peptide quantification. Mol. Cell. Proteomics 12, 549–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Blanco M. A., LeRoy G., Khan Z., Aleckovic M., Zee B. M., Garcia B. A., and Kang Y. (2012) Global secretome analysis identifies novel mediators of bone metastasis. Cell Res. 22, 1339–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deshmukh A. S., Cox J., Jensen L. J., Meissner F., and Mann M. (2015) Secretome Analysis of Lipid-Induced Insulin Resistance in Skeletal Muscle Cells by a Combined Experimental and Bioinformatics Workflow. J. Proteome Res. 14, 4885–4895 [DOI] [PubMed] [Google Scholar]
- 29. McKinsey E. L., Parrish J. K., Irwin A. E., Niemeyer B. F., Kern H. B., Birks D. K., and Jedlicka P. (2011) A novel oncogenic mechanism in Ewing sarcoma involving IGF pathway targeting by EWS/Fli1-regulated microRNAs. Oncogene 30, 4910–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prieur A., Tirode F., Cohen P., and Delattre O. (2004) EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol. Cell. Biol. 24, 7275–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scarpa S., D'Orazi G., Modesti M., and Modesti A. (1987) Ewing's sarcoma lines synthesize laminin and fibronectin. Virchows Arch. A Pathol. Anat. Histopathol. 410, 375–381 [DOI] [PubMed] [Google Scholar]
- 32. Scarpa S., Modesti A., and Triche T. J. (1987) Extracellular matrix synthesis by undifferentiated childhood tumor cell lines. Am. J. Pathol. 129, 74–85 [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang G., Young B. B., Ezura Y., Favata M., Soslowsky L. J., Chakravarti S., and Birk D. E. (2005) Development of tendon structure and function: regulation of collagen fibrillogenesis. J. Musculoskelet Neuronal Interact. 5, 5–21 [PubMed] [Google Scholar]
- 34. Brekken R. A., and Sage E. H. (2001) SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 19, 816–827 [DOI] [PubMed] [Google Scholar]
- 35. Zhang G., Chen S., Goldoni S., Calder B. W., Simpson H. C., Owens R. T., McQuillan D. J., Young M. F., Iozzo R. V., and Birk D. E. (2009) Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J. Biol. Chem. 284, 8888–8897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nacu N., Luzina I. G., Highsmith K., Lockatell V., Pochetuhen K., Cooper Z. A., Gillmeister M. P., Todd N. W., and Atamas S. P. (2008) Macrophages Produce TGF-beta-Induced (-ig-h3) following Ingestion of Apoptotic Cells and Regulate MMP14 Levels and Collagen Turnover in Fibroblasts. J. Immunol. 180, 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hashimoto K., Noshiro M., Ohno S., Kawamoto T., Satakeda H., Akagawa Y., Nakashima K., Okimura A., Ishida H., Okamoto T., Pan H., Shen M., Yan W., and Kato Y. (1997) Characterization of a cartilage-derived 66-kDa protein (RGD-CAP/beta ig-h3) that binds to collagen. Biochim. Biophys. Acta 1355, 303–314 [DOI] [PubMed] [Google Scholar]
- 38. Tumbarello D. A., Andrews M. R., and Brenton J. D. (2016) SPARC Regulates Transforming Growth Factor Beta Induced (TGFBI) Extracellular Matrix Deposition and Paclitaxel Response in Ovarian Cancer Cells. PLoS ONE 11, e0162698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hall-Glenn F., Aivazi A., Akopyan L., Ong J. R., Baxter R. R., Benya P. D., Goldschmeding R., van Nieuwenhoven F. A., Hunziker E. B., and Lyons K. M. (2013) CCN2/CTGF is required for matrix organization and to protect growth plate chondrocytes from cellular stress. J. Cell Commun. Signal. 7, 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klatt A. R., Becker A. K., Neacsu C. D., Paulsson M., and Wagener R. (2011) The matrilins: modulators of extracellular matrix assembly. Int. J. Biochem. Cell Biol. 43, 320–330 [DOI] [PubMed] [Google Scholar]
- 41. White J. M. (2003) ADAMs: modulators of cell–cell and cell–matrix interactions. Curr. Opinion Cell Biol. 15, 598–606 [DOI] [PubMed] [Google Scholar]
- 42. Jara P., Calyeca J., Romero Y., Placido L., Yu G., Kaminski N., Maldonado V., Cisneros J., Selman M., and Pardo A. (2015) Matrix metalloproteinase (MMP)-19-deficient fibroblasts display a profibrotic phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L511–L522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chakravarti S., Zhang G., Chervoneva I., Roberts L., and Birk D. E. (2006) Collagen fibril assembly during postnatal development and dysfunctional regulation in the lumican-deficient murine cornea. Dev. Dyn. 235, 2493–2506 [DOI] [PubMed] [Google Scholar]
- 44. Faissner A., Kruse J., Kuhn K., and Schachner M. (1990) Binding of the J1 adhesion molecules to extracellular matrix constituents. J. Neurochem. 54, 1004–1015 [DOI] [PubMed] [Google Scholar]
- 45. Kusubata M., Hirota A., Ebihara T., Kuwaba K., Matsubara Y., Sasaki T., Kusakabe M., Tsukada T., Irie S., and Koyama Y. (1999) Spatiotemporal changes of fibronectin, tenascin-C, fibulin-1, and fibulin-2 in the skin during the development of chronic contact dermatitis. J. Invest. Dermatol. 113, 906–912 [DOI] [PubMed] [Google Scholar]
- 46. Midwood K. S., and Orend G. (2009) The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 3, 287–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mendoza-Naranjo A., El-Naggar A., Wai D. H., Mistry P., Lazic N., Ayala F. R., da Cunha I. W., Rodriguez-Viciana P., Cheng H., Tavares Guerreiro Fregnani J. H., Reynolds P., Arceci R. J., Nicholson A., Triche T. J., Soares F. A., Flanagan A. M., Wang Y. Z., Strauss S. J., and Sorensen P. H. (2013) ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol. Med. 5, 1087–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Provenzano P. P., Eliceiri K. W., Campbell J. M., Inman D. R., White J. G., and Keely P. J. (2006) Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang W., Chiquet-Ehrismann R., Moyano J. V., Garcia-Pardo A., and Orend G. (2001) Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 61, 8586–8594 [PubMed] [Google Scholar]
- 50. Oskarsson T., Acharyya S., Zhang X. H., Vanharanta S., Tavazoie S. F., Morris P. G., Downey R. J., Manova-Todorova K., Brogi E., and Massague J. (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li M., Peng F., Li G., Fu Y., Huang Y., Chen Z., and Chen Y. (2016) Proteomic analysis of stromal proteins in different stages of colorectal cancer establishes Tenascin-C as a stromal biomarker for colorectal cancer metastasis. Oncotarget 7, 37226–37237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Toretsky J. A., Kalebic T., Blakesley V., LeRoith D., and Helman L. J. (1997) The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 272, 30822–30827 [DOI] [PubMed] [Google Scholar]
- 53. Conover C. A., Oxvig C., Overgaard M. T., Christiansen M., and Giudice L. C. (1999) Evidence that the insulin-like growth factor binding protein-4 protease in human ovarian follicular fluid is pregnancy associated plasma protein-A. J. Clin. Endocrinol. Metab. 84, 4742–4745 [DOI] [PubMed] [Google Scholar]
- 54. Juergens H., Daw N. C., Geoerger B., Ferrari S., Villarroel M., Aerts I., Whelan J., Dirksen U., Hixon M. L., Yin D., Wang T., Green S., Paccagnella L., and Gualberto A. (2011) Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 29, 4534–4540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Malempati S., Weigel B., Ingle A. M., Ahern C. H., Carroll J. M., Roberts C. T., Reid J. M., Schmechel S., Voss S. D., Cho S. Y., Chen H. X., Krailo M. D., Adamson P. C., and Blaney S. M. (2012) Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children's Oncology Group. J. Clin. Oncol. 30, 256–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pappo A. S., Patel S. R., Crowley J., Reinke D. K., Kuenkele K. P., Chawla S. P., Toner G. C., Maki R. G., Meyers P. A., Chugh R., Ganjoo K. N., Schuetze S. M., Juergens H., Leahy M. G., Geoerger B., Benjamin R. S., Helman L. J., and Baker L. H. (2011) R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J. Clin. Oncol. 29, 4541–4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arnaldez F. I., and Helman L. J. (2014) New strategies in ewing sarcoma: lost in translation? Clin. Cancer Res. 20, 3050–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hynes R. O. (2009) The Extracellular Matrix: Not Just Pretty Fibrils. Science 326, 1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maeda T., Sakabe T., Sunaga A., Sakai K., Rivera A. L., Keene D. R., Sasaki T., Stavnezer E., Iannotti J., Schweitzer R., Ilic D., Baskaran H., and Sakai T. (2011) Conversion of mechanical force into TGF-beta-mediated biochemical signals. Curr. Biol. 21, 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kauppila S., Stenback F., Risteli J., Jukkola A., and Risteli L. (1998) Aberrant type I and type III collagen gene expression in human breast cancer in vivo. J. Pathol. 186, 262–268 [DOI] [PubMed] [Google Scholar]
- 61. Hayashi M., Baker A., Goldstein S. D., Albert C. M., Jackson K. W., McCarty G., Kahlert U. D., and Loeb D. M. (2017) Inhibition of porcupine prolongs metastasis free survival in a mouse xenograft model of Ewing sarcoma. Oncotarget. 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eisinger-Mathason T. S., Zhang M., Qiu Q., Skuli N., Nakazawa M. S., Karakasheva T., Mucaj V., Shay J. E., Stangenberg L., Sadri N., Pure E., Yoon S. S., Kirsch D. G., and Simon M. C. (2013) Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 3, 1190–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available via ProteomeXchange - https://www.ebi.ac.uk/pride/archive/ with identifier: PXD007909.