

Abstract
The strictly anaerobic bacterium C. difficile has become one of the most problematic hospital acquired pathogens and a major burden for health care systems. Although antibiotics work effectively in most C. difficile infections (CDIs), their detrimental effect on the intestinal microbiome paves the way for recurrent episodes of CDI. To develop alternative, non-antibiotics-based treatment strategies, deeper knowledge on the physiology of C. difficile, stress adaptation mechanisms and regulation of virulence factors is mandatory. The focus of this work was to tackle the thiol proteome of C. difficile and its stress-induced alterations, because recent research has reported that the amino acid cysteine plays a central role in the metabolism of this pathogen. We have developed a novel cysteine labeling approach to determine the redox state of protein thiols on a global scale. Applicability of this technique was demonstrated by inducing disulfide stress using the chemical diamide. The method can be transferred to any kind of redox challenge and was applied in this work to assess the effect of bile acids on the thiol proteome of C. difficile. We present redox-quantification for more than 1,500 thiol peptides and discuss the general difficulty of redox analyses of peptides possessing more than a single cysteine residue. The presented method will be especially useful not only when determining redox status, but also for providing information on protein quantity. Additionally, our comprehensive data set reveals protein cysteine sites particularly susceptible to oxidation and builds a groundwork for redox proteomics studies in C. difficile.
Clostridium difficile, the cause of pseudomembranous colitis, was identified as a human intestinal pathogen almost 30 years ago (1). However, only during the last decade more and more attention has been paid to the anaerobic bacterium because it has become one of the most problematic pathogens in hospitals by means of incidence and costs for the health care system (2). Although antibiotics are still effective in fighting most C. difficile infections (CDIs)1, resistant spores of the pathogen will persist in antibiotic treatment and germinate when the concentration of antibiotics decreases leading to very frequent relapses (3). During infection, intestinal pathogens must deal with several adverse conditions - such as changing osmolarity, changing pH and high concentrations of bile acids. Alterations in gene expression of C. difficile as a response to environmental stress have been investigated in several proteomics and transcriptomics studies (4–7). However, the specific inspection of cysteine-containing proteins in C. difficile and how their redox status might change upon stress has not yet been reported. The amino acid cysteine adopts many important cellular functions. It can contribute to the stabilization of the 3-D structure of proteins by forming disulfide bridges or by complexing with metal ions. It can represent the active site of enzymes or function as a molecular switch undergoing cycles of oxidation and reduction (8). The cost of this versatility is its vulnerability to oxidative damage. Concerning the strict anaerobic life style of C. difficile, low concentrations of oxygen, as they occur in the intestines, already represent a challenge for the bacterium's redox balance. But also, other stress factors can affect the redox status of protein thiols. Cremers et al. for instance, reported that bile acids instigate disulfide stress in Escherichia coli (9). Determination of the redox status of cysteine residues is not a trivial task. Because of its high reactivity, the thiol group of cysteines can change its oxidation state during sample preparation. It can undergo (1) reversible oxidation, e.g. to sulfenic acid or to a disulfide bridge with another thiol group, or it can be (2) irreversibly oxidized, e.g. to sulfinic or sulfonic acid. Several protocols have been published on how to define the oxidized and reduced proportion of a cysteine thiol. Leichert et al. introduced a protocol in which cysteine thiols are alkylated during cell lysis and thereby protected from further modification. Subsequently, reversibly oxidized thiols are reduced and radioactively labeled in a second alkylation step, and proteins are eventually separated on 2D-gels that are exposed to phosphorimaging screens to cull the oxidation state of cysteine-containing proteins (10). A similar approach was followed by Hochgräfe et al. employing a fluorescent labeling of cysteines but also relying on a protein separation on 2D-gels (11). The differential cysteine labeling procedure was further evolved in a technique called OxICAT which again follows the principle of stepwise alkylation, but makes use of light and heavy ICAT (isotope coded affinity tag) reagents (12). The labeled cysteine-containing peptides are enriched and analyzed by LC-MS/MS with a quantification of reduced versus oxidized peptides on MS-level. The biggest advantage of this gel-free method is certainly the determination of the exact site of cysteine oxidation in the protein, whereas resolution of the gel-based approach was limited to protein level. Only two different ICAT reagents are available, limiting experimental design to duplex analyses. Introduction of the TMT methodology for the labeling of cysteine thiols allowed for a six-channel analyses. The six isobaric TMT reagents do not increase sample complexity on MS level, because quantification is achieved on MS/MS level after fragmentation of the tags and allow for comprehensive thiol proteome studies (13, 14). Here we will focus on our novel method, however, for a comprehensive read on the plethora of methods in cysteine redox proteomics, we would like to refer to two excellent reviews (15, 16).
This work is the first to specifically target the thiol proteome of C. difficile. We employed a novel model of the broadly applied differential cysteine labeling approach, which we named diaCys for differential isotopic alkylation of cysteines. Our data not only provides a global picture of the redox status of protein thiols under control conditions, but also pinpoints cysteine residues that are susceptible to oxidation after shock with the disulfide instigator diamide, and evaluates the potential of bile acids to induce disulfide stress in C. difficile.
EXPERIMENTAL PROCEDURES
Strains, Media, and Growth Conditions
C. difficile 630Δerm (17) was inoculated to an A600 of 0.05 and grown anaerobically at 37 °C in brain heart infusion medium (BHI, Oxoid, Hampshire, UK). At an A600 of 0.4 the culture was split into three subcultures. One was shocked with sublethal concentrations of diamide (2 mm), the second with 0.03% of a mixture of the sodium salts of the four major human bile acids (18), [44% cholate (Sigma-Aldrich, Taufkirchen, Germany), 34% chenodeoxycholate (Sigma-Aldrich), 20% deoxycholate (Sigma-Aldrich), 2% lithocholate (Steraloids Inc., Newport, RI, USA)] and the third subculture was left untreated (control). After 15 min, 5 ml of each culture were harvested anaerobically on ice, cells were pelleted by centrifugation and washed twice in ice-cold, oxygen-free PBS (pH 7.2) buffer. For the iodoTMT approach 1.1 mL of the control and diamide shock samples were harvested and cells pellets were washed three times with oxygen-free PBS buffer.
Differential Cysteine Labeling (diaCys) and MS Sample Preparation
For the diaCys protocol, cell pellets were resuspended in 600 μL lysis buffer (8 m urea, 10 mm EDTA, 1% CHAPS, 200 mm TrisHCl pH 8.0) containing 50 mm IAM (Sigma-Aldrich). The third of three independent replicates of each condition (control, diamide, bile) was dissolved in 600 μL lysis buffer containing heavy IAM (Sigma-Aldrich), (label switch). Cells were lyzed by ultrasonication with permanent cooling (Bandelin, Berlin, Germany, Sonopuls HD 3200 with MS73, 6 cycles of 1 min 60% amplitude) and left for 30 min at 30 °C in the dark. Cell lysates were centrifuged (5 min, 20,000 × g, 4 °C) to remove cell debris and mixed with 4 volumes of ice-cold acetone. Proteins were precipitated overnight at −20 °C and pelleted the next morning by centrifugation (20,000 × g, 45 min, at room temperature). Pellets were washed twice with 80% (v/v) acetone and once with pure acetone. Air-dried protein pellets were each solubilized in 100 μL lysis buffer provided with 5 mm TCEP and 50 mm heavy IAM (replicates 1 and 2) or 50 mm light IAM (replicate 3). Reduction and alkylation proceeded for 30 min at 30 °C in the dark before proteins were acetone precipitated once more. Dry protein pellets were solubilized in 50 μL of buffer (8 m urea, 10 mm EDTA, 50 mm TrisHCl pH 8.0) and protein concentration determined via Bradford (Roti-NanoQuant, Carl Roth, Karlsruhe, Germany). Thirty micrograms of protein were separated via SDS-PAGE (Criterion™ TGX™ 4–20% Precast Midi Gel, Bio-Rad, Munich, Germany) and Coomassie-stained. Gel lanes were cut into 10 slices, proteins trypsinized in-gel, peptides eluted and desalted as described previously (19).
iodoTMT Labeling and MS Sample Preparation
Three independent replicates of control and diamide shock samples were dissolved in 600 μL UHE (8 m urea, 20 mm HEPES, 10 mm EDTA, pH 8.0) containing the content of one vial of iodoTMT (Thermo Fisher Scientific, Waltham, Massachusetts), (1st alkylation of cysteines). Cells were lysed via ultrasonication and incubated for 1 h at 30 °C in the dark. Samples were centrifuged (10 min, 20,000 × g, RT) and soluble proteins were acetone precipitated overnight. Protein pellets were resuspended in 200 μL UHT buffer (8 m urea, 20 mm HEPES, 5 mm TCEP, pH 8.0) and protein concentration was determined. 200 μg of each sample was provided with the content of one vial of iodoTMT (second alkylation of cysteines) and incubated at 30 °C in the dark for 1 h. IodoTMT tags were distributed on to the samples as follows (Table I):
Table I.
| Sample | 1st alkylation | 2nd alkylation |
|---|---|---|
| control 1 | 126 | 127 |
| control 2 | 127 | 128 |
| control 3 | 128 | 129 |
| diamide 1 | 129 | 126 |
| diamide 2 | 130 | 131 |
| diamide 3 | 131 | 130 |
Samples “control 1,” “diamide 2,” and “control 3” were combined to give one sample, so were samples “control 2,” “diamide 1,” and “diamide 3,” and proteins of each of the two resulting mixtures were precipitated with acetone overnight. Protein pellets were resuspended in 1 ml of 50 mm ammonium bicarbonate buffer each and trypsin was added for protein digestion (protein/trypsin ratio 100:1). After 2 h at 37 °C in a thermo mixer (800 rpm) the same amount of trypsin was added a second time to a final protein/trypsin ratio of 50:1. Trypsinization proceeded overnight and stopped the next morning by the addition of trifluoric acid (final 1%). Samples were frozen at −80 °C and freeze-dried. Dry pellets were dissolved in 600 μL TBS (50 mm Tris, 150 mm NaCl, pH 7.6). 600 μL of Immobilized anti-TMT antibody resin (ThermoFisher Scientific) was washed three times with the same volume of TBS buffer. The antibody resin was subsequently combined with the peptide sample and incubated overnight at 4 °C on a rotary shaker. Samples were centrifuged for 2 min at 100 × g. Pellets were incubated in 1 mL TBS, 0.1% NP-40 for 30 min at 22 °C, centrifuged (2 min, 100 × g) and the pellets were washed twice with 1 mL TBS, 0.1% NP-40 and twice with ddH2O. Pellets were dissolved in 400 μL TMT elution buffer (ThermoFisher Scientific) and incubated for 10 min at 65 °C. Pellets were centrifuged 2 min at 100 × g, supernatants collected and pellets eluted a second time. Elutions were combined, frozen at −80 °C and freeze-dried. Dry pellets were dissolved in 20 μL 5% acetonitrile and 0.1% TFA, another 20 μL of 0.1% TFA was added and peptides were desalted by stage tipping as described previously (20). Peptide material of each of the two samples was sufficient to be analyzed in two technical replicates by LC-MS/MS.
Bovine serum albumin (BSA, Sigma-Aldrich) and diamide treated BSA were differentially cysteine labeled by the diaCys approach and by iodoTMT (omitting the antibody purification step), tryptically digested, desalted and analyzed by MS as the corresponding C. difficile protein samples.
Western Blotting of iodoTMTzero
C. difficile was harvested at an A600 of 0.4 and cells were washed three times with ice cold and oxygen-free PBS. Cells were lyzed in UHE buffer (8 m urea, 200 mm HEPES, 10 mm EDTA, pH 8.0) by ultrasonication and cell debris removed by centrifugation. After determination of protein, 200 μg of protein were filled up to a volume of 600 μL with UHE buffer and reduced in 5 mm TCEP for 15 min at 30 °C before IAM was added in the following final concentrations: 0 mm, 0.35 mm, 0.74 mm, 1.5 mm, 3 mm 5 mm, 10 mm and 50 mm. Proteins were alkylated for 30 min in the dark at 30 °C and subsequently precipitated with acetone. Dry protein pellets were dissolved in 50 μL UHE buffer each and protein concentration determined. Twenty micrograms of each sample were supplied with 1/10 of the content of one iodoTMTzero (Thermo Scientific) vial and incubated at 30 °C for one hour. Proteins and a protein ladder (Precision Plus Protein Kaleidoscope Prestained Protein Standard, Bio-Rad) were separated via SDS-PAGE (gradient 4–15%, Bio-Rad) for 1 h at constant voltage of 120 V, and blotted onto a PVDF membrane for 1 h at 100 V. The membrane was incubated for 90 min in PBS buffer containing 0.05% Tween (PBST) and 5% skimmed milk powder. After three washing steps in PBST, the first antibody (1:1,000 of TMT Monoclonal Antibody for Western blotting, (Invitrogen, Thermo Fisher Scientific) in 150 mm NaCl, 5% BSA, 0.02% Azide, 50 mm TrisHCl pH 7.6) was bound to the membrane overnight at 4 °C. The membrane was washed three times in PBST, before incubation with the second antibody (goat anti-Mouse IgG, Alexa Fluor Plus 680, Invitrogen) diluted 1:15,000 in PBST, 5% skimmed milk powder, 0.02% SDS for 1 h at RT. The membrane was washed three times in PBST. Signal was detected on an Odyssey CLx (LI-COR, Lincoln, NE) at 700 nm and quantitated by Image Studio ver. 2.0.
LC-MS/MS- Analysis and Raw Data Processing
Liquid chromatographic separation of peptides was done by an EASY-nLC 1000 system (Thermo Scientific) and subsequent peptide detection by an Orbitrap Elite Hybrid mass spectrometer (Thermo Scientific) of samples of the diaCys approach was performed as described elsewhere (19). IodoTMT samples were loaded onto the LC system with 10 μL of buffer A (0.1% (v/v) acetic acid) at a constant flow of 500 nL/min without trapping. Peptides were eluted using a non-linear 160 min gradient from 1% to 99% buffer B (0.1% (v/v) acetic acid in acetonitrile) at a flow of 300 nL/min and directly injected into an Orbitrap Velos (Thermo Scientific). MS survey scans at a resolution of r = 60,000 were acquired in the m/z range of 300 to 1600 with activated lockmass correction. The 10 most abundant precursors with a minimum signal of 10,000 and a minimum charge of 2 were selected for HCD fragmentation at a normalized collision energy of 40% and an activation time of 100 ms. Precursors were excluded for 60 s with an exclusion mass width of ±10 ppm. Database searching and quantification was performed within the framework of the MaxQuant software suite (ver. 1.5.7.0) using the Andromeda algorithm (21). Only fully tryptic peptides with no more than 2 missed cleavages were considered. Spectra were searched against the Uniprot Peptoclostridium difficile 630Δerm database (UP000032805 downloaded 3rd of August 2015) containing 3764 protein entries and common contaminations. Precursor ion mass tolerance was 10 ppm. Fragment ion tolerance for diaCys samples was 0.5 Da and 20 ppm for HCD fragments of iodoTMT samples. DiaCys samples were searched twice, firstly as label experiment of cysteine (light IAM C2H4INO and heavy IAM 13C2H2D2INO) and secondly with a variable IAM modification of cysteine. A variable oxidation of methionine was allowed in all searches. Information on the abundance of reduced and oxidized species of cysteine peptides was obtained from the MaxQuant intensities of corresponding peptides. Protein quantification was based on LFQ intensities based on at least two peptides. A false discovery rate of no more than 1% on peptide and protein level was allowed. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (22) with data set identifier PXD007278.
Further Data Evaluation
Treemaps of C. difficile 630Δerm peptides were constructed using the Paver software (DECODON GmbH, Greifswald, Germany) based on an assignment of corresponding proteins to TIGRFAM roles (23) as described recently (19). The Paver software was fed with percental changes in the redox state of peptide specific cysteine residues instead of changes in protein abundance, i.e. the color code of the tree maps indicates changes in redox state. To estimate the location of a specific cysteine in the protein structure with respect to its surface accessibility, two different bioinformatics tools were employed. Primary protein sequences were submitted to the NetSurfP software ver. 1.1 (24) which predicts the surface accessibility of every single amino acid in the protein sequence. The SWISS MODEL tool was also provided with the primary sequences of proteins of interest and modeled protein structures based on homology to other proteins of known 3-D structure (25). Surface accessibility of cysteines was then deduced from the image of the SWISS MODEL output.
Experimental Design and Statistical Rationale
The diaCys approach was conducted in three independent biological replicates with a switch of light and heavy IAM modification in the third replicate. The iodoTMT experiment was also performed in three biological replicates. Statistical significance was assessed by an unpaired Student's t test. Two biological replicates of iodoTMTzero Western Blots were performed.
RESULTS
Thiol Proteome of C. difficile
Cysteine is one of the low abundant amino acids in proteins. However, the cysteine content in the proteome of C. difficile is over 1.2%, almost twice as high as in the closely related Bacillus subtilis, a preferentially aerobically growing species. An avoidance of cysteine in bacteria pursuing an aerobic lifestyle appears expedient with respect to the potential of reactive oxygen species (ROS) to damage cysteine containing proteins. Thus, Daniels et al. postulated that the exclusion of cysteine in Gram-positive bacteria is an adaptation strategy to aerobic conditions (26). Gram-negative bacteria with their protective outer membrane do not exhibit a reduced cysteine content. We compared the proteome amino acid composition of six pathogenic firmicutes with a low GC-content and found a lower abundance of cysteine in the three aerobic bacteria compared with the three strict anaerobes (supplemental Fig. S1). The overrepresentation of cysteine in anaerobic bacteria suggests a more frequent occurrence of the amino acid in catalytic centers of enzymes or in molecular switches.
The proteome of C. difficile Δerm features a total of 14,527 cysteine residues. Almost one quarter of the thiol proteome (3303 different cysteines) could be experimentally proven in our work (supplemental Table S1). As depicted in supplemental Fig. S2 there was no strong bias against cysteine containing peptides in our analysis. With respect to the total number of detected peptides only one out of eight peptides contained one or more cysteine residues with most peptides containing only a single cysteine (supplemental Fig. S3).
Redox State of the Thiol Proteome
The diaCys approach, which implements a differential cysteine labeling with light and heavy IAM, was used to track oxidative changes in the thiol proteome of C. difficile challenged with diamide or a mixture of bile acids (Fig. 1). For the peptides containing only one cysteine (OCPs) the oxidation status can be determined if light and heavy versions of this peptide are detected and quantified. In one diaCys experiment ∼1500 OCPs could be redox-quantified (supplemental Table S2). Almost half of the peptides under control conditions exist in a mostly reduced state (<10% oxidized) and only about 10% are oxidized by more than 30% (Fig. 2). Whereas peptides in the bile acids shock experiment exhibit a similar distribution, a major shift toward oxidized peptides could be determined after diamide shock. The number of peptides in a reduced state (>90% reduced) drops to about 10%, with more than 40% of all peptides having an oxidation state of 30% or higher. Thus, the electrophilic substance diamide has a massive oxidizing impact on the global thiol redox state of protein cysteines in C. difficile.
Fig. 1.

The diaCys approach makes use of a stepwise alkylation of cysteines. In a first alkylation step concomitant with cell lysis all reduced thiol groups are carbamidomethylated with iodoacetamide (green circle). After reduction of reversibly oxidized cysteine residues, nascent thiols are alkylated with iodoacetamide carrying heavy isotopes (red squares). The ratio of light and heavily modified cysteine can be deduced from the MS spectrum, and differences between control and stress samples calculated.
Fig. 2.
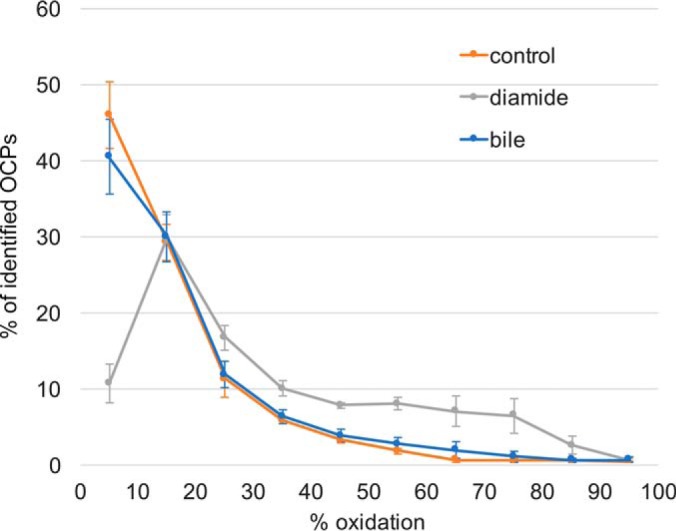
Redox-quantification of OCPs. Peptides were sorted (10% steps) according to their extent of oxidation, with the average of three biological replicates plotted for all conditions (control, bile acids and diamide shock). Whereas diamide induces a dramatic drop in the amount of reduced cysteine, bile acids do not have a global impact on C. difficile′s thiol proteome.
In a parallel approach to the diaCys method, we employed the iodoTMT technology and differentially labeled cysteines of control and diamide shocked samples. Although cysteine-containing peptides are enriched during the iodoTMT protocol, performance of the iodoTMT experiment with respect to identification of cysteine-containing peptides did not reach that of the diaCys method (2060 versus 3332, Fig. 3A). However, technical performances of the iodoTMT was sound and comparable to published data on iodoTMT-based redox proteomics (27–29). Redox states of OCPs according to the iodoTMT approach were calculated for control conditions and after diamide shock. Although a shift toward higher oxidation rates was observed after diamide shock, a very high oxidation rate of cysteine residues was already observed under control conditions and generally much higher than determined by the diaCys approach (Fig. 3B and supplemental Fig. S4, supplemental Table S3). We presumed that an incomplete alkylation of reduced cysteine thiols in the first alkylation step could account for this. To substantiate this hypothesis, we set up an experiment in which we reduced all cysteine residues in our sample and alkylated them with IAM in the first alkylation step but applying the same molarity as in an iodoTMT experiment. For the following second alkylation, we used the iodoTMT zero label, which should bind to all remaining free thiols and could be detected via Western blotting (supplemental Fig. S5). The experiment proved that the amount of iodoTMT in one vial was insufficient to completely label all reduced cysteines of 200 μg of protein in our experimental setup. We assume that a high intracellular concentration of low molecular weight thiols, resulting from C. difficile′s intensive fermentation of cysteine (30), scavenges alkylating agents. This did not represent a problem in the diaCys approach. Because of its affordability IAM could be applied in a concentration more than 50 times as high than in the iodoTMT experiment assuring complete labeling of all thiols. To once more verify technical validity of the iodoTMT data, cysteines of the standard protein BSA were differentially labeled by both, iodoTMT and diaCys. Although fewer cysteine containing peptides were identified in the iodoTMT experiment compared with diaCys, the redox state of cysteines, which were redox-quantified by both approaches, was comparable showing a very high oxidation of all cysteines in the purchased standard protein (supplemental Table S4).
Fig. 3.

Cysteine containing peptides assigned to method of identification. A, Number of peptides categorized according to their number of cysteines and identification method: blue, diaCys; gray, iodoTMT; orange, both approaches. B, Redox states of OCPs that could be determined by iodoTMT and the diaCys approach.
Diamide Induces Extensive Disulfide Stress in C. difficile
In order to determine the change in redox state of a specific cysteine residue, one can calculate the difference between its redox states at different conditions (e.g. % ox diamide - % ox control). Only OCPs which were redox-quantitated in at least two out of three biological replicates of control and stress samples were considered. Doing so, the change in redox state could be determined for 1143 OCPs for diamide shock versus control and for 1137 OCPs for bile acids shock versus control (supplemental Table S5). These peptides correspond to 550 and 553 proteins, respectively. In Fig. 4A the strong oxidizing effect of diamide is illustrated. More than a quarter of the redox-quantified OCPs are reversibly oxidized with a p value of <0.01 after diamide shock compared with control conditions (supplemental Table S5). Also, many of the cysteines are extensively oxidized, with an increase of up to 80%. Bile acids shock did not have such a global impact on the C. difficile thiol proteome, and bile acids do not instigate a general disulfide stress from our experimental setup (Fig. 4B). Six OCPs were detected to be more oxidized after bile acids shock and seven OCPs proved to be more reduced (p value < 0.01). The specific impact of bile acids on these redox-changed cysteines needs to be further investigated. As expected, there was no extensive alteration in protein quantity after this short shock of diamide or bile acids (Fig. 4C, 4D, supplemental Table S6) as determined by label-free protein quantification (LFQ).
Fig. 4.

Volcano plots of changes in redox state of cysteines in OCPs after diamide (A) and bile acids shock (B). Each spot represents one peptide. Peptides to the right of the y axis were oxidized, to the left were reduced. The higher the y-value for a specific spot, the more significant its determined change in redox state (-log10 of p value of 0.01 = 2). Proteins were quantified via LFQ (MaxQuant), the log2 value of the changes in protein abundance (average of three independent experiments) for diamide versus control (C) and bile acids versus control (D) are visualized in volcano plots.
One specific cysteine residue was occasionally determined in more than one peptide because of missed cleavage sites of trypsin or the occurrence of oxidized methionine. Thereby the redox state of the same cysteine residue in different peptides correlated very well in most cases emphasizing the reliability of the diaCys assay (supplemental Table S2). By averaging redox states of all peptides, in which a specific cysteine was identified, differences in redox state for diamide versus control could be calculated for a total of 941 specific cysteine residues. In the bile acids versus control experiment redox states of 929 unique cysteines could be tracked. The change in redox states for all these cysteine residues was visualized in Voronoi treemaps in Fig. 5A and 5B, respectively, and corresponding proteins were classified into TIGRfam main roles. A more detailed labeling of the Voronoi redox treemaps with TIGRfam sub roles, protein names and the exact positions of cysteine residues in the protein sequence is provided in the supplemental data (supplemental Fig. S6a–S6d). It becomes obvious that proteins containing oxidation-susceptible cysteines scatter over all functional categories. The 30 peptides showing the strongest oxidation after diamide shock are listed separately in supplemental Table S7. Most of these oxidized cysteines are exposed at the protein surface according to the prediction software NetSurfP and a protein structure homology modeling performed by the SWISS MODEL workspace.
Fig. 5.

Changes in redox state of protein cysteine residues. A single cell in the Voronoi treemaps depicts the change in redox state of one specific cysteine residue. The change in redox state of 941 cysteines in the diamide versus control experiment (A) and of 929 cysteines in the bile acids versus control experiment (B) could be determined. Red colors represent oxidation, blue colors represent reduction and gray cells were not determined. Cells of the treemaps are arranged according to TIGRfam functional categories of the corresponding proteins with the TIGRfam main roles given in the map. A more detailed labeling with TIGRfam sub roles, protein names and exact positions of cysteine residues can be found supplementary (supplemental Fig. S6a–S6d).
Site-specific Mapping of Cysteine Oxidation
Many of the identified proteins feature more than one cysteine residue. The diaCys approach works site-specifically and allows for redox quantification of several of their cysteines. As an example, all five cysteines of the phosphoglycerate kinase Pgk (A0A031WJX1) could be redox-quantified with four of them showing no increased oxidation after diamide shock, but Cys15, which is surface-exposed, is oxidized by 60% (supplemental Fig. S7A). Redox-regulation of phosphoglycerate kinase was already shown for Synechocystis and in mitochondria of green algae (31, 32). Future investigation will clarify if thiol oxidation of Pgk in C. difficile also has a regulatory effect. Of the five redox-quantified cysteine residues of the Acetyl-CoA decarbonylase AcsD (A0A031WGJ0), two were determined to be oxidized by 56 and 45%, respectively (supplemental Fig. S7b). Considering the predicted three-dimensional structure of the protein, a formation of a disulfide bridge of the two cysteine residues is conceivable. As a third example, only one of the six redox-quantified cysteines of alcohol dehydrogenase AdhE1 (A0A0A8U1W3) increased its redox state strongly by 68% (supplemental Fig. S7c). In all three exemplified proteins, the oxidized cysteine residues are exposed at the protein surface whereas cysteine residues with an unchanged redox state are mostly buried in the interior of the protein.
Peptides with Multiple Cysteine Sites
Applying the MaxQuant software, redox quantification of OCPs could be reliably performed by setting the carbamidomethylation of cysteines as “label” comparable to a SILAC experiment (21). This allowed for a better coverage of both heavy and light species of cysteine peptides. However, in a peptide with two cysteines both could be modified by light carbamidomethylation or by heavy carbamidomethylation, but also a mixed peptide with one cysteine lightly labeled and the other one heavily labeled could exist. With an increasing number of cysteine residues, more possible different species of a peptide could occur (Fig. 6). The detection of peptides with multiple cysteines and blent carbamidomethylation is not supported by the MaxQuant “label” search. Therefore, we searched the same raw data in a separate MaxQuant database search, with light and heavy carbamidomethylation as a variable cysteine modification. Thus, an identification of mixed modified peptides was also enabled, as for the peptide IGFLCECGGVFR of the small uncharacterized protein A0A031WDP1. According to the search with variable cysteine modification the fully reduced peptide species is three times as abundant as the fully oxidized under control conditions (Table II). After diamide shock, proportions are reversed with the oxidized species being 2 to 3 times as abundant as the reduced peptide. The ratio between fully reduced and oxidized peptide species can also be deduced from the data evaluation with heavy and light cysteine set as label and corresponds to the ratio determined by the variable cysteine modification search. However, information on the presence and abundance of a mixed modified species is lost. There are also examples of peptides with more than one cysteine for which exclusively heavy and light species could be detected, but no blent modification. One such peptide is MDCIFCK of a small histidine triad nucleotide-binding protein (A0A031WHD1). Only fully light and heavy peptides have been identified, also for the methionine oxidized variant, presuming formation of an internal disulfide bridge between the two cysteines, but with no other forms of oxidation. Again, the results of both different search strategies correlate well. The amount of the fully reduced peptide species drops to one third after diamide shock (Table II).
Fig. 6.

Peptides with one or more cysteine residues. Peptides featuring a single cysteine (OCP) are present with a maximum of two species of different masses in MS spectra. However, with an increasing number of cysteine residues the possibilities of how light (L) and heavy (H) carbamidomethylation is distributed increase (2n with n = number of cysteine residues in the peptide).
Table II. Two examples of peptides with two cysteine residues are shown. Ratios of peptide abundance with light and heavy carbamidomethylation are given for the three replicates (A, B, and C) for control and diamide shock. Ratios have been calculated from intensities obtained by a database search with variable carbamidomethylation and from a search with carbamidomethylation set as “label.” Peptide IGFLCECGGVFR of protein A0A031WDP was identified in three different species: cysteines appear to be both oxidized, both reduced and in the search with variable carbamidomethylation also appear as mixed form with one cysteine oxidized and the other one reduced. For peptide MDCIFCK of protein A0A031WHD1 no blent version was identified.
| protein peptide sequence | redox state | control A variable | control A label | control B variable | control B label | control C variable | control C label | diamide A variable | diamide A label | diamide B variable | diamide B label | diamide C variable | diamide C label |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A0A031WDP1 | reduced | 4.34 | 16.94 | 3.30 | 3.22 | 2.77 | 2.77 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| IGFLCECGGVFR | mixed | 0.02 | n.d. | 0.02 | n.d. | n.d. | n.d. | 0.02 | n.d. | 0.02 | n.d. | 1.45 | n.d. |
| oxidized | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.07 | 1.06 | 2.06 | 1.61 | 3.52 | 3.61 | |
| A0A031WHD1 | |||||||||||||
| MDCIFCK | reduced | 15.23 | 9.63 | 7.87 | 7.87 | 5.92 | 5.22 | 5.64 | 5.64 | 2.98 | 2.72 | 1.59 | 1.59 |
| oxidized | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |
| M(ox)DCIFCK | reduced | 17.89 | 6.16 | n.d. | 2.82 | n.d. | 6.43 | 5.64 | 5.46 | 2.98 | 1.61 | 1.59 | 1.42 |
| oxidized | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
DISCUSSION
This work is the first to characterize the thiol proteome of the cysteine-rich anaerobic pathogen C. difficile and allowed the absolute determination of the extent of reversible oxidation of over 1500 thiol peptides. In contrast to other thiol labeling methods (e.g. iodoTMT, OxICAT), the novel diaCys approach is not hampered by the presence of higher concentrations of low molecular weight (LMW) thiols as cysteine or by its degradation products. Potentially, the diaCys method could even be employed for labeling of extracellular proteins directly in the growth medium, as the affordability of the labels IAM and heavy IAM allows the use of them in great excess to ensure complete labeling and to enable the labeling of protein amounts bigger than 100–200 μg. Although the diaCys approach does not involve an enrichment of cysteine containing peptides, the number of such peptides identified in this study can compete with what was achieved in other redox proteomics studies. In 2010 Sinha et al. introduced the d-switch method which is very similar to our diaCys approach but was not used on a global proteome scale (33). They applied light and heavy N-ethylmaleinimide for differential labeling of nitrosylated cysteines and identified 11 nitrosylated peptides from human cell lines. Wojdyla et al. reported the relative redox-quantification of 114 sulfenylated and nitrosylated peptides of E. coli (27) making use of the iodoTMT method. Even though iodoTMT facilitated redox-quantification of many more peptides from C. difficile in this work, this technology did not prove to be suitable for redox proteomics in C. difficile, most likely because of the presence of interfering low molecular weight thiols. Recently, substantial progress regarding the number of redox-quantified peptides in bacteria has been made by the Antelmann group, who employed the OxICAT approach originally developed by Leichert et al. (12) and could redox-quantify 228 cysteine residues in hypochlorite stressed Staphylococcus aureus (34) and 1098 cysteines in Mycobacterium smegmatis (35). Deng et al. also made use of isotopically labeled cysteine reactive tags that could be enriched and thereby captured 307 cysteine residues in Pseudomonas aeruginosa and S. aureus (36). Here, the introduced diaCys approach yielded redox-quantification of more than 1500 peptides, providing one of the highest coverages for a thiol proteome so far. This leads us to hypothesize that an enrichment of cysteine-containing peptides does not necessarily facilitate their identification but may instead lead to sample loss. Possibly, the usage of bulky cysteine tags for subsequent enrichment does not support labeling of poorly accessibly cysteines which are then lost in the analysis. We assume that in the case of low complexity organisms such as bacteria, sample fractionation, speed and sensitivity of current mass spectrometers seem to be sufficient in an in-depth analysis of thiol peptides, despite the clear majority of non-thiol peptides. In addition, the omission of a cysteine peptide enrichment step facilitates a more reliable label-free protein quantification which is not only based on cysteine peptides but on all peptides that have been detected for a specific protein. Thus, redox quantification of the thiol proteome and global determination of protein abundance can be performed simultaneously and does not require a double tagging of peptides as presented by others (37).
In our protocol, we used TCEP, which non-selectively reduces reversibly oxidized cysteine residues to thiols. Therefore, no statement on the nature of the oxidative events we have observed in the thiol proteome can be made. However, the diaCys approach should be adaptable to use reductants other than TCEP, e.g. arsenite, to specifically reduce sulfenic acid (38), or ascorbate, for the reduction of nitrosocysteine (39). However, irreversible cysteine modifications are not amenable to be discovered by our approach.
The MaxQuant software has proved to be very useful for data evaluation. Searching for the differentially labeled cysteine samples as “label experiment” increased the number of thiol peptides, of which pairs (light and heavy species) could be identified, as well as the dynamic range of the redox quantification. Evaluation of peptides featuring more than one cysteine residue, however, is not as straight forward and presents an issue with all hitherto introduced cysteine labeling methods. This is because of lack of a preexisting automated bioinformatics workflow to evaluate peptides with several cysteine residues. A database search in which cysteine modification is set to variable proved to be a helpful trick to approach determination of redox states of every cysteine in such delicate peptides. For some of these peptides a manual inspection of MS/MS spectra could possibly provide more information on the specific redox state of each of the cysteines. Alternatively, other digestion strategies than trypsin could be applied to separate cysteines of interest into different peptides.
In a global study on the thiol proteome of Mycobacterium smegmatis Hillion et al. found 40% of the redox-quantified peptides to be oxidized by less than 10% and less than one quarter of the peptides to be oxidized by more than 25% (35). This corresponds very well with our data showing that most of cysteine peptides in C. difficile exists in a predominately reduced state (Fig. 2). The Voronoi treemap of Fig. 5a clearly illustrates the widespread oxidizing effect diamide had on the thiol proteome of C. difficile (293 peptides oxidized with p value < 0.01; 477 peptides oxidized with p value < 0.05). The affected proteins were observed to be spread over all functional categories. Diamide as a chemical instigator of disulfide stress was used in other bacteria and cysteine oxidation in proteins tracked on 2-D gels. In one of these studies Leichert et al. determined the methionine synthase in E. coli to be oxidized after diamide shock (10). Also, in our study the protein showed an increase of 60% oxidation on one of its cysteine residues (p value 0.016). Hochgräfe et al. exposed B. subtilis to diamide and published a list of 40 proteins found to be oxidized on 2-D gels (11). For ten of these oxidized proteins, homologs existing in C. difficile exhibit significantly oxidized cysteine peptides after diamide shock (A0A0A8U887, A0A031WJX1, A0A031WB55, A0A031WJB8, A0A031WEK6, A0A031WC37, A0A031WBB5, A0A031W9A7, A0A031WDE6, A0A0A8TYM1) with a p value of < 0.01 (Table S5). One of these proteins is a 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (A0A0A8U887). In the B. subtilis study the protein was determined to be oxidized as a spot on a 2-D gel, however this provided no information regarding the site of oxidation. In the present C. difficile study an oxidation of three different cysteines residues, on the protein A0A0A8U887, could be pinpointed with an increase in oxidation of 75%, 40 and 7% (all p value < 0.01), respectively. Also, for protein A0A031WB55, a deoxyribose-phosphate aldolase, three distinct cysteine residues were determined to be oxidized by 55%, 53 and 38%, respectively (all p value < 0.01). Accordingly, in B. subtilis the deoxyribose-phosphate aldolase Dra was one of the proteins oxidized after diamide shock. The good correspondence of our global redox quantification of the thiol proteome under control conditions with published bacterial data sets, and the great conformity of the results of diamide shock in B. subtilis and C. difficile substantiate applicability of the diaCys method, introduced here, and the reliability of the redox quantification of all the proteins which have not been described yet.
One aim of this study was to shed light on the thiol oxidizing effect of bile acids that has been previously reported (9). We could not prove bile acids to induce disulfide formation in C. difficile. We speculate that the denaturing action of bile acids on proteins causes subsequent oxidation of cysteine residues that have originally been buried inside the protein structure, but were exposed at the surface during protein denaturation. Such cysteines could then be oxidized in the presence of oxygen or other electrophiles. However, in our experimental setup we work under exclusion of oxygen. Cysteines may become surface-exposed after bile acids shock, but are not attacked by reactive oxygen species, which would explain why bile acids did not instigate disulfide formation in our experiments.
Cysteine and other thiols play a central role in the metabolism of C. difficile. The bacterium requires large amounts of cysteine for optimal growth (30), with its production of toxins dependent on cysteine availability (40, 41). Furthermore, some C. difficile strains show an astonishing tolerance to oxygen for virtually strict anaerobic bacteria (42), which is probably closely linked to their thiol metabolism. The impairment of cysteine metabolism and thiol proteome could evolve into new strategies to treat CDIs, and the introduced diaCys approach represents a tool to monitor the impact of such thiol targeting therapies.
In this work, we not only introduced a simple and robust cysteine labeling strategy which represents a useful alternative for researchers in the field of redox proteomics, but also present a detailed description of C. difficile′s thiol proteome and how it is disturbed by electrophilic stress.
DATA AVAILABILITY
MS data are available at www.ebi.ac.uk/pride/archive with project ID: PXD007278.
Supplementary Material
Acknowledgments
We thank Maria Conway for proofreading the manuscript.
Footnotes
* This work was supported by grants of the “Bundesministerium für Bildung und Forschung” (03Z1CN21) to F. H., of the “Niedersächsisches Ministerium für Wissenschaft und Kultur” (VWZN2889) to K. R., of the “Ministerium für Bildung, Wissenschaft und Kultur Mecklenburg-Vorpommern” (UG16001) to K. R. and of the University of Greifswald to S. S.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CDI
- Clostridium difficile infection
- BHI
- brain heart infusion
- IAM
- iodoacetamide
- OCP
- one cysteine peptide
- PBS
- phosphate buffered saline
- PBST
- phosphate buffered saline + Tween
- RT
- room temperature.
REFERENCES
- 1. Bartlett J. G., Chang T. W., Gurwith M., Gorbach S. L., and Onderdonk A. B. (1978) Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N. Engl. J. Med. 298, 531–534 [DOI] [PubMed] [Google Scholar]
- 2. Mergenhagen K. A., Wojciechowski A. L., and Paladino J. A. (2014) A review of the economics of treating Clostridium difficile infection. Pharmacoeconomics 32, 639–650 [DOI] [PubMed] [Google Scholar]
- 3. Johnson S. (2009) Recurrent Clostridium difficile infection: a review of risk factors, treatments, and outcomes. J. Infect. 58, 403–410 [DOI] [PubMed] [Google Scholar]
- 4. Emerson J. E., Stabler R. A., Wren B. W., and Fairweather N. F. (2008) Microarray analysis of the transcriptional responses of Clostridium difficile to environmental and antibiotic stress. J. Med. Microbiol. 57, 757–764 [DOI] [PubMed] [Google Scholar]
- 5. Chen J.-W., Scaria J., Mao C., Sobral B., Zhang S., Lawley T., and Chang Y.-F. (2013) Proteomic comparison of historic and recently emerged hypervirulent Clostridium difficile strains. J. Proteome Res. 12, 1151–1161 [DOI] [PubMed] [Google Scholar]
- 6. Ternan N. G., Jain S., Graham R. L. J., and McMullan G. (2014) Semiquantitative analysis of clinical heat stress in Clostridium difficile strain 630 using a GeLC/MS workflow with emPAI quantitation. PLoS ONE 9, e88960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chong P. M., Lynch T., McCorrister S., Kibsey P., Miller M., Gravel D., Westmacott G. R., Mulvey M. R., Canadian Nosocomial Infection Surveillance Program (CNISP). (2014) Proteomic analysis of a NAP1 Clostridium difficile clinical isolate resistant to metronidazole. PLoS ONE 9, e82622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Go Y.-M., Chandler J. D., and Jones D. P. (2015) The cysteine proteome. Free Radic. Biol. Med. 84, 227–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cremers C. M., Knoefler D., Vitvitsky V., Banerjee R., and Jakob U. (2014) Bile salts act as effective protein-unfolding agents and instigators of disulfide stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, E1610–E1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leichert L. I., and Jakob U. (2004) Protein thiol modifications visualized in vivo. PLoS Biol. 2, e333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hochgräfe F., Mostertz J., Albrecht D., and Hecker M. (2005) Fluorescence thiol modification assay: oxidatively modified proteins in Bacillus subtilis. Mol. Microbiol. 58, 409–425 [DOI] [PubMed] [Google Scholar]
- 12. Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., and Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pan K.-T., Chen Y.-Y., Pu T.-H., Chao Y.-S., Yang C.-Y., Bomgarden R. D., Rogers J. C., Meng T.-C., and Khoo K.-H. (2014) Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxid. Redox Signal. 20, 1365–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo J., Nguyen A. Y., Dai Z., Su D., Gaffrey M. J., Moore R. J., Jacobs J. M., Monroe M. E., Smith R. D., Koppenaal D. W., Pakrasi H. B., and Qian W.-J. (2014) Proteome-wide light/dark modulation of thiol oxidation in cyanobacteria revealed by quantitative site-specific redox proteomics. Mol. Cell. Proteomics 13, 3270–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wojdyla K., and Rogowska-Wrzesinska A. (2015) Differential alkylation-based redox proteomics–Lessons learnt. Redox Biol. 6, 240–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang J., Carroll K. S., and Liebler D. C. (2016) The Expanding Landscape of the Thiol Redox Proteome. Mol. Cell. Proteomics 15, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hussain H. A., Roberts A. P., and Mullany P. (2005) Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Δerm) and demonstration that the conjugative transposon Tn916ΔE enters the genome of this strain at multiple sites. J. Med. Microbiol. 54, 137–141 [DOI] [PubMed] [Google Scholar]
- 18. Macdonald I. A., Bokkenheuser V. D., Winter J., McLernon A. M., and Mosbach E. H. (1983) Degradation of steroids in the human gut. J. Lipid Res. 24, 675–700 [PubMed] [Google Scholar]
- 19. Otto A., Maass S., Lassek C., Becher D., Hecker M., Riedel K., and Sievers S. (2016) The protein inventory of Clostridium difficile grown in complex and minimal medium. Proteomics Clin. Appl. 10, 1068–1072 [DOI] [PubMed] [Google Scholar]
- 20. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 21. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Publishing Group 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 22. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Punta M., Coggill P. C., Eberhardt R. Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., Heger A., Holm L., Sonnhammer E. L. L., Eddy S. R., Bateman A., and Finn R. D. (2012) The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petersen B., Petersen T. N., Andersen P., Nielsen M., and Lundegaard C. (2009) A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., and Schwede T. (2009) Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4, 1–13 [DOI] [PubMed] [Google Scholar]
- 26. Daniels R., Mellroth P., Bernsel A., Neiers F., Normark S., Heijne von G, Henriques-Normark B. (2010) Disulfide bond formation and cysteine exclusion in gram-positive bacteria. J. Biol. Chem. 285, 3300–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wojdyla K., Williamson J., Roepstorff P., and Rogowska-Wrzesinska A. (2015) The SNO/SOH TMT strategy for combinatorial analysis of reversible cysteine oxidations. J. Proteomics 113, 415–434 [DOI] [PubMed] [Google Scholar]
- 28. Araki K., Kusano H., Sasaki N., Tanaka R., Hatta T., Fukui K., and Natsume T. (2016) Redox sensitivities of global cellular cysteine residues under reductive and oxidative stress. J. Proteome Res. 15, 2548–2559 [DOI] [PubMed] [Google Scholar]
- 29. Yin Z., Balmant K., Geng S., Zhu N., Zhang T., Dufresne C., Dai S., and Chen S. (2017) Bicarbonate induced redox proteome changes in Arabidopsis suspension cells. Front. Plant Sci. 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neumann-Schaal M., Hofmann J. D., Will S. E., and Schomburg D. (2015) Time-resolved amino acid uptake of Clostridium difficile 630Δerm and concomitant fermentation product and toxin formation. BMC Microbiol. 15, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsukamoto Y., Fukushima Y., Hara S., and Hisabori T. (2013) Redox control of the activity of phosphoglycerate kinase in Synechocystis sp. PCC6803. Plant Cell Physiol. 54, 484–491 [DOI] [PubMed] [Google Scholar]
- 32. Morisse S., Michelet L., Bedhomme M., Marchand C. H., Calvaresi M., Trost P., Fermani S., Zaffagnini M., and Lemaire S. D. (2014) Thioredoxin-dependent redox regulation of chloroplastic phosphoglycerate kinase from Chlamydomonas reinhardtii. J. Biol. Chem. 289, 30012–30024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sinha V., Wijewickrama G. T., Chandrasena R. E. P., Xu H., Edirisinghe P. D., Schiefer I. T., and Thatcher G. R. J. (2010) Proteomic and mass spectroscopic quantitation of protein S-nitrosation differentiates NO-donors. ACS Chem. Biol. 5, 667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Imber M., Huyen N. T. T., Pietrzyk-Brzezinska A. J., Loi V. V., Hillion M., Bernhardt J., Thärichen L., Kolšek K., Saleh M., Hamilton C. J., Adrian L., Gräter F., Wahl M. C., and Antelmann H. (2017) Protein S-Bacillithiolation Functions in Thiol Protection and Redox Regulation of the Glyceraldehyde-3-Phosphate Dehydrogenase Gap in Staphylococcus aureus Under Hypochlorite Stress. Antioxid. Redox Signal, ars.2016.6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hillion M., Bernhardt J., Busche T., Rossius M., Maass S., Becher D., Rawat M., Wirtz M., Hell R., Rückert C., Kalinowski J., and Antelmann H. (2017) Monitoring global protein thiol-oxidation and protein S-mycothiolation in Mycobacterium smegmatis under hypochlorite stress. Sci. Rep. 7, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deng X., Weerapana E., Ulanovskaya O., Sun F., Liang H., Ji Q., Ye Y., Fu Y., Zhou L., Li J., Zhang H., Wang C., Alvarez S., Hicks L. M., Lan L., Wu M., Cravatt B. F., and He C. (2013) Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe 13, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parker J., Balmant K., Zhu F., Zhu N., and Chen S. (2015) cysTMTRAQ-An integrative method for unbiased thiol-based redox proteomics. Mol. Cell. Proteomics 14, 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saurin A. T., Neubert H., Brennan J. P., and Eaton P. (2004) Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc. Natl. Acad. Sci. U.S.A. 101, 17982–17987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaffrey S. R., Erdjument-Bromage H., Ferris C. D., Tempst P., and Snyder S. H. (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193–197 [DOI] [PubMed] [Google Scholar]
- 40. Karlsson S., Lindberg A., Norin E., Burman L. G., and Akerlund T. (2000) Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infection Immunity 68, 5881–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dubois T., Dancer-Thibonnier M., Monot M., Hamiot A., Bouillaut L., Soutourina O., Martin-Verstraete I., and Dupuy B. (2016) Control of Clostridium difficile Physiopathology in Response to Cysteine Availability. Infection Immunity 84, 2389–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Edwards A. N., Karim S. T., Pascual R. A., Jowhar L. M., Anderson S. E., and McBride S. M. (2016) Chemical and Stress Resistances of Clostridium difficile Spores and Vegetative Cells. Front. Microbiol. 7, 1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MS data are available at www.ebi.ac.uk/pride/archive with project ID: PXD007278.