

Abstract
Background
Postnatal overfeeding activates tissue glucocorticoid (GC) activity by up-regulating 11β-hydroxysteroid dehydrogenase 1 (11β-HSD1) and increasing sensitivity to high-fat (HF) diet-induced non-alcoholic fatty liver disease (NAFLD). The present study aimed to evaluate the effects of postnatal overfeeding on GC regulation and lipogenesis in the liver and to observe the impact of GC on hepatocyte lipid metabolism.
Methods
In vivo, Male Sprague-Dawley rat pup litters were adjusted to litter sizes of three (small litter, SL) or ten (normal litter, NL) on postnatal day 3 and then given standard chow from postnatal week 3 (W3) to W13. In vitro, HepG2 cells were stimulated by GC, mifepristone (Mi) or GC + Mi within 48 h, followed by sodium oleate (OA) intervention (or not) for 24 h. Intracellular lipid droplets, triglyceride (TG) concentrations and gene expression related to lipid metabolism were measured in hepatic tissues or HepG2 cells.
Results
In vivo, weight gain in the body and liver and TG concentrations in the liver were significantly increased in the SL rats compared to the NL rats at W3 and W13 (p < 0.05); mRNA expression of hepatic 11β-HSD1, acetyl-CoA carboxylase 1 (ACC), stearoyl-CoA desaturase-1 (SCD1), fatty acid synthase (FASN) and their nuclear transcription factor, sterol regulatory element binding protein-1c (SREBP-1c) (p < 0.05), was also increased. In vitro, intracellular lipid droplets and TG content in HepG2 cells increased under stimulation with GC or OA (p < 0.05); the increase was more significant following treatment with GC and OA together (p < 0.05). The ACC, SCD1, FASN and SREBP-1c mRNA expression changes were highly similar to the changes in TG content in cells. All the changes induced by GC disappeared when the glucocorticoid receptor (GR) was blocked by Mi.
Conclusions
Postnatal overfeeding induced GC overexposure through 11β-HSD1 up-regulation in the liver. GC activated hepatic de novo lipogenesis (DNL) via GR and led to hepatic lipid accumulation, which increased the risk of NAFLD during adulthood.
Keywords: Glucocorticoid, NAFLD, Postnatal overfeeding, 11β-HSD1, Glucocorticoid receptor
Background
Non-alcoholic fatty liver disease (NAFLD) and other components of metabolic syndrome (MS) have become increasingly comorbid with the increasing prevalence of obesity in both children and adults [1, 2]. Unlike what happens in adults, the onset of paediatric NAFLD is asymptomatic until it progresses into hepatic fibrosis and cirrhosis [3, 4]. In order to develop strategies to effectively prevent NAFLD and MS, it is important to better understand the mechanisms by which obesity increases susceptibility to NAFLD.
NAFLD is characterized by excessive triglyceride (TG) accumulation in the absence of significant alcohol consumption [1]. It is primarily caused by the imbalance of hepatic lipid homeostasis between the acquisition and removal of TG/fatty acid, which involves increased fatty acid/TG uptake, enhanced de novo lipogenesis (DNL), impaired fatty acid β-oxidation, and/or decreased lipid export in the liver. Several rate-limiting enzymes and transcription factors participate in hepatic lipid metabolism [5]. Hepatic lipoprotein lipase (LPL) and liver-type fatty acid-binding protein (L-FABP) are thought to play a central role in hepatic lipid uptake [6, 7]. The activation of DNL to TG synthesis in the liver involves sterol regulatory element binding protein-1c (SREBP-1c) and lipogenic enzymes including fatty acid synthase (FASN), acetyl-CoA carboxylase 1 (ACC) and stearoyl-CoA desaturase-1 (SCD1) [8, 9]. In addition, fatty acid β-oxidation and lipid export in the liver involve peroxisome proliferator-activated receptor α (PPARα), carnitine palmitoyltransferase 1 (CPT1) and microsomal triglyceride transfer protein (MTP) [10–15]. The activities of these enzyme systems are regulated by nutrition, the endocrine system and inflammation [16–18].
Glucocorticoid (GC), such as corticosterone and cortisol, affects fat accumulation and lipid and glucose metabolism [19, 20]. At the tissue level, GC exposure is determined not only by circulating levels, but also by the tissue-specific GC-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) and the GC-inactivating enzymes 5α-reductase type 1 (5αR1) and 5β-reductase (5βR) [21, 22]. Previous studies have shown that the overexpression of 11β-HSD1 in tissue amplifies local GC action, which leads to increased accumulation of adipose tissue and metabolic disorders in both humans and rodents [23–26]. GC is involved in every stage of the pathogenesis of NAFLD [27]. In animal models, GC increases lipid biosynthesis within the liver that can lead to hepatic steatosis and increase circulating TG levels [28, 29].
The results of both experimental studies using animal models and clinical investigations have indicated that the early nutrition environment is associated with the development of obesity and MS in later life [30–33] and that GC is a possible mediator of the permanent programming of obesity, insulin resistance, and other metabolic dysregulations [34–36]. Previously, we reported that small litter (SL) rearing induced obesity in adult rats. The animals also had hyperinsulinemia, elevated circulating corticosterone levels, peripheral tissue-specific alterations in 11β-HSD1 expression and activity and 5αR1 and 5βR expression starting at puberty [37]. In addition, SL rats also displayed increased ACC activation in the livers and were more prone to develop NAFLD when challenged with high-fat (HF) diets [38]. Our hypothesis was that local GC activity plays the crucial role in the pathogenesis of hepatic steatosis by regulating lipid synthesis enzymes. Therefore, in this study we examined the expression patterns of 11β-HSD1 and 5αR1 and 5βR in the livers of SL rats, as well as those of lipid metabolism-related genes involved in hepatic DNL and fatty acid β-oxidation and lipid export. Moreover, we wanted to determine the action of GC on hepatic lipid metabolism. We first treated HepG2 cells with GC and then with OA in vitro to mimic hepatic GC overexposure in vivo.
Methods
Animals and experimental design
All animal studies were performed following the guidelines established by the University Committee on the Use and Care of Animals and were overseen by the Unit for Laboratory Animal Medicine at Nanjing Medical University (IACUC: 14030102). Male Sprague-Dawley rats were used. They were maintained under a controlled 12/12 h light/dark cycle in temperature (22 ± 2 °C) conditions with free access to food and water.
The experimental setup was similar to that described in Boullu-Ciocca [39]. In rats, the weaning period is postnatal week 3, puberty is postnatal weeks 6-8 and adulthood is week 9 and afterward [40]. In our previous studies, we showed that metabolism disorders in SL rats took place during postnatal weeks 13-16 [38, 41]. Therefore, postnatal weeks 3 and 13 were selected as two experimental points of this study to examine the effects of early nutrition on adult health. On postnatal day 3 (P3), male pups were randomly redistributed to litter sizes of three (SLs) or ten (normal litters (NLs)) to induce early postnatal overfeeding or normal nutrition, respectively [42]. After weaning (P21, W3), the NL and SL rats were fed a standard diet (NL or SL group) until postnatal week 13 (W13). All rats were housed 3-4 per cage after weaning. Body weight and food intake were monitored weekly throughout life. The animals were killed at W3 and W13 after an overnight fast.
Tissue collection
The rats were anaesthetized with chloral hydrate (300 mg/kg body weight, i.p.) after an overnight fast (12 h). Body weight was recorded. Each rat’s liver was dissected and weighed, and the hepatosomatic index (HSI) was calculated as (liver weight/body weight) * 100% [43]. All tissue samples were snap-frozen in liquid nitrogen and stored at − 70 °C until gene expression analysis.
Hepatic lipid assays
Concentrations of TG in the liver and cells were determined using TG assay kits (E1013, Applygen, Beijing, China). The hepatic TG concentration was expressed relative to 1 g of liver protein. Hepatic protein concentrations were determined using a Pierce BCA protein assay kit with bovine serum albumin as the standard (Thermo Fisher Scientific, Rockford, IL, USA).
Cell culture
HepG2 cells, obtained from Keygen Biotech (Nanjing, China, ATCC HB-8065), were maintained in DMEM medium containing 10% FBS and 1% P/S at 37 °C with 5% CO2 (Thermo Scientific, CO2 incubator) in 75 cm2 flasks. Cells were plated in 6-well plates at 2*105 cells per well. The following day, confluent cells were starved for 6 h without FBS. Then, the cells were treated with 2.0 ml of fresh supplemented culture medium containing dexamethasone (active GC, D4902, Sigma), mifepristone (glucocorticoid receptor (GR) antagonist, Mi, M8046, Sigma), both GC and Mi, or vehicle (culture medium) for 48 h, followed by exposure (or not) to sodium oleate (OA, O7501, Sigma), which is rich in fatty acids, for 24 h. To evaluate the possible effects of GC on gene expression related to lipid metabolism, HepG2 cells were incubated with GC at different concentrations (0, 50, 100, 125, 250, 500 and 1000 nM; n = 3 for each concentration) and time (24, 36 and 48 h) and to ascertain the maximal response. The effects of GC (125 nM) combined with Mi at different concentrations (0, 0.1, 1, 5, 10 μM) were then used (n = 3) to evaluate the individual and combined effects on the hepatic lipid homeostasis. The TG content in the cells was determined using commercial kits (E1013, Applygen, Beijing, China).
Oil red O staining
At the end of incubation, the cultured cells were washed with PBS and fixed with 4% formaldehyde for 30 min at room temperature. Then, the cells were stained using Oil red O working solutions containing 6 ml of Oil red O stock solution (0.5 g in 100 ml of isopropanol) and 4 ml of ddH2O at 37 °C for 30 min. Staining was visualized by bright-field microscopy (BX51, OLYMPUS, Japan).
Total RNA extraction and real-time PCR
Total RNA was extracted from cells or liver tissues using Trizol (Invitrogen) according to the manufacturer’s instructions and quantified spectrophotometrically at OD260. The integrity of the total RNA was assessed using agarose gel electrophoresis, and cDNA was synthesized using M-MLV reverse transcriptase (TAKARA) with 0.5 μg of the RNA sample as recommended by the manufacturer. Genes of interest were analysed by real-time PCR using the SYBR GREEN ABI Prism 7500 sequence detector for the target genes, including SREBP-1c, ACC, SCD1, FASN, PPARα, LPL, L-FABP, CPT1 and MTP (Table 1). Expression of the target genes was normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Table 1).
Table 1.
Primer sequences used for mRNA quantification by real-time PCR
| Forward primer 5′–3′ | Forward primer 5′–3′ | |
|---|---|---|
| ACC rat | TGAAGGGCTACCTCTAATG | TCACAACCCAAGAACCAC |
| SCD1 rat | CTCCCTACCTCCACCCCTAT | AACCAACCCTCTCGTTCAGT |
| FASN rat | AAGAGTGGGAGAGCCTGTTC | AGTTCACCAAGCCTACCACA |
| LPL rat | GCTTCCCCTTACTGGTTCC | AACTGGCAGGCAATGAGACT |
| L-FABP rat | AAGGGAAGGACATCAAGGGG | CACTGCCTTGACCTTTTCCC |
| CPT1 rat | ACGAAGAACATTGTGAGCGG | GAGGACCTTGACCATAGCCA |
| MTP rat | AGCAACATGCCTACTTCTTACAC | TCACGGGTTCACTTTCACTG |
| SREBP-1c rat | CGCTACCGTTCCTCTATCA | CTCCTCCACTGCCACAAG |
| PPARα rat | AGCCATTCTGCGACATCA | CGTCTGACTCGGTCTTCTTG |
| GAPDH rat | GGCTCTCTGCTCCTCCCTGTTCTA | CGTCCGATACGGCCAAATCCGT |
| ACC hum | CACGCTCAAGTCACCAAGAA | GCAAATGGGAGGCAATAAGA |
| SCD1 hum | GTTCGTTGCCACTTTCTTGC | TGGTAGTTGTGGAAGCCCTC |
| FASN hum | CGGTGTTTGAGTTCGTGGAG | CGGGGATAGAGGTGCTGAG |
| LPL hum | TTCTCGTTGGCAGGGTTGAT | CTGACACTGTTTTCACGCCA |
| L-FABP hum | TTCAAGTTCACCATCACCGC | TTATGTCGCCGTTGAGTTCG |
| CPT1 hum | CAACTCACATTCAGGCAGCA | CGATGTGCTTGCTGTCTCTC |
| MTP hum | CTGCTCAGACCTCAGACTCA | TCTCTGATGTCACTGCTACCA |
| SREBP-1c hum | TTCCCAGCCCCTCAGATAC | GAGAAGCACCAAGGAGACGA |
| PPARα hum | CCAGCATCCTCTCTCCAACT | AGGAAAACGAAGACCCAAGAT |
| GAPDH hum | GTCGGAGTCAACGGATTTGG | CATGGGTGGAATCATATTGGA |
Statistical methods
Data are expressed as Means ± SEM. Two-ways analysis of variance (ANOVA) tests were used to analyse body weight gain. Two-sided Student’s t-test was used to analyse liver weight, hepatic lipid content, mRNA expression and the effects of different treatments in cell culture. Statistical significance was accepted at p < 0.05.
Results
Food intake and body weight
The food intake of the SL rats increased significantly only at W3 to W5 compared to the NL rats (p < 0.01, Table 2), and there were no significant differences between groups after that time (p > 0.05, Table 2). Body weight increased with age in both groups (p < 0.001), and the SL rats gained more than the NL rats (p < 0.001); there was a significant interaction for weight gain in the SL rats with age (p < 0.001, Fig. 1a).
Table 2.
Food intake at W3, 4, 5, 6, 8, and 13 in NL and SL rats
| NL | SL | |
|---|---|---|
| Food intake (g/day) | ||
| 3 wk | 9.1 ± 0.2 | 14.3 ± 0.5*** |
| 4 wk | 10.6 ± 0.1 | 18.9 ± 0.3*** |
| 5 wk | 14.1 ± 0.7 | 18.0 ± 0.5** |
| 6 wk | 22.1 ± 0.2 | 22.0 ± 0.3 |
| 8 wk | 26.6 ± 0.2 | 26.4 ± 0.1 |
| 13 wk | 27.5 ± 0.6 | 27.8 ± 0.2 |
Data are expressed as the mean ± SEM. Significant differences between groups of rats at corresponding time points were analyzed by two-sided Student’s t-test
**p < 0.01, ***p < 0.001 vs. NL rats. n = 6 in each NL and SL group
Fig. 1.
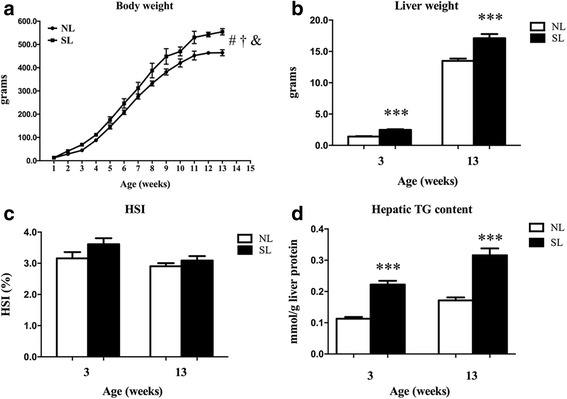
Body weight (a) from postnatal week 1 to week 13; liver weight (b), hepatosomatic index (c) and hepatic TG content (d) in normal litters (NLs) and small litters (SLs) at week 3 (W3) and week 13 (W13). Data are expressed as the mean ± SEM. Body weight gain was analyzed by two-way ANOVA. #F = 1980, p < 0.001 for effect of age; †F = 363, p < 0.001 for effect of SL; &F = 11, p < 0.001 for interaction of SL and age. Significant differences between groups at W3 or W13 were analyzed by two-sided Student’s t-test. ***p < 0.001 vs. NL rats. n = 6 in each NL and SL group
Liver weight and hepatic TG content
The liver weight was higher in the SL rats compared to the NL rats at W3 and W13 (p < 0.001, Fig. 1b), but there was no significant difference in HSI between groups (p > 0.05, Fig. 1c). Hepatic TG content was higher in the SL rats compared to the NL rats at W3 and W13 (p < 0.001, Fig. 1d).
11β-HSD1, 5αR1 and 5βR mRNA expression in the liver at W3 and W13
Hepatic 11β-HSD1 mRNA expression was higher in the SL rats compared to the NL rats at W3 (p < 0.001, Fig. 2a) and W13 (p < 0.05, Fig. 2b). Compared to the NL rats, hepatic 5αR1 and 5βR mRNA expression was higher in the SL rats at W3 (p < 0.01, Fig. 2a) but decreased significantly in the SL rats compared to the NL rats at W13 (p < 0.01, Fig. 2b).
Fig. 2.
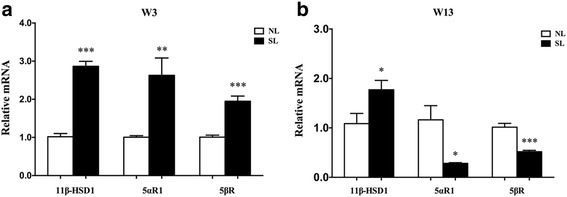
mRNA expression of 11β-HSD1, 5αR1 and 5βR at W3 (a) and W13 (b). Data are expressed as the mean ± SEM. Significant differences between groups at W3 or W13 were analyzed by two-sided Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. NL rats. n = 6 in each NL and SL group
mRNA expression of rate-limiting enzymes in hepatic tissue at W3 and W13
Hepatic ACC, SCD1, FASN and SREBP-1c mRNA expression was significantly increased in the SL rats compared to the NL rats at W3 and W13 (p < 0.05, Fig. 3), whereas mRNA expression of LPL and L-FABP mRNA only increased in the SL rats at W3 (p < 0.05, Fig. 3a), but not at W13 (p > 0.05, Fig. 3b). There were no significant differences in the expression of PPARα, CPT1 or MTP between the two groups at W3 or W13 (p > 0.05, Fig. 3).
Fig. 3.
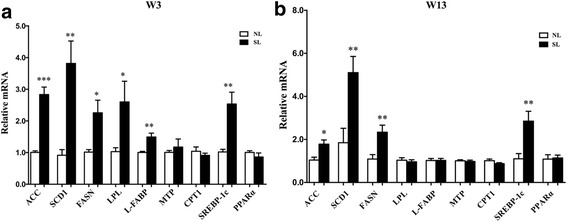
mRNA expression of the genes involved in hepatic lipid metabolism at W3 (a) and W13 (b). Data are expressed as the mean ± SEM. Significant differences between groups at W3 or W13 were analyzed by two-sided Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001 vs. NL rats. n = 6 in each NL and SL group
Effects of GC or/and OA on lipid accumulation in HepG2 cells
Oil red O staining showed that little lipid droplets existed in the normal HepG2 cells, but these intracellular lipid droplets were obviously increased in the cells after treatment with GC or OA; there was more significant lipid accumulation after treatment with GC + OA (Fig. 4a). In addition, Mi treatment attenuated the increase of the lipid accumulation induced by GC or GC + OA (Fig. 4a). Like the lipid droplet accumulation, the TG content in the HepG2 cells increased after treatment with GC or OA or GC + OA and decreased when Mi was added compared to the GC or GC + OA treatment (p < 0.05, Fig. 4b).
Fig. 4.
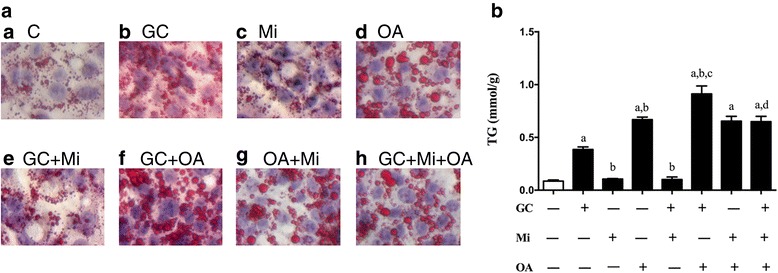
Oil red O staining of cells after treatment with GC, Mi, or GC + Mi within the first 48 h, followed by sodium oleate (OA) intervention (or not) for 24 h (a). The TG contents are shown in (b). Data are expressed as the mean ± SEM. Effects of different treatments were analyzed by two-sided Student’s t-test. ap < 0.05 vs. Control, bp < 0.05 vs. GC; cp < 0.05 vs. OA; dp < 0.05 vs. GC + OA. n = 3 in each treatment
Gene expression in response to GC and OA in HepG2 cells
To determine whether the regulation of hepatic lipid accumulation by GC was mediated by metabolism enzymes, we next examined ACC, SCD1, FASN and SREBP-1c mRNA expression in the HepG2 cells. As expected, the level of ACC mRNA was dependent on the dose and timing of the GC stimulation (p < 0.05, Fig. 5a, b); the optimum concentration and timing were 125 nM for 48 h. SCD1, FASN and SREBP-1c mRNA expression increased in GC stimulation (p < 0.05, Figs. 6a, b, g), but CPT1 expression decreased (p < 0.05, Fig. 6f), and all these alterations were more significant in the GC + OA treatment (p < 0.05, Fig. 7b, c, g, h), as well as ACC (p < 0.05, Fig. 7a). Mi alone (p > 0.05, Fig. 6) and the Mi + OA treatment (p > 0.05, Fig. 7) did not have any effect on the expression of these genes, but Mi attenuated GC or GC + OA induced up-regulation of ACC, SCD1, FASN and SREBP-1c expression (p < 0.05, Figs. 5c, 6, 7) and down-regulation of CPT1 expression (p < 0.05, Figs. 6, 7).
Fig. 5.
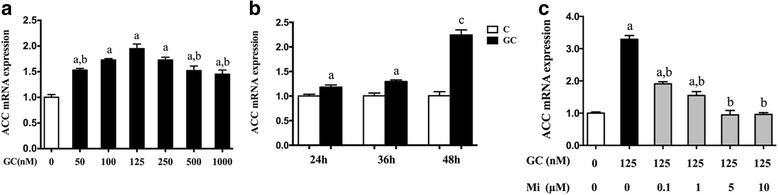
ACC mRNA expression was dependent on the GC treatment concentrations (a), timing (b) and treatment with GC (125 nM) and Mi at different concentrations (c). Data are expressed as the mean ± SEM. Effects of different treatments were analyzed by two-sided Student’s t-test. ap < 0.05 vs. control, bp < 0.05 vs. GC (125 nM), cp < 0.01 vs. control. n = 3 in each treatment
Fig. 6.
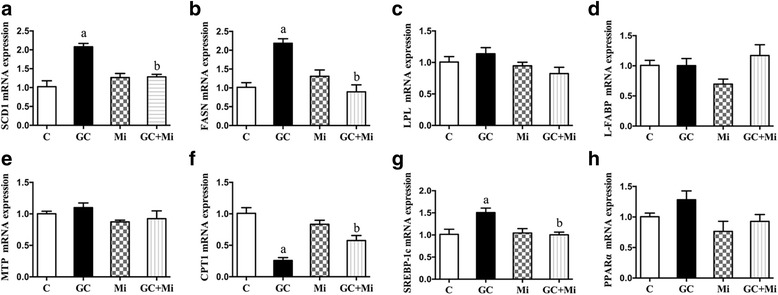
mRNA expression of SCD1 (a), FASN (b), LPL (c), L-FABP (d), MTP (e), CPT1 (f), SREBP-1c (g) and PPARα (h) in GC or Mi or GC + Mi treatment. Data are expressed as the mean ± SEM. Effects of different treatments were analyzed by two-sided Student’s t-test. ap < 0.05 vs. control, bp < 0.05 vs. GC. n = 3 in each treatment
Fig. 7.
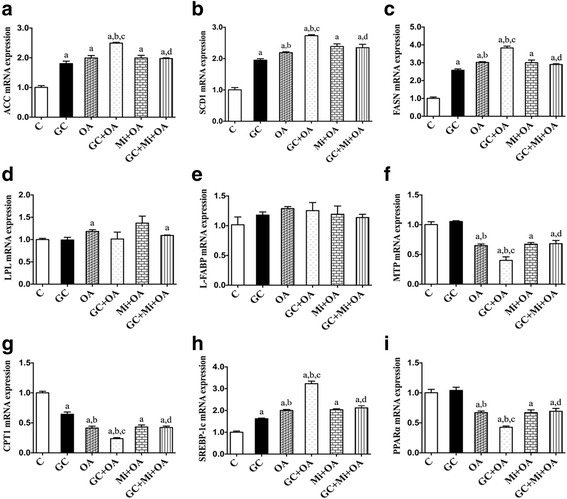
mRNA expression of ACC (a), SCD1 (b), FASN (c), LPL (d), L-FABP (e), MTP (f), CPT1 (g), SREBP-1c (h) and PPARα (i) in GC or OA or GC + OA or Mi + OA or GC + Mi + OA treatments. Data are expressed as the mean ± SEM. Effects of different treatments were analyzed by two-sided Student’s t-test. ap < 0.05 vs. control, bp < 0.05 vs. GC, cp < 0.05 vs. OA, dp < 0.05 vs. GC + OA. n = 3 in each treatment
Discussion
It has become increasingly recognized that the metabolic programming effects of nutritional modifications in early postnatal life are independently related to the development of obesity and MS in later life [31]. Over-nutrition during lactation induces a persistent increase in body weight, hyperinsulinemia, hyperleptinemia and MS in adults, including NAFLD [30, 31]. Consistent with our previous reports, we confirmed that early neonatal overfeeding induced increased mRNA expression of 11β-HSD1, decreased expression of 5αR1 and 5βR and abnormal lipid metabolism in the livers of the SL rats compared to NL rats. The new finding of this study was that exposure to GC increased hepatocyte lipid accumulation by up-regulating the gene mRNA expression of hepatic DNL through GR. We suggest that early postnatal overfeeding induced by SL rearing leads to peripheral GC metabolism activity, which might contribute to the increase in hepatic lipid synthesis in adult rats.
Previous studies have shown the presence of higher 11β-HSD1 mRNA and/or activity in the adipose tissue of obese rodents [24, 25] or humans [23, 26]. 11β-HSD1 is known to be positively associated with features of MS in adults [20]. Transgenic mice with adipose- or liver-specific 11β-HSD1 overexpression exhibit elevated intra-adipose and portal corticosterone levels, abdominal obesity, dyslipidaemia, insulin resistance and hypertension [44, 45]. In our studies, SL adult rats exhibited obesity and increased hepatic 11β-HSD1 overexpression but decreased 5αR1 and 5βR expression, indicating that there are more active GC in hepatic tissue. The high concentrations of GC in the liver could have important effects on lipid metabolism [28, 45, 46]. In the present study, small litters displayed significant increases in liver mass and TG contents compared to NL rats.
GC can lead to hepatic steatosis by decreasing lipid export and oxidation [47, 48], increasing cholesterol synthesis and fatty acid uptake [49, 50] or increasing lipid biosynthesis [28, 29]. In addition to the changes of 11β-HSD1, 5αR1 and 5βR in the liver, we also found that postnatal overfeeding induced a significant increase in DNL by SREBP-1c, ACC, SCD1 and FASN overexpression in the liver from weaning to adulthood, which might be an important mechanism underlying the development and progression of NAFLD in adulthood, that is, overexposure to GC through 11β-HSD1 up-regulation in the liver.
In line with our hypothesis that overexposure to GC induces an increase in DNL in hepatocytes, we found that both lipid accumulation and TG content in HepG2 cells were significantly increased by GC treatment via DNL increase through SREBP-1c, ACC, SCD1 and FASN overexpression. Therefore, the augmented active GC induced by the increase in 11β-HSD1 might be an important factor responsible for the increased DNL in the livers of SL-reared rats. Because the effects of GC were mainly mediated via the GR, which is a member of the steroid hormone receptor superfamily [51, 52], we used Mi, the GR antagonist [53, 54], and confirmed that most of the hepatic lipid metabolism changes induced by GC were inhibited by Mi. Thus, we suggest that GC could increase lipid accumulation by increasing DNL through its receptor in the hepatocytes.
Although postnatal overfeeding can alter lipid metabolism in the liver, a high-fat diet is central to the onset of NAFLD [55, 56]. In our previous studies, we found that neonatal overfeeding in rats induced by SL rearing increased their vulnerability to a HF diet from post-suckling to adulthood and promoted early onset and exaggeration of HF diet-induced NAFLD [38]. Moreover, we found that SL and a high-fat diet exhibited a significant interaction with regard to 11β-HSD1 expression, but hepatic 11β-HSD1 expression was not observed in NL-HF rats [57]. We suggest that the the increased activity of the GC induced by 11β-HSD1 and a HF diet have a significant interaction on lipid metabolism in the liver. In the present study, we found that the GC + OA treatment in vitro resulted in the most significant lipid accumulation and DNL increase in HepG2 cells compared to separate GC or OA treatments.
Previous studies have shown that during energy overconsumption, LPL and L-FABP expression increased in the liver [58, 59], but CPT1and MTP decreased [60, 61]; all these alterations could contribute to the occurrence of NAFLD [27, 62]. In the present study, we found that CPT1 decreased after GC treatment in vitro, but it did not change at W13 in the SL rats. Notably, our previous observation indicated that CPT1 decreased at W16 in the SL rats [38], suggesting that long-term overexposure to GC also affected lipid oxidation in the hepatocytes. Furthermore, the transient elevation of LPL and L-FABP mRNA expression in the SL rats (at W3) might be due to the excessive food intake; it did not change after weaning in vivo or after GC overexposure in vitro. There was also no change in MTP or PPARα caused by GC overexposure either in vivo or in vitro. Therefore, we suggest that GC overexposure in the SL rat model and HepG2 cells augmented the hepatic lipid accumulation mainly through DNL increase.
Conclusions
Postnatal overfeeding induced GC overexposure through 11β-HSD1 up-regulation in the liver, and the GC activated the hepatic DNL by GR. This resulted in hepatic lipid accumulation, leading to an increased risk of NAFLD during adulthood. More animal and clinical studies are needed to examine the prolonged effects of manipulating the availability of pre-receptor GC and the mechanisms of GR activation in the liver. Specifically, we suggest that targeting pre-receptor GC activation in the liver may provide a novel approach to the treatment of NAFLD, particularly in childhood.
Acknowledgments
Funding
This work was financially supported by the National Natural Science Foundation of China (81273064, 81773421), the Innovation Team of Jiangsu Health (CXTDA2017035) and Jiangsu Province Social Development Research (BE2015607).
Availability of data and materials
Data are all contained within the article.
Abbreviations
- 11β-HSD1
11β-hydroxysteroid dehydrogenase 1
- 5αR1
5α-reductase type 1
- 5βR
5β-reductase
- ACC
Acetyl-CoA carboxylase 1
- ANOVA
Analysis of variance
- CPT1
Carnitine palmitoyltransferase
- DNL
De novo lipogenesis
- FASN
Fatty acid synthase
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GC
Glucocorticoid
- GR
Glucocorticoid receptor
- HF
High-fat
- HSI
Hepatosomatic index
- L-FABP
Liver fatty acid-binding protein
- LPL
Lipoprotein lipase
- Mi
Mifepristone
- MS
Metabolic syndrome
- MTP
Microsomal triglyceride transfer protein
- NAFLD
Non-alcoholic fatty liver disease
- NL
Normal litter
- OA
Sodium oleate
- P3
Postnatal day 3
- PPARα
Peroxisome proliferator-activated receptor α
- SCD1
Stearoyl-CoA desaturase-1
- SL
Small litter
- SREBP-1c
Sterol regulatory element binding protein-1c
- TG
Triglyceride
- W13
Postnatal week 13
- W3
Postnatal week 3
Authors’ contributions
FY and XL conceived and designed the experiments. FY and CM performed the animal experiment. FY and YD performed the cell experiment. YD and CM performed the analyses. FY and XL wrote the paper. YD and CM reviewed the manuscript. All authors approved the manuscript.
Ethics approval and consent to participate
All animal studies were performed following the guidelines established by the University Committee on the Use and Care of Animals and were overseen by the Unit for Laboratory Animal Medicine at Nanjing Medical University (IACUC: 14030102).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Fan Yang, Email: yangfan87241586@126.com.
Yanyan Dai, Email: kkxx82myt@163.com.
Cuiting Min, Email: min_cuiting@163.com.
Xiaonan Li, Email: xiaonan6189@163.com.
References
- 1.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 2.Symonds ME, Sebert SP, Hyatt MA, Budge H. Nutritional programming of the metabolic syndrome. Nat Rev Endocrinol. 2009;5:604–610. doi: 10.1038/nrendo.2009.195. [DOI] [PubMed] [Google Scholar]
- 3.Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut. 2009;58:1538–1544. doi: 10.1136/gut.2008.171280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goyal NP, Schwimmer JB. The progression and natural history of pediatric nonalcoholic fatty liver disease. Clin Liver Dis. 2016;20:325–338. doi: 10.1016/j.cld.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Zhao Y, Peng L, Yang LC, Xu XD, Li WJ, Luo XM, Jin X. Wedelolactone regulates lipid metabolism and improves hepatic steatosis partly by AMPK activation and up-regulation of expression of PPARalpha/LPL and LDLR. PLoS One. 2015;10:e0132720. doi: 10.1371/journal.pone.0132720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hostetler HA, McIntosh AL, Atshaves BP, Storey SM, Payne HR, Kier AB, Schroeder F. L-FABP directly interacts with PPARalpha in cultured primary hepatocytes. J Lipid Res. 2009;50:1663–1675. doi: 10.1194/jlr.M900058-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding RB, Bao J, Deng CX. Emerging roles of SIRT1 in fatty liver diseases. Int J Biol Sci. 2017;13:852–867. doi: 10.7150/ijbs.19370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carneiro L, Asrih M, Repond C, Sempoux C, Stehle JC, Leloup C, Jornayvaz FR, Pellerin L. AMPK activation caused by reduced liver lactate metabolism protects against hepatic steatosis in MCT1 haploinsufficient mice. Mol Metab. 2017;6:1625–1633. doi: 10.1016/j.molmet.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Xiao G, Trujillo C, Chang V, Blanco L, Joseph SB, Bassilian S, Saad MF, Tontonoz P, Lee WN, Kurland IJ. Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J Biol Chem. 2002;277:50237–50244. doi: 10.1074/jbc.M201208200. [DOI] [PubMed] [Google Scholar]
- 12.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha) J Biol Chem. 1998;273:5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 13.Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Fast DG, Attie AD. The enzymatic and non-enzymatic roles of protein-disulfide isomerase in apolipoprotein B secretion. J Biol Chem. 1997;272:27644–27651. doi: 10.1074/jbc.272.44.27644. [DOI] [PubMed] [Google Scholar]
- 15.Stefano JT, de Oliveira CP, Correa-Giannella ML, de Lima VM, de Sa SV, de Oliveira EP, de Mello ES, Giannella-Neto D, Alves VA, Carrilho FJ. Nonalcoholic steatohepatitis (NASH) in ob/ob mice treated with yo jyo hen shi ko (YHK): effects on peroxisome proliferator-activated receptors (PPARs) and microsomal triglyceride transfer protein (MTP) Dig Dis Sci. 2007;52:3448–3454. doi: 10.1007/s10620-007-9810-8. [DOI] [PubMed] [Google Scholar]
- 16.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han H, Qiu F, Zhao H, Tang H, Li X, Shi D. Dietary flaxseed oil prevents western-type diet-induced nonalcoholic fatty liver disease in apolipoprotein-E knockout mice. Oxidative Med Cell Longev. 2017;2017:3256241. doi: 10.1155/2017/3256241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasiri M, Nikolaou N, Parajes S, Krone NP, Valsamakis G, Mastorakos G, Hughes B, Taylor A, Bujalska IJ, Gathercole LL, Tomlinson JW. 5alpha-reductase type 2 regulates glucocorticoid action and metabolic phenotype in human hepatocytes. Endocrinology. 2015;156:2863–2871. doi: 10.1210/en.2015-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taves MD, Gomez-Sanchez CE, Soma KK. Extra-adrenal glucocorticoids and mineralocorticoids: evidence for local synthesis, regulation, and function. Am J Physiol Endocrinol Metab. 2011;301:E11–E24. doi: 10.1152/ajpendo.00100.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 21.Sheppard KE. Corticosteroid receptors, 11 beta-hydroxysteroid dehydrogenase, and the heart. Vitam Horm. 2003;66:77–112. doi: 10.1016/S0083-6729(03)01003-3. [DOI] [PubMed] [Google Scholar]
- 22.Chapman K, Holmes M, Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93:1139–1206. doi: 10.1152/physrev.00020.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrus P, Rosqvist F, Edholm D, Mejhert N, Arner P, Dahlman I, Ryden M, Sundbom M, Riserus U. Saturated fatty acids in human visceral adipose tissue are associated with increased 11- beta-hydroxysteroid-dehydrogenase type 1 expression. Lipids Health Dis. 2015;14:42–46. doi: 10.1186/s12944-015-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Zhou Y, Zhu AY, Li XQ, Mundt SS, Gao L, Lisnock J, Hernandez M, Alonso-Galicia M, Springer MS, O'Neill EA, Daugherty BL, Puig O. Metabolic syndrome in mice induced by expressing a transcriptional activator in adipose tissue. Transgenic Res. 2012;21:633–644. doi: 10.1007/s11248-011-9562-2. [DOI] [PubMed] [Google Scholar]
- 25.Man TY, Michailidou Z, Gokcel A, Ramage L, Chapman KE, Kenyon CJ, Seckl JR, Morton NM. Dietary manipulation reveals an unexpected inverse relationship between fat mass and adipose 11beta-hydroxysteroid dehydrogenase type 1. Am J Physiol Endocrinol Metab. 2011;300:E1076–E1084. doi: 10.1152/ajpendo.00531.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson AJ, Andrew R, Homer NZ, Jones GC, Smith K, Livingstone DE, Walker BR, Stimson RH. Metformin increases cortisol regeneration by 11betaHSD1 in obese men with and without type 2 diabetes mellitus. J Clin Endocrinol Metab. 2016;101:3787–3793. doi: 10.1210/jc.2016-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woods CP, Hazlehurst JM, Tomlinson JW. Glucocorticoids and non-alcoholic fatty liver disease. J Steroid Biochem Mol Biol. 2015;154:94–103. doi: 10.1016/j.jsbmb.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 28.Staels B, van Tol A, Chan L, Verhoeven G, Auwerx J. Variable effects of different corticosteroids on plasma lipids, apolipoproteins, and hepatic apolipoprotein mRNA levels in rats. Arterioscler Thromb. 1991;11:760–769. doi: 10.1161/01.ATV.11.3.760. [DOI] [PubMed] [Google Scholar]
- 29.Glenny HP, Brindley DN. The effects of cortisol, corticotropin and thyroxine on the synthesis of glycerolipids and on the phosphatidate phosphohydrolase activity in rat liver. Biochem J. 1978;176:777–784. doi: 10.1042/bj1760777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ojha S, Saroha V, Symonds ME, Budge H. Excess nutrient supply in early life and its later metabolic consequences. Clin Exp Pharmacol Physiol. 2013;40:817–823. doi: 10.1111/1440-1681.12061. [DOI] [PubMed] [Google Scholar]
- 31.Patel MS, Srinivasan M. Metabolic programming in the immediate postnatal life. Ann Nutr Metab. 2011;58(Suppl 2):18–28. doi: 10.1159/000328040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glavas MM, Kirigiti MA, Xiao XQ, Enriori PJ, Fisher SK, Evans AE, Grayson BE, Cowley MA, Smith MS, Grove KL. Early overnutrition results in early-onset arcuate leptin resistance and increased sensitivity to high-fat diet. Endocrinology. 2010;151:1598–1610. doi: 10.1210/en.2009-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srinivasan M, Aalinkeel R, Song F, Mitrani P, Pandya JD, Strutt B, Hill DJ, Patel MS. Maternal hyperinsulinemia predisposes rat fetuses for hyperinsulinemia, and adult-onset obesity and maternal mild food restriction reverses this phenotype. Am J Physiol Endocrinol Metab. 2006;290:E129–E134. doi: 10.1152/ajpendo.00248.2005. [DOI] [PubMed] [Google Scholar]
- 34.Khulan B, Drake AJ. Glucocorticoids as mediators of developmental programming effects. Best Pract Res Clin Endocrinol Metab. 2012;26:689–700. doi: 10.1016/j.beem.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Lindsay RS, Lindsay RM, Waddell BJ, Seckl JR. Prenatal glucocorticoid exposure leads to offspring hyperglycaemia in the rat: studies with the 11 beta-hydroxysteroid dehydrogenase inhibitor carbenoxolone. Diabetologia. 1996;39:1299–1305. doi: 10.1007/s001250050573. [DOI] [PubMed] [Google Scholar]
- 36.Lesage J, Del-Favero F, Leonhardt M, Louvart H, Maccari S, Vieau D, Darnaudery M. Prenatal stress induces intrauterine growth restriction and programmes glucose intolerance and feeding behaviour disturbances in the aged rat. J Endocrinol. 2004;181:291–296. doi: 10.1677/joe.0.1810291. [DOI] [PubMed] [Google Scholar]
- 37.Hou M, Liu Y, Zhu L, Sun B, Guo M, Buren J, Li X. Neonatal overfeeding induced by small litter rearing causes altered glucocorticoid metabolism in rats. PLoS One. 2011;6:e25726. doi: 10.1371/journal.pone.0025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji C, Dai Y, Jiang W, Liu J, Hou M, Wang J, Buren J, Li X. Postnatal overfeeding promotes early onset and exaggeration of high-fat diet-induced nonalcoholic fatty liver disease through disordered hepatic lipid metabolism in rats. J Nutr Biochem. 2014;25:1108–1116. doi: 10.1016/j.jnutbio.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Boullu-Ciocca S, Achard V, Tassistro V, Dutour A, Grino M. Postnatal programming of glucocorticoid metabolism in rats modulates high-fat diet-induced regulation of visceral adipose tissue glucocorticoid exposure and sensitivity and adiponectin and proinflammatory adipokines gene expression in adulthood. Diabetes. 2008;57:669–677. doi: 10.2337/db07-1316. [DOI] [PubMed] [Google Scholar]
- 40.Tirelli E, Laviola G, Adriani W. Ontogenesis of behavioral sensitization and conditioned place preference induced by psychostimulants in laboratory rodents. Neurosci Biobehav Rev. 2003;27:163–178. doi: 10.1016/S0149-7634(03)00018-6. [DOI] [PubMed] [Google Scholar]
- 41.Dai Y, Yang F, Zhou N, Sha L, Zhou S, Wang J, Li X. A post-weaning fish oil dietary intervention reverses adverse metabolic outcomes and 11beta-hydroxysteroid dehydrogenase type 1 expression in postnatal overfed rats. Br J Nutr. 2016;116:1519–1529. doi: 10.1017/S0007114516003718. [DOI] [PubMed] [Google Scholar]
- 42.Plagemann A, Harder T, Schellong K, Schulz S, Stupin JH. Early postnatal life as a critical time window for determination of long-term metabolic health. Best Pract Res Clin Endocrinol Metab. 2012;26:641–653. doi: 10.1016/j.beem.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 43.Wootton RJ, Evans GW, Mills L. Annual cycle in female three-spined sticklebacks (Gasterosteus aculeatus L.) from an upland and lowland population. J Fish Biol. 2010;12:331–343. doi: 10.1111/j.1095-8649.1978.tb04178.x. [DOI] [Google Scholar]
- 44.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 45.Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, Seckl JR, Mullins JJ. Metabolic syndrome without obesity: hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci U S A. 2004;101:7088–7093. doi: 10.1073/pnas.0305524101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Guia RM, Rose AJ, Sommerfeld A, Seibert O, Strzoda D, Zota A, Feuchter Y, Krones-Herzig A, Sijmonsma T, Kirilov M, Sticht C, Gretz N, Dallinga-Thie G, Diederichs S, Kloting N, Bluher M, Berriel Diaz M, Herzig S. microRNA-379 couples glucocorticoid hormones to dysfunctional lipid homeostasis. EMBO J. 2015;34:344–360. doi: 10.15252/embj.201490464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bocharov AV, Huang W, Vishniakova TG, Zaitseva EV, Frolova EG, Rampal P, Bertolotti R. Glucocorticoids upregulate high-affinity, high-density lipoprotein binding sites in rat hepatocytes. Metabolism. 1995;44:730–738. doi: 10.1016/0026-0495(95)90185-X. [DOI] [PubMed] [Google Scholar]
- 48.Letteron P, Brahimi-Bourouina N, Robin MA, Moreau A, Feldmann G, Pessayre D. Glucocorticoids inhibit mitochondrial matrix acyl-CoA dehydrogenases and fatty acid beta-oxidation. Am J Phys. 1997;272:G1141–G1150. doi: 10.1152/ajpgi.1997.272.5.G1141. [DOI] [PubMed] [Google Scholar]
- 49.Carr BR, Simpson ER. Cholesterol synthesis by human fetal hepatocytes: effects of hormones. J Clin Endocrinol Metab. 1984;58:1111–1116. doi: 10.1210/jcem-58-6-1111. [DOI] [PubMed] [Google Scholar]
- 50.D'Souza AM, Beaudry JL, Szigiato AA, Trumble SJ, Snook LA, Bonen A, Giacca A, Riddell MC. Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am J Physiol Gastrointest Liver Physiol. 2012;302:G850–G863. doi: 10.1152/ajpgi.00378.2011. [DOI] [PubMed] [Google Scholar]
- 51.Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uchoa ET, Aguilera G, Herman JP, Fiedler JL, Deak T, de Sousa MB. Novel aspects of glucocorticoid actions. J Neuroendocrinol. 2014;26:557–572. doi: 10.1111/jne.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Molitch ME. Diagnosis and treatment of pituitary adenomas: a review. JAMA. 2017;317:516–524. doi: 10.1001/jama.2016.19699. [DOI] [PubMed] [Google Scholar]
- 54.Jiang Z, Qian L, Zou H, Jia Y, Ni Y, Yang X, Jiang Z, Zhao R. Porcine glucocorticoid receptor (NR3C1) gene: tissue-specificity of transcriptional strength and glucocorticoid responsiveness of alternative promoters. J Steroid Biochem Mol Biol. 2014;141:87–93. doi: 10.1016/j.jsbmb.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 55.Morgan K, Uyuni A, Nandgiri G, Mao L, Castaneda L, Kathirvel E, French SW, Morgan TR. Altered expression of transcription factors and genes regulating lipogenesis in liver and adipose tissue of mice with high fat diet-induced obesity and nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2008;20:843–854. doi: 10.1097/MEG.0b013e3282f9b203. [DOI] [PubMed] [Google Scholar]
- 56.Nakamura A, Terauchi Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int J Mol Sci. 2013;14:21240–21257. doi: 10.3390/ijms141121240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hou M, Ji C, Wang J, Liu Y, Sun B, Guo M, Buren J, Li X. The effects of dietary fatty acid composition in the post-sucking period on metabolic alterations in adulthood: can omega3 polyunsaturated fatty acids prevent adverse programming outcomes? J Endocrinol. 2012;215:119–127. doi: 10.1530/JOE-12-0191. [DOI] [PubMed] [Google Scholar]
- 58.Ahn J, Lee H, Chung CH, Ha T. High fat diet induced downregulation of microRNA-467b increased lipoprotein lipase in hepatic steatosis. Biochem Biophys Res Commun. 2011;414:664–669. doi: 10.1016/j.bbrc.2011.09.120. [DOI] [PubMed] [Google Scholar]
- 59.Yang SY, Zhao NJ, Li XJ, Zhang HJ, Chen KJ, Li CD. Ping-tang recipe (平糖方) improves insulin resistance and attenuates hepatic steatosis in high-fat diet-induced obese rats. Chin J Integr Med. 2012;18:262–268. doi: 10.1007/s11655-012-1023-0. [DOI] [PubMed] [Google Scholar]
- 60.Nakade Y, Sakamoto K, Yamauchi T, Inoue T, Kobayashi Y, Yamamoto T, Ishii N, Ohashi T, Sumida Y, Ito K, Nakao H, Fukuzawa Y, Umezawa K, Yoneda M. Conophylline inhibits non-alcoholic steatohepatitis in mice. PLoS One. 2017;12:e0178436. doi: 10.1371/journal.pone.0178436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jia Y, Yee JK, Wang C, Nikolaenko L, Diaz-Arjonilla M, Cohen JN, French SW, Liu PY, Lue Y, Lee WP, Swerdloff RS. Testosterone protects high fat/low carbohydrate diet induced non-alcoholic fatty liver disease in castrated male rats mainly via modulating ER stress. Am J Physiol Endocrinol Metab. 2018;314:E366–E376. doi: 10.1152/ajpendo.00124.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Higuchi N, Kato M, Tanaka M, Miyazaki M, Takao S, Kohjima M, Kotoh K, Enjoji M, Nakamuta M, Takayanagi R. Effects of insulin resistance and hepatic lipid accumulation on hepatic mRNA expression levels of apoB, MTP and L-FABP in non-alcoholic fatty liver disease. Exp Ther Med. 2011;2:1077–1081. doi: 10.3892/etm.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are all contained within the article.