

Abstract
Single-electron reduction of C=O and C=N bonds of aldehydes, ketones, and imines results in the formation of ketyl and α-aminoalkyl anion radicals, respectively. These reactive intermediates are characterized by an altered electronic character with respect to their parent molecules and undergo a diverse range of synthetically useful transformations, which are not available to even-electron species. This Review summarizes the reactions of ketyl and α-aminyl radicals generated from carbonyl derivatives under transition-metal photoredox-catalysed conditions. We primarily focus on recent developments in the field, as well as give a brief overview of catalytic enantioselective transformations that provide means to achieve precise stereocontrol over the reactivity of ion radicals.
Introduction
Aldehydes, ketones, and imines are important intermediates for the assembly of complex molecules. Conventional synthetic protocols usually take advantage of the strong electrostatic polarization of carbon-heteroatom double bond of carbonyl (C=O) and iminyl (C=N) groups, which places a partial positive charge at the carbon atom, rendering the atom electrophilic and thus susceptible to undergo a nucleophilic attack. Methods that alter this natural reactivity pattern of carbonyl derivatives and enable them to engage in carbon-carbon bond forming reactions with non-nucleophilic partners have provided a paradigm shift in organic synthesis.1, 2 Among the strategies reported in the literature, the formation of ketyl radicals via single electron reduction of carbonyl derivatives has emerged as an appealing route to access a wide range of valuable molecular architectures. 3 However, a widespread application of ketyl radicals in synthesis has been hindered by (i) the highly negative reduction potential 4 of aldehydes (E1/2red = −1.93 V vs. SCE for benzaldehyde),5 ketones (E1/2red = −2.11 V vs. SCE for acetophenone), 5 and imines (E1/2red = −1.91 V vs. SCE for N-benzylideneaniline) 5 (Scheme 1) and (ii) the requirement to employ toxic, air- and moisture-sensitive reducing agents, and harsh reaction conditions to generate the ketyl and α-aminoalkyl anion radical intermediates. Accordingly, early examples of reductive coupling reactions between carbonyl derivatives and alkenes/alkynes were performed using very strong reductants such as alkali 6 and alkali earth 7 metals, tin, 8-13 zinc,14, 15 titanium,16 and samarium reagents,17-22 as well as under electrochemical 23-26 and photochemical 27-32 conditions. Due to the severe drawbacks of these strategies, such as the requirement for a strong reductant and generation of a stoichiometric amount of organometallic by-products, there has been a growing demand for development of new methods to access ketyl and α-aminoalkyl anion radicals under mild and eco-compatible reaction conditions. In order to address these issues, a number of reductive coupling reactions of carbonyl derivatives involving a catalytic amount of early transition (e.g., Ti, V) or lanthanide metals (e.g., Sm) have been developed over the years and applied in natural product synthesis. 3, 33-38 Nonetheless, complementary strategies to generate ketyl and α-aminoalkyl anion radical intermediates and to harness their unusual reactivity are still highly sought after.
Scheme 1.

Electrochemical series of selected carbonyl derivatives; potentials are reported against SCE.68
Over the past few decades, visible light photoredox catalysis has emerged as an attractive alternative to traditional ways of generating radical intermediates. 39-43 The use of photoredox catalysts, which, upon photoexcitation with visible light, can engage in single electron transfer (SET) processes with organic substrates, obviates the need for radical initiators and a stoichiometric amount of strong reducing agents. Since RuII(bpy)3Cl2 was first reported to engage in a SET with aromatic carbonyl compounds in the late 1970s,44 visible light organometallic photoredox catalysis has become widely recognized as an efficient way to generate ketyl 45-62 and α-aminoalkyl anion radicals under mild reaction conditions.58, 63-65
Since most of the photoexcited catalysts or the reduced forms thereof are not sufficiently reducing (E1/2red = −1.33 V vs. SCE for Ru)66 to reduce a carbonyl or iminyl species (E1/2red = −1.93 V vs. SCE for benzaldehyde) (Scheme 1),5 the majority of the reported examples of photoredox-catalysed formation of ketyl or α-aminoalkyl anion radicals require Brønsted acids,49, 53, 54 Lewis acids,47, 49, 50, 52, 55, 57, 62 or acids generated in situ,56, 59-61 as activators to promote the electron transfer step. With a Brønsted acid additive, proton-coupled electron transfer (PCET),67 which involves a simultaneous transfer of a proton and an electron to an organic substrate in a concerted process, enables a single-electron reduction of compounds with very negative reduction potentials. In reactions that exploit PCET, an interaction between a carbonyl/iminyl group and an acid activator lowers the energy barrier during the SET process, and thus facilitates the formation of a ketyl/α-aminoalkyl anion radical intermediates, which would otherwise be inaccessible via separate proton- and electron-transfer steps.53, 54, 69-71
Scope of the review
Over the past decade, ketyl and α-aminoalkyl anion radicals generated from aldehydes, ketones, and imines under photoredox conditions have been shown to undergo a diverse array of new C–C bond forming transformations, which provided convenient synthetic routes to medicinally relevant building blocks.72 Most remarkably, the area of catalytic asymmetric radical coupling reactions has enjoyed tremendous growth, and thus a number of these ketyl/α-amino radical reactions have been accomplished in an enantioselective fashion. The purpose of this Review is to provide an overview of the important precedents that laid the foundation for photocatalytic coupling reactions of carbonyl and iminyl derivatives, and the recent (2008-2017) progress in the field. Radical-radical coupling reactions, radical additions to π-systems, as well as other reactions initiated by a single-electron reduction of carbonyl or iminyl moieties along with their proposed mechanisms, are discussed herein.
Functionalization of α-C–X bond of ketones via ketyl radical intermediates
In late 1970s, Kellogg and coworkers published their seminal studies describing the reduction of phenacyl onium salts (sulfonium, ammonium, phosphonium) with N-substituted 1,4-dihydropyridines in the presence of RuII(bpy)2Cl2 (Scheme 2).44, 73 While the reaction was very sluggish in the dark or in the absence of a photoredox catalyst, a significant rate enhancement was observed upon addition of a catalytic amount of RuII(bpy)3Cl2 to the reaction mixture. In addition to having higher rates, the reactions performed in the presence of RuII(bpy)3Cl2 were characterized by their cleanness and a lack of competing rearrangement of N-substituted 1,4-dihydropyridines.
Scheme 2.

Photoredox-catalysed reduction of phenacyl onium salts with N-substituted 1,4-dihydropyridines.
Following the initial Kellogg's report, the ability of photoredox catalysts to reduce ketone-activated α-C–X bonds, where X = Cl, Br, N, O, S, P, has been widely explored.61, 73-78 Single-electron transfer to a α-carbonyl compound generates an anion radical, which can be represented either as a carbon radical bonded directly to a negatively charged oxygen 3.2, or its mesomeric structure containing a radical positioned on oxygen attached to a negatively charged carbon atom 3.3 (Scheme 3). This anion radical is known to undergo mesolysis – a fragmentation to afford an anion, X−, and the α-carbonyl radical 3.4. The α-carbonyl radical can then either abstract a hydrogen atom to yield a reduced product, or undergo a further transformation, such as a coupling reaction with another radical or an addition to a π-bond.
Scheme 3.

Proposed mechanism for a single-electron reduction of α-heteroatom substituted carbonyl compounds.
An interesting example of a photoredox reduction of α-halocarbonyl reduction was reported in 1990 by Fukuzumi and co-workers.77 Phenacyl halides (bromides and chlorides) were efficiently converted to acetophenones using RuII(bpy)3Cl2 as a photocatalyst and 10-methyl-9,10-dihydroacridine as the stoichiometric reductant (Scheme 4). The authors also observed that the addition of perchloric acid improved the product yields.
Scheme 4.

Reductive dehalogenation of phenacyl bromides in the absence and presence of an acid additive.77
Based on the quenching experiments, the authors proposed that in the absence of acid, the reaction proceeds via reductive quenching of *RuII(bpy)32+ by ArcH2 (5.4) (E1/2red= +0.80 V vs. SCE)79 generating RuI(bpy)3+ reductant and cation radical ArcH2•+ (5.5) (Scheme 5). The Ru(I) species may then reduce phenacyl bromide (5.1) to form α-carbonyl radical 5.2 and RuII(bpy)32+ species. The α-carbonyl radical can then abstract a hydrogen atom from cation radical ArcH2•+ (5.5) to generate the final product 5.3 and by-product ArcH+ (5.6). Conversely, in the presence of HClO4, *RuII(bpy)32+ reduces either protonated or acid–activated (via PCET) phenacyl halides producing strongly oxidizing RuIII(bpy)33+in situ. Ru(III) can then oxidize ArcH2 (5.4) (or ArcH3+ at a high concentration of the acid) to form ArcH2•+ (5.5) and regenerate Ru(II). The acetophenone product (5.3) is generated upon one-electron reduction of a ketyl radical 5.9 by ArcH• (5.7) followed by the loss of bromide and keto-enol tautomerization.
Scheme 5.
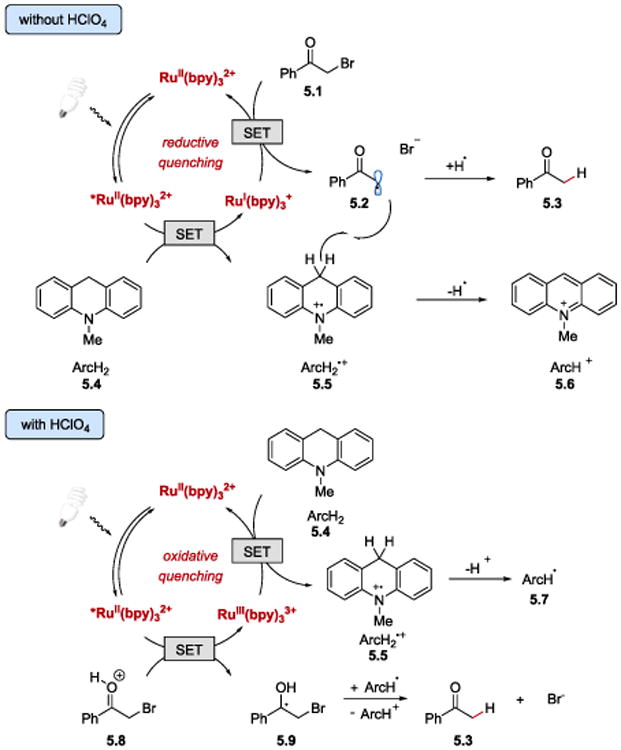
Electron transfer pathways in photoredox reduction of phenacyl halides by dihydroacridine derivatives with and without perchloric acid.77
The ability of photoredox catalysts to reduce α-heteroatom substituted ketones has also been utilized to achieve reductive opening of epoxides and aziridines (Scheme 6).61 Due to the highly negative redox potentials of epoxides (E1/2red = −2.27 V vs. Ag/AgI for stilbene oxide in DMF),80 which precludes a direct electron transfer to the oxirane from Ru(I) or Ir(II), the authors installed an α-carbonyl moiety as a relay. Positioning an epoxide or an aziridine next to the redox-active carbonyl group enabled their reductive opening with RuII(bpy)32+/IrIII(ppy)2(dtbppy)+ photocatalysts and Hantzsch ester (HEH) as a stoichiometric reductant. By performing the reaction in the presence of allyl sulfone, it was possible to obtain products of a reduction/allylation sequence.
Scheme 6.

Photocatalytic reductive ring opening of epoxides and aziridines and reductive allylation reaction.61
Reductive opening of epoxides and aziridines 7.1 proceeds via a reductive quenching cycle of *RuII(bpy)32+ (Scheme 7). SET from HEH• (7.7) to *RuII(bpy)32+ forms a strong reductant, RuI(bpy)3+. Reduction of epoxychalcone or α-ketoaziridine 7.1 by RuI(bpy)3+ delivers a ketyl radical 7.2 and regenerates RuII(bpy)32+ catalyst. Subsequently, ketyl radical 7.2 undergoes C–O or C–N bond cleavage to form anion radical 7.3. Protonation by PyH+ (7.8) and a hydrogen atom abstraction from HEH (7.6) affords the β-hydroxy or β-aminoketone product 7.5. Instead of undergoing hydrogen atom abstraction, the intermediate α-carbonyl radical 7.3 can engage in allylation process with 7.10 to form a new C–C bond. This tandem ring-opening/allylation reaction affords β-hydroxy-α-allylketones 7.11 in good yields and high diastereoselectivity. It was found that the reduction of both epoxides and aziridines required an aryl substituent on the carbonyl group in order to enable the formation of a ketyl radical intermediate.
Scheme 7.
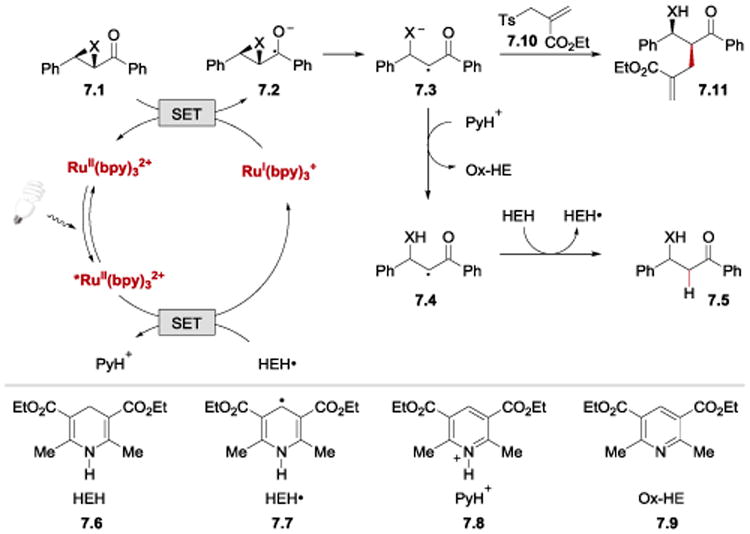
Plausible mechanism of the photocatalytic reductive ring opening of epoxides and aziridines and reductive allylation.61, 81
In 2016, Ma and Chen coupled a variety of carbonyl-group activated tertiary alkyl bromides with 4-alkyl Hantzsch esters and 4-alkyl Hantzsch nitriles under photocatalytic conditions to synthesize congested ketones, including ketones with all-carbon quaternary centers (Scheme 8).82 In their approach, 4-substituted Hantzsch derivatives were used as alkyl radical precursors, which can release the Csp3-centered alkyl radicals upon a single electron oxidation.
Scheme 8.

Photoredox catalysed synthesis of congested ketones with 4-alkyl Hantzsch esters and 4-alkyl Hantzsch nitriles as precursors for alkyl radicals.82
It was proposed that the reaction starts with the photoexcitation of fac-IrIII(ppy)3. Oxidation of 4-alkyl Hantzsch ester (9.4) by *fac-IrIII(ppy)3 in the presence of base generates radical 9.5 and Ir(II). Aromatization-driven C–C bond cleavage of radical 9.5 83 gives benzylic radical 9.6 (Ar = Ph, E1/2red= −1.43 V vs. SCE)84 and a pyridine byproduct 9.8. SET from the Ir(II) species (E1/2III/II = −2.19 V vs SCE)85 to α-bromoketone 9.1 (R = Ph, E1/2red= −1.65 V vs SCE)82 regenerates Ir(III) and furnishes anion radical 9.2, which subsequently undergoes mesolysis to α-carbonyl radical 9.3 and bromide. Radical recombination between 9.3 and 9.6 affords the product 9.7. An alternative pathway, in which anion radical 9.2 directly recombines with 9.6, is also viable based on computational studies.
In 2017, Landais and co-workers utilized photoredox-catalysis to achieve intramolecular carboarylation of cyclopropenes with phenacyl bromides (Scheme 10).86 Naphthalenones were obtained in moderate yields from both α-bromoacetophenones and heteroaryl α-bromoketones.
Scheme 10.

Photoredox-catalysed intramolecular carboarylation of cyclopropenes.86
The reaction has been proposed to proceed via an oxidative quenching cycle of Ir(III) as depicted in Scheme 11. The α-carbonyl radical 11.2 formed upon reduction of α-bromoacetophenone (11.1) by the excited state photocatalyst, *fac-IrIII(ppy)3 (E1/2IV/*III = −1.73 V vs SCE),85, 87 undergoes addition to cyclopropene 11.3 to generate cyclopropyl radical 11.4. Intramolecular addition of 11.4 to the arene moiety provides cyclohexadienyl radical 11.5, which is then oxidized by Ir(IV) species to 11.6, and deprotonated under basic conditions to afford cyclopropane 11.7. Naphthalenone 11.8 is then obtained upon deprotonation of 11.7 α to the ketone followed by the opening of the cyclopropane ring and protonation.
Scheme 11.
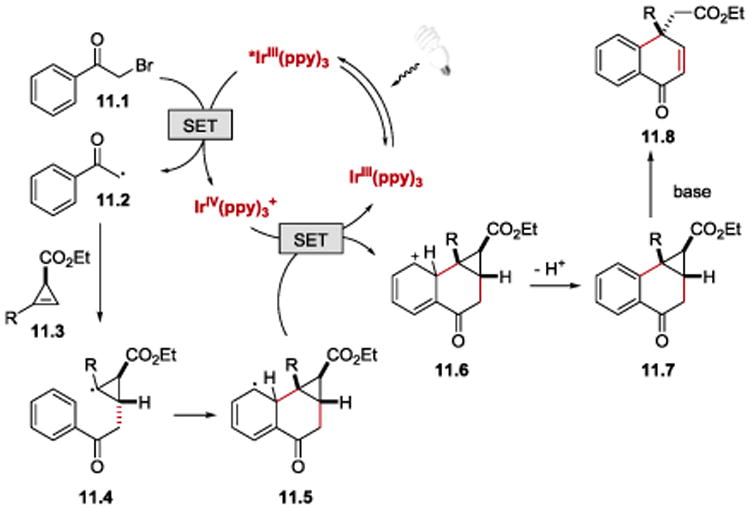
Proposed mechanism for the photoredox-catalysed reaction between phenacyl bromides and cyclopropenes.86
Radical-radical coupling reactions
The earliest literature example which invokes the formation of a ketyl radical and its subsequent recombination with another radical species under photoredox conditions was reported by Pac in 1983. 88 Pac and co-workers described their studies on the reduction of (hetero)aromatic aldehydes and ketones under visible light irradiation with N-benzyl-1,4-dihydronicotinamide (BNAH) as a stoichiometric reductant and RuII(bpy)3Cl2 as a photon-absorbing catalyst (Scheme 12). The authors found that the course of the reaction was strongly dependent on the structural and electronic parameters of the carbonyl compounds used. While benzaldehydes and trifluoroacetophenone were preferentially converted to coupling adducts 12.2, di-2-pyridylketone was almost quantitatively reduced to the corresponding alcohol 12.1 without formation of any adduct. The authors argued that the exclusive reduction of di-2-pyridylketone to di-2-pyridinylmethanol can be attributed to the two pyridyl groups attached to the carbonyl group. The pyridyl substituents are strongly electron-withdrawing and thus facilitate the one-electron reduction of the intermediate radical HO-C•(2-py)2. In addition, the steric hindrance of radical HO-C•(2-py)2, bearing two large pyridyl groups, inhibits the radical coupling reaction and prevents the formation of the adduct 12.2.
Scheme 12.

Ru(bpy)32+-catalysed reactions of carbonyl compounds with BNAH. aYield based on BNAH used.88
It was proposed that the reaction proceeds as outlined in Scheme 13.88, 89 Reduction of excited *RuII(bpy)32+ (E1/2*II/I = +0.77 V vs SCE)66 by BNAH (13.3) (E1/2red = +0.639 V vs SCE)83 gives RuI(bpy)3+ and BNAH•+ (13.4). RuI(bpy)3+ (E1/2red= −1.33 V vs SCE)66 reduces the carbonyl compound 13.1 presumably via PCET to a ketyl radical 13.2. The ketyl radical 13.2 can then recombine with BNA• (13.5) (formed upon deprotonation of BNAH•+) to afford 13.8 or undergo a competitive one-electron reduction by BNA• (13.5) or BNAH (13.3) to generate 13.7. Recombination of two BNA• radicals (13.5) leads to the formation of a side product 13.9.
Scheme 13.
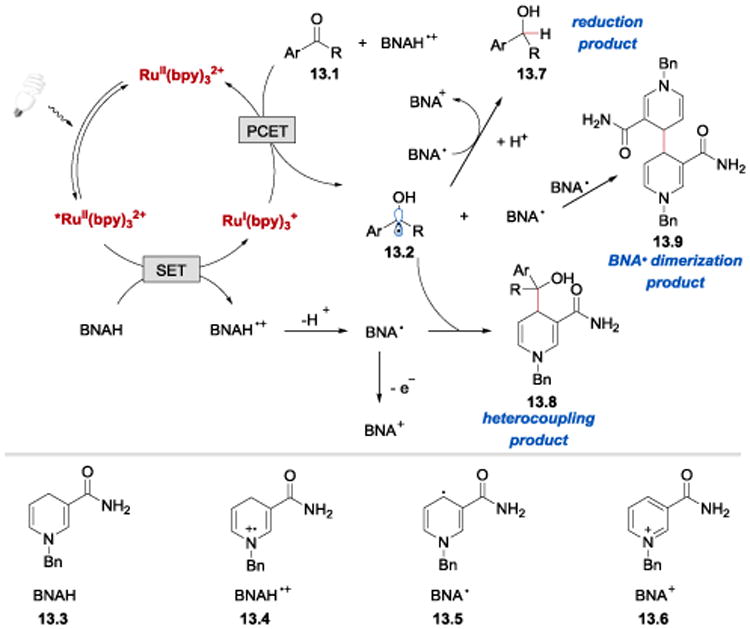
Proposed mechanism of the photoredox-catalysed transformations of carbonyl compounds with BNAH.88-90
Following initial studies by Pac et al. on photoredox-catalysed reactions of aromatic aldehydes and ketones with BNAH, ketyl and α-aminoalkyl anion radicals generated from carbonyl derivatives upon a single-electron reduction have been employed in a number of radical homo-and heterocoupling reactions. For instance, in 2013, MacMillan and co-workers developed a strategy for direct β-functionalization of cyclic ketones with aryl ketones (Scheme 14) by merging photoredox and organocatalysis.52 The products are formed in good yields and moderate to good diastereoselectivities upon coupling of benzophenone with cyclohexanones bearing both alkyl and aryl substituents at the 3-, and 4-positions (Scheme 14). The reaction also works well with cyclopentanone (65% yield); however, lower yields (10-20%) of the desired β-alkyloxy products are observed when 7-membered ketones are employed as substrates. In terms of ketyl radical precursors, the reaction tolerates a range of substituted benzophenones. Aryl–alkyl ketones, however, being more difficult to reduce than biaryl ketones (E1/2red = −2.11 V vs SCE for acetophenone),5 do not undergo the desired coupling reaction when IrIII(ppy)3 is employed as the photocatalyst. Expansion of the scope of ketyl-radical partners and coupling of both electron-rich and electron-poor acetophenone derivatives have been facilitated by the use of IrIII(p-OMe-ppy)3.
Scheme 14.

Direct β-functionalization of cyclic ketones with biaryl and aryl-alkyl ketones via the merger of photoredox and organocatalysis. aReaction performed with 1.0 equiv of LiAsF6. bReaction performed with Ir(p-OMe-ppy)3, in MeCN (0.17 M). 52
MacMillan et. al. proposed that the photocatalytic synthesis of γ-hydroxyketones proceeds as illustrated in Scheme 15. Irradiation of IrIII(ppy)3 with visible light produces *IrIII(ppy)3, which is sufficiently reducing (E1/2IV/*III = −1.73 V vs SCE)85, 87 to engage in a SET process in the presence of acetic acid – the reduction potential of benzophenone (E1/2red = −1.83 V vs SCE)91 is elevated in acidic medium through hydrogen bonding interactions, which renders the C=O bond reduction thermodynamically feasible. An electron transfer from *IrIII(ppy)3 to the aryl ketone 15.1 affords a strongly oxidizing IrIV(ppy)3+ and the corresponding ketyl radical 15.2. IrIV(ppy)3+ (E1/2IV/III = +0.77 V vs SCE)85 then oxidizes an electron-rich enamine 15.5 (E1/2red = +0.385 V vs SCE for 15.5)92 formed upon condensation of amine catalyst 15.4 with the ketone coupling partner 15.3. A subsequent proton loss from the oxidized enamine 15.6 generates an enaminyl radical 15.7, which readily recombines with the ketyl radical 15.2 to generate γ-hydroxyketone enamine 15.8. Enamine hydrolysis affords the desired γ-hydroxyketone product 15.9, regenerates the secondary amine catalyst 15.4 and completes the organocatalytic cycle. With IrIII(p-OMe-ppy)3 the reaction proceeds via an alternative pathway, in which oxidation of the enamine precedes the reduction of acetophenone, as evidenced by Stern-Volmer quenching experiments: while the emission of *IrIII(ppy)3 is readily quenched by benzophenone, neither acetophenone or acetophenone in the presence of acetic acid can quench the excited state of IrIII(p-OMe-ppy)3. In contrast, emission of *IrIII(p-OMe-ppy)3 is efficiently quenched by the enamine generated from cyclohexanone and azepane.
Scheme 15.
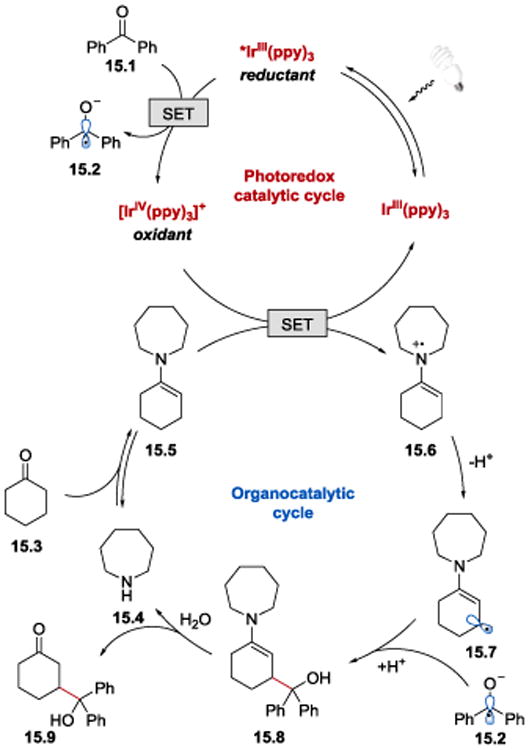
Proposed mechanism for the synthesis of γ-hydroxyketones under photoredox conditions.52
In 2015, MacMillan et. al. reported that in analogy to γ-hydroxyketones, racemic γ-aminoketones can be synthesized via coupling of β-enaminyl radicals with α-amino radicals under photoredox conditions with IrIII(ppy)2(dtbbpy)PF6 catalyst.63 The reaction is applicable to a wide range of imine coupling partners: aldimines, diaryl and aryl-alkyl ketimines furnish the desired γ-aminoketones in high yields (Scheme 16). With regard to the β-enaminyl radical precursors, cyclohexanone derivatives with substituents at 2-, 3-, and 4-positions as well as cyclopentanones readily undergo the β-aminoalkyl ketone-forming reaction.
Scheme 16.

Photoredox-catalysed synthesis of γ-aminoketones. Diastereomeric ratios were between 1:1 and 1.5:1. aLiBF4 (1.0 equiv) was added. bPyrrolidine (20 mol%) was used in place of azepane. cMorpholine (40 mol%) was used in place of azepane.63
The reaction is suggested to proceed via reductive quenching of *IrIII(ppy)2(dtbbpy)+ (E1/2*III/II = +0.66 V vs SCE)93 by DABCO (17.3) (E1/2red = +0.69 V vs SCE)94 (Scheme 17). In this process, DABCO cation radical (DABCO·+) (17.4) and the strongly reducing IrII(ppy)2(dtbbpy)(E1/2III/II = −1.51 V vs SCE)93 are generated. Subsequent reduction of 17.4 by the electron-rich enamine 17.7(E1/2red = +0.385 V vs SCE for 17.7)92 affords enaminylcation radical 17.8 and regenerates DABCO, which acts as an electron transfer agent. A SET from IrII(ppy)2(dtbbpy) to a protonated imine 17.1 forges α-amino radical 17.2, which undergoes radical-radical coupling with β-enaminyl radical 17.9, formed upon deprotonation of 17.8. Hydrolysis of the resulting γ-aminoketone enamine 17.10 furnishes the final γ-aminoketone product 17.11. The proposed mechanism is corroborated by (i) Stern-Volmer quenching studies, according to which DABCO is a better quencher of the excited photocatalyst than enamine; and (ii) significant drop in the reaction efficiency upon lowering the loading of DABCO.
Scheme 17.
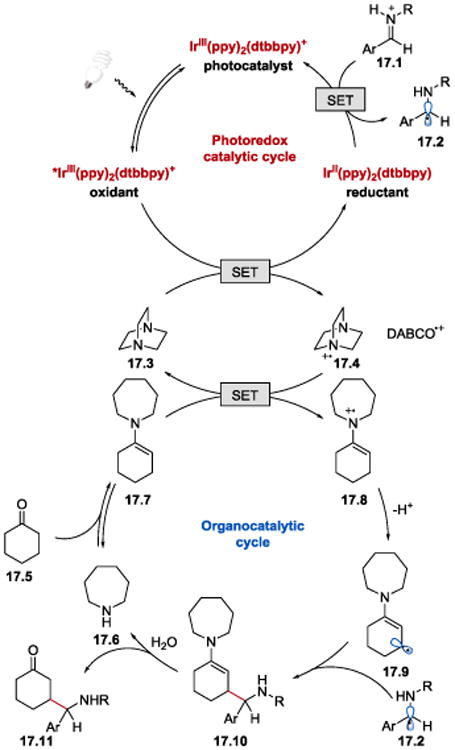
Proposed mechanism for the synthesis of γ-aminoketones under photoredox conditions.63
In 2014, MacMillan and coworkers demonstrated that α-amino radicals generated from imines can efficiently couple with benzylic ether radicals to furnish a variety of β-amino ether products.95 Various oxygen-protecting groups, cyclic ethers, and heteroaromatic-containing ethers are well tolerated under the optimized conditions With regard to the aldimine substrate, high yields can be obtained with electron-rich, electron-poor, as well as heteroaromatic substituents on both the aldehyde- and imine-derived moieties of the imine (Scheme 18).
Scheme 18.

Coupling of benzylic ethers with imines: imine substrate scope.95
It was proposed that the key to achieve the desired reactivity is the combination of an iridium photocatalyst with a thiol organocatalyst (Scheme 20). Upon irradiation with blue light, IrIII(ppy)2(dtbbpy)+ undergoes photoexcitation to give a long-lived (τ = 557 ns)96 excited state, *IrIII(ppy)2(dtbbpy)+. This *IrIII species can act as an oxidant (E1/2*III/II = +0.66 V vs SCE)93 and accept an electron from the thiol organocatalyst methyl thioglycolate (20.3) to form IrII(ppy)2(dtbbpy) and thiyl radical 20.4. The authors argued that this electron transfer step is facilitated by the weakly basic additive, LiOAc, and proceeds via a concerted PCET event. Abstraction of a hydrogen atom (HAT) from benzyl ether 20.5 affords benzylic ether radical 20.6 and regenerates the thiol catalyst. A C–C bond forming coupling reaction between a benzylic ether radical 20.6 and an α-aminoanion radical 20.2, generated upon a single electron reduction of imine 20.1 (E1/2red = -1.91 V vs SCE for N-benzylideneaniline)5 by IrII(ppy)2(dtbbpy) (E1/2III/II = −1.51 V vs SCE)93, yields the final of β-amino ether product 20.7.
Scheme 20.
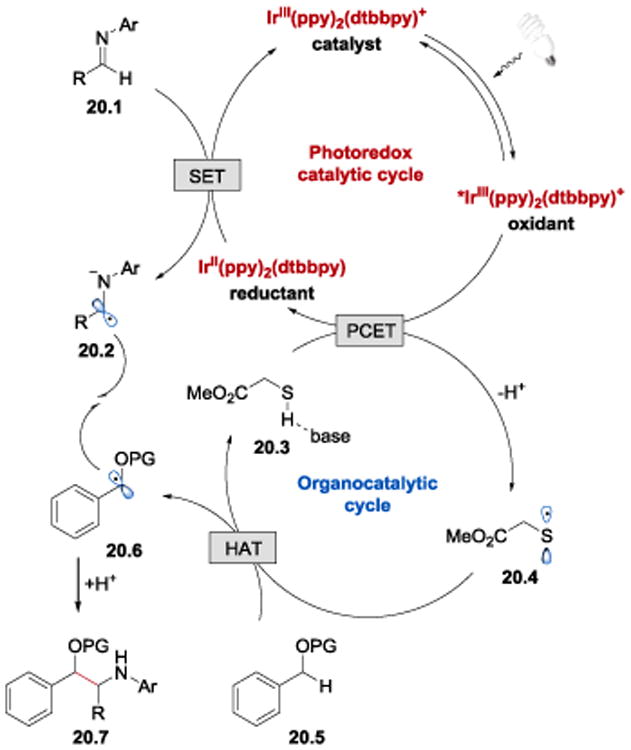
Proposed mechanism of the coupling reaction between benzylic ethers and imines.95
Ketyl and α-aminoalkyl anion radicals are known to undergo C–C bond-forming homocoupling reactions to furnish pinacol and imino-pinacol products, respectively. In 2015, Rueping and co-workers reported that such a transformation can proceed efficiently under visible light photoredox conditions (Scheme 21 and Scheme 22).56 In case of the pinacol coupling of aldehydes and ketones, the protocol is applicable to the synthesis of diols derived from benzaldehydes bearing electron-donating and electron-withdrawing groups, as well as both electron-poor aromatic and aliphatic ketones. In this transformation, IrIII[F(CF3)ppy]2(bpy)PF6 is employed as a photoredox catalyst, while tributylamine plays a dual role – it not only acts as an electron donor, but also serves as a precursor to a radical cation, which activates the carbonyl substrates in the reduction step.
Scheme 21.

Photoredox-catalysed pinacol coupling of aldehydes and ketones. a1.5 equiv of NBu3 was used.56
Scheme 22.

Photoredox-catalysed imino-pinacol coupling.56
Under slightly modified reaction conditions, imines were found to undergo photoredox-catalysed imino-pinacol coupling reaction to yield symmetric diamines in good yields (Scheme 22).
The postulated mechanism for the photoredox-catalysed reductive coupling of carbonyl derivatives developed by Rueping et al. is depicted in Scheme 23. The reaction is initiated by visible light-induced photoexcitation of IrIII[F(CF3)ppy]2(bpy)+ to its excited state, *IrIII[F(CF3)ppy]2(bpy)+. Reductive quenching of *IrIII[F(CF3)ppy]2(bpy)+ by NBu3 affords IrII[F(CF3)ppy]2(bpy) (E1/2III/II = −1.31 V vs SCE)56 and cation radical 23.2. The resulting Lewis acidic cation radical 23.2 can interact with the weakly basic C=O (or C=N) bond of a carbonyl compound through a two-center/three-electron bond. This interaction facilitates the reduction of species 23.4 by IrII[F(CF3)ppy]2(bpy) to a ketyl radical by lowering the energy barrier for the SET process. Alternatively, the carbonyl group can be activated by the α-ammonium radical 23.3 generated from 23.2 via a [1,2]-H shift. The so-formed 23.5 can then engage in hydrogen-bonding interaction with a C=O bond and thus enable ketyl radical 23.6 formation. Homocoupling of two 23.6 followed by protonation affords the desired product 23.7.
Scheme 23.
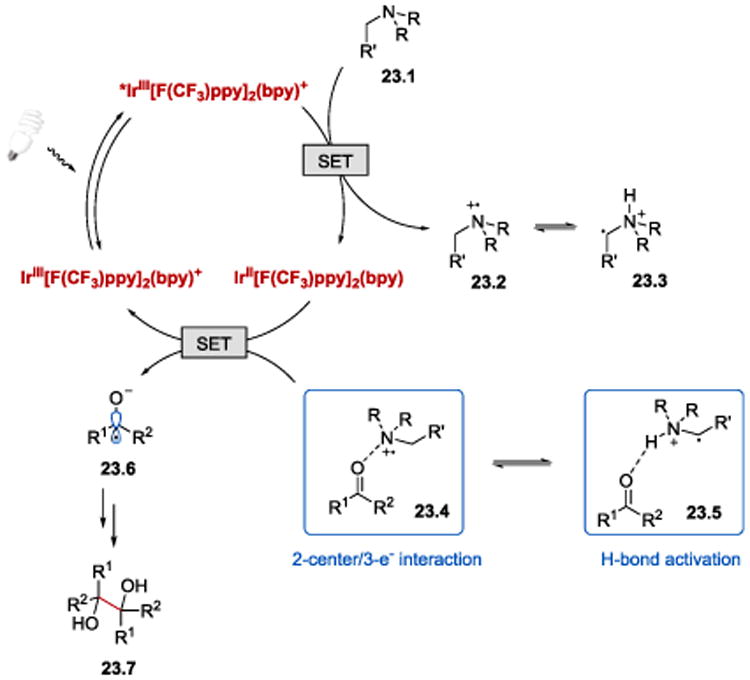
Postulated catalytic cycle for the photoredox-catalysed pinacol coupling of aldehydes and ketones.56
In 2015, Ooi et al.64 utilized an elegant dual catalytic system comprising a photoredox catalyst and a chiral Brønsted acid to achieve an asymmetric α-coupling of N-sulfonyl imines with N-arylaminomethanes (Scheme 24). Unsymmetrical chiral vicinal diamines were obtained in high yields and excellent enantioselectivities (up to 97% ee) from a wide range of aryl and heteroaryl N-sulfonyl aldimines. With respect to the aminomethyl radical precursor, both N,N-diarylaminomethanes and N-alkyl-N-arylaminomethanes were suitable reaction partners for the α-coupling process. Development of the catalytic enantioselective coupling protocol by Ooi has been a remarkable milestone in the field of asymmetric radical chemistry as the precise control of the high intrinsic reactivity of odd-electron species is considered a formidable challenge in organic synthesis.
Scheme 24.

Asymmetric α-coupling of imines with N-arylaminomethanes under synergistic chiral Brønsted acid/photoredox catalysis. a0.05 M, 34 h.64
The reaction has been proposed to proceed via a reductive quenching pathway of the iridium photocatalyst as depicted in Scheme 25. The process is initiated by irradiation of IrIII(ppy)2(Me2phen)+ with visible light, followed by a single electron reduction of *IrIII(ppy)2(Me2phen)+ by Ph2NMe (25.1). A SET from IrII(ppy)2(Me2phen) (E1/2III/II = −1.58 V vs SCE)64 to N-sulfonyl imine 25.4 (E1/2red = −1.45 V vs SCE for N-benzylidenemethanesulfonamide)64 produces α-aminoanion radical 25.5 and regenerates the active photocatalyst. Catalytic ion pair formation between P-spiro chiral tetraaminophosphonium cation 25.6 and 25.5 enables an enantioselective radical coupling reaction between 25.5 and aminomethyl radical 25.3 to afford the desired diamine product 25.7.
Scheme 25.
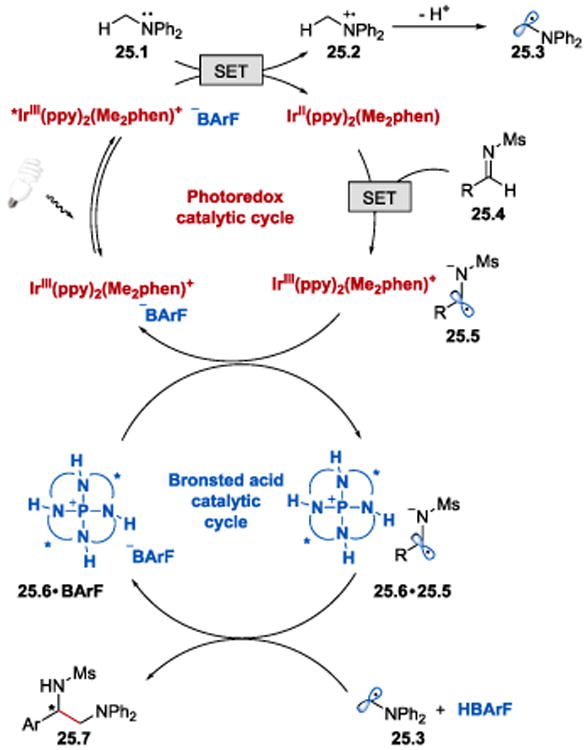
Proposed mechanism of asymmetric α-coupling of imines with N-arylaminomethanes.64
A conceptually similar reaction was reported by Rueping et al. in 2016.58 In this case, the coupling between aldimines and tertiary amines yielding unsymmetrical vicinal diamines was accomplished with the use of IrIII(ppy)2(dtbbpy)PF6 as a photoredox catalyst (Scheme 26). With respect to the amine-derived part of the imine moiety, the reaction tolerated both electron-rich and electron-poor (hetero)aromatic substrates with different substitution patterns. In terms of the aldehyde-derived R1-substituent, imines containing aryl, heteroaryl, and carbonyl group underwent coupling reaction in good yields. Under Rueping's conditions, a range of N,N-disubstituted aniline derivatives were converted into the desired diamine products in moderate to good yields. In addition, coupling of a two secondary α-amino radical fragments was enabled by an introduction of an electrofugal groups (TMS or CO2H) onto the methylene moiety of the amine partner.
Scheme 26.

α-Coupling of imines with anilines. aR3 = H bR3 = TMS58
Rueping et. al. also demonstrated that their reductive coupling protocol is applicable to the synthesis of vicinal aminoalcohols via recombination of ketyl and α-amino radicals (Scheme 27). Differently-substituted benzaldehydes as well as pyridinecarboxaldehyde successfully provided the desired products. The authors also noted that addition of catalytic amounts of benzoic acid had a beneficial influence on the reactions: it enabled the coupling to proceed faster and with lower catalyst loading (0.5 mol%).
Scheme 27.

Coupling of aldehydes with N,N-dimethyl-p-toluidine.58
A proposed mechanism for the Rueping's synthesis of 1,2-aminoalcohols is illustrated in Scheme 28. Reductive quenching of *IrIII(ppy)2(dtbbpy)+ by amine 28.1, followed by a single-electron reduction of aldehyde 28.4 by IrII(ppy)2(dtbbpy) generates the ketyl radical 28.5, which couples with the α-amino radical 28.3 to provide the final product 28.7 upon protonation of 28.6.
Scheme 28.
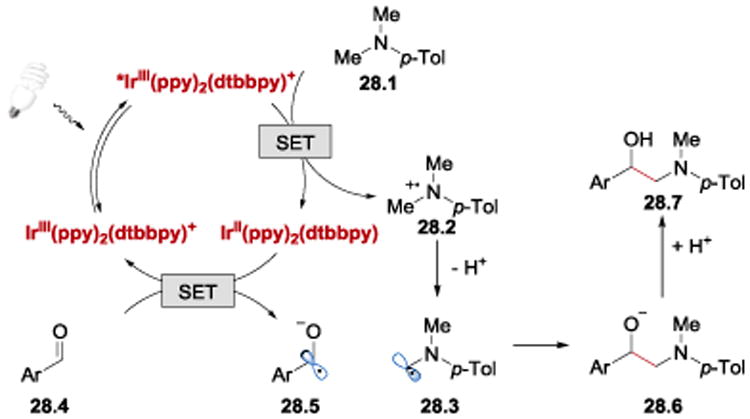
Proposed photoredox catalytic cycle for the synthesis of unsymmetrical 1,2-aminoalcohols.58
In 2016, Meggers and co-workers employed a chiral metal photocatalyst (^-IrS) to develop a novel catalytic enantio- and diastereoselective synthesis of 1,2-amino alcohols from trifluoromethyl ketones and tertiary amines (Scheme 29).62 A variety of aromatic amines can be used in this transformation to afford the desired coupling products in high yields and excellent enantioselectivities (up to 99% ee). The scope of this reaction, however, is limited to heteroaryl trifluoromethyl ketones that are sufficiently electron-deficient to undergo a single-electron reduction process and possess two coordinating directing groups through which they can bind to the chiral metal center.
Scheme 29.

Photoredox-catalysed enantio- and diastereoselective synthesis of 1,2-amino alcohols. a5 mol% of ^-IrS was used.62
The authors postulated that the mechanism involves a SET from a tertiary amine 30.4 to the photoexcited *Ir(III)-bound ketone 30.3, which generates an amino cation radical 30.6 and an iridium-coordinated ketyl radical 30.5 (Scheme 30).62 Subsequent proton transfer and radical-radical cross-coupling between 30.7 and 30.8 affords the Ir(III)-bound 1,2-amino alcohol product 30.9, which is then replaced by new substrate 30.1. In this transformation, the chiral iridium complex acts both as a photoredox catalyst, and a Lewis acid that activates ketones for the reduction process and controls the stereochemistry of the radical-radical cross-coupling step.
Scheme 30.
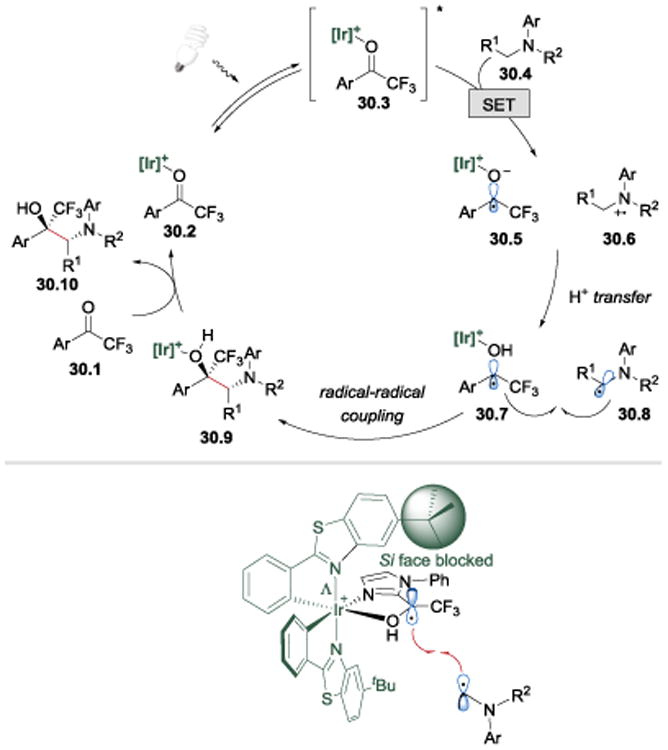
Postulated mechanism for the visible-light-activated catalytic asymmetric process and a proposed stereochemical model for the asymmetric induction.62
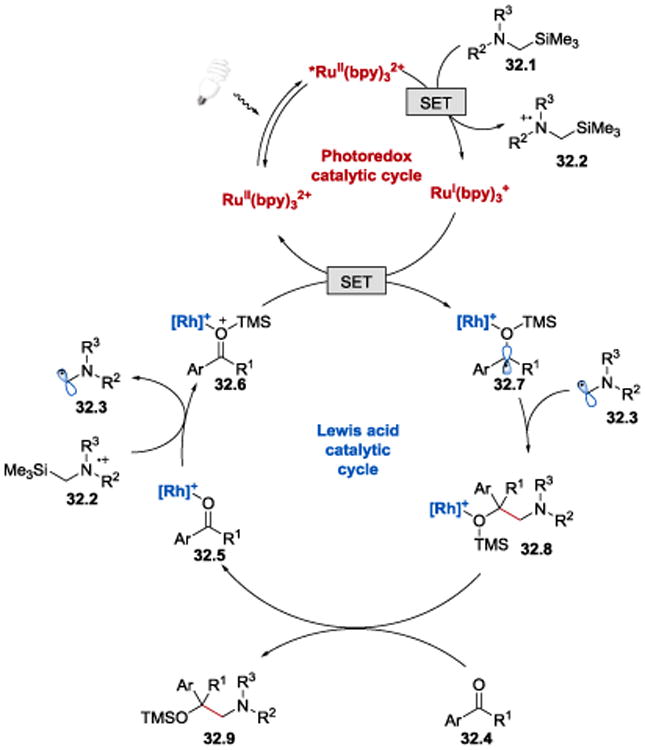
In their subsequent work, Meggers et al. extended the scope of the coupling reaction from ketones containing the α-CF3 group to a range of 2-acyl imidazoles (Scheme 31).97 The chiral-at-the metal rhodium-based Lewis acid ^-RhS was employed in combination with Ru(bpy)3(PF6)2 photoredox catalyst to facilitate the reaction between ketones and α-silylamines. Chiral 1,2-aminoalcohols were obtained in high yields and excellent enantioselectivities from both aromatic and aliphatic 2-acyl imidazoles and differently substituted α-trimethylsilylalkylamines.
Scheme 31.

Enantioselective synthesis of 1,2-aminoalcohols under Rh/Ru photoredox catalysis.97
The process begins with absorption of visible light by RuII(bpy)32+ to give photoexcited *RuII(bpy)32+. *RuII(bpy)32+ (E1/2*II/I = +0.77 V vs SCE)66 is sufficiently oxidizing to accept an electron from α-silylamine 32.1 (E1/2red = +0.41 V vs SCE for N-phenyl-N-((trimethylsilyl)methyl)aniline),98 generating RuI(bpy)3+ species and a cation radical 32.2, which undergoes rapid silyl transfer with Rh-bounded ketone 32.5 to afford an α-aminomethyl radical 32.3 and an electron deficient silylated intermediate 32.6. A SET from RuI(bpy)3+ (E1/2II/I = −1.33 V vs SCE)66 to 32.6 forms a rhodium coordinated, silylated ketyl radical 32.7, which subsequently undergoes a radical-radical coupling reaction with the α-aminomethyl radical 32.3 to afford Rh-bound coupled product 32.8. Release of the 1,2-aminoalcohol 32.9 and coordination of a new substrate 32.4 completes the catalytic cycle.
In 2017, Xia group reported that ketyl and α-aminoalkyl radicals can undergo intermolecular coupling reaction with anion radicals derived from dicyanobenzenes or isonicotinonitrile to afford arylation products (Scheme 33).99 This photocatalytic protocol is applicable to a broad range of aromatic aldehydes, ketones, and imines, and allows access to secondary/tertiary alcohols and amines under mild reaction conditions in moderate to excellent yields.
Scheme 33.

Photocatalytic arylation of aldehydes and imines.99
In analogy to the Rueping's protocol,56 it has been proposed that the C=X bond reduction is enabled by the interaction with the α-ammonium radical 34.3 (or the Lewis acidic cation radical 34.2) (Scheme 34). PCET from IrII(ppy)2(dtbbpy) (E1/2III/II = −1.51 V vs SCE)93 to 34.4 (or, alternatively, to 34.5) leads to the formation of ketyl or α-aminoalkyl radical 34.6, which undergoes intermolecular radical-radical cross-coupling with 34.8, formed in the second photoredox cycle upon a single electron reduction of 34.7 byIrII(ppy)2(dtbbpy). Subsequent elimination of the cyanide anion from 34.9 affords the desired product 34.10.
Scheme 34.
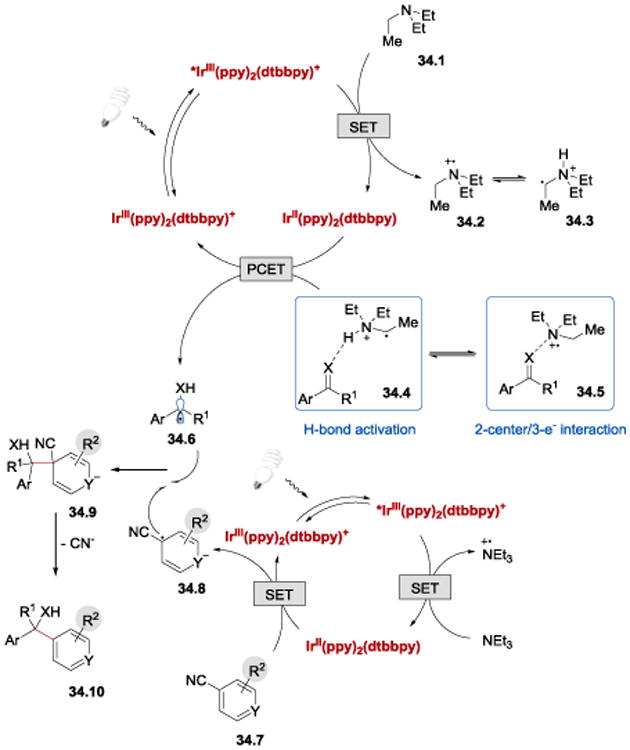
Proposed mechanism for the photocatalytic arylation of aldehydes and imines.99
Reactions of enones and cyclopropylketones via ketyl radical intermediates
The reactivity of conjugated ketyl radical intermediates generated from enones in the presence or Lewis/Brønsted acid activators was extensively studied by the Yoon group. Enones, whose reduction potential is elevated by coordination to an acid, have been reported to engage in a number of photocatalytic cycloaddition reactions. For example, in 2008, Yoon demonstrated that (hetero)aryl enones readily react with pendant Michael acceptors in the presence of the Lewis acid to yield cis products of intramolecular [2+2] cycloaddition (Scheme 35).47 Aliphatic enones and enoates, which are more difficult to reduce, do not form the desired products under the reaction conditions. Subsequently, Yoon developed intermolecular versions of this reaction, which enabled an access to crossed [2+2] heterodimers.48, 100 Aryl enones were efficiently coupled with suitable α,β-unsaturated carbonyl derivatives to yield trans cycloaddition products (Scheme 36). The competing homodimerization was successfully suppressed provided that aryl enones were both (i) more readily reducible, and (ii) less reactive than their corresponding Michael acceptor coupling partners.
Scheme 35.

Photocatalytic [2+2] enone cycloaddition.47
Scheme 36.

Photocatalytic intermolecular [2+2] enone cycloaddition. aReaction was performed with 6 equiv of Michael acceptor.48
Yoon proposed that the cycloaddition reaction proceeds via a reductive quenching cycle of *RuII(bpy)32+ as depicted in Scheme 37. RuI(bpy)3+, formed upon reduction of *RuII(bpy)32+ by Hünig's base, transfers an electron to the lithium-activated enone 37.2 to furnish an enone anion radical 37.3, initiating the [2+2] cycloaddition process with 37.4 and regenerating the RuII(bpy)32+ photocatalyst. Since aryl enones are significantly easier to be reduced than less-conjugated enone substrates, 101 the coupling reaction can be performed with high chemoselectivity.
Scheme 37.
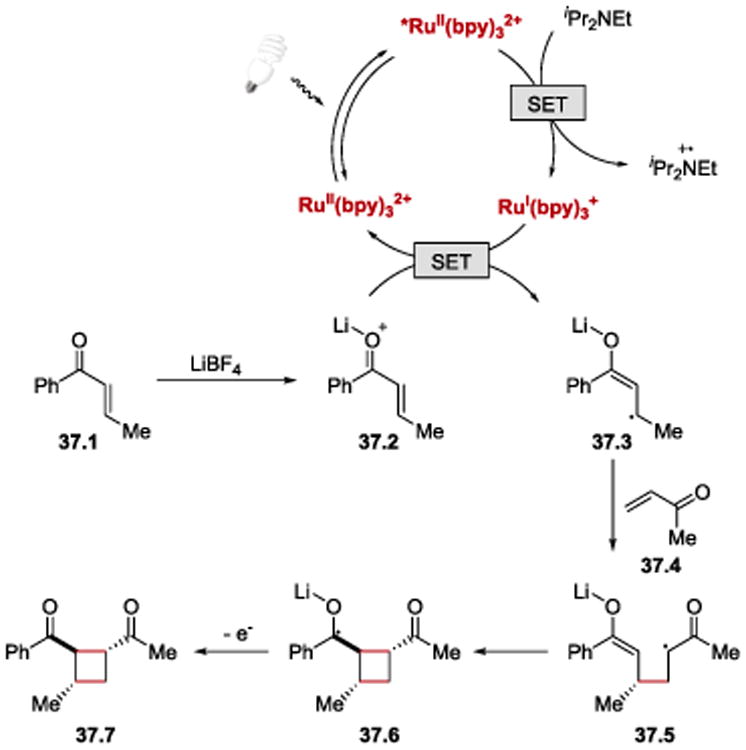
Proposed mechanism of photocatalytic [2+2] enone cycloaddition.100
In 2014, Yoon reported an asymmetric variant of the intermolecular [2+2] enone cycloaddition (Scheme 38).55 Since no background reaction takes place in the absence of a Lewis acid due to the need for enone activation towards a single-electron reduction, high enantioselectivities of 1,2-trans cyclobutane products could be obtained by using a combination of a Lewis acid, Eu(OTf)3, and a chiral ligand 38.1. The cyclization reaction proceeded in good yields for enones bearing both electron-rich and electron poor aryl rings, heteroaryl enones, as well as γ-substituted enones.
Scheme 38.

Photocatalytic enantioselective intermolecular [2+2] enone cycloaddition.55
While bis(enones) with a three-carbon tether were shown to undergo [2+2] cycloaddition,47, 100 reaction of bis(enones) with a longer aliphatic tether length under similar photoreductive conditions led to the formation of [4+2] hetero Diels-Alder cycloadducts (Scheme 39).50 Notably, Yoon's photoredox conditions facilitated coupling between an electron-deficient diene and an electron-poor dienophile, which is normally difficult to achieve upon thermal activation. In this process, Mg(ClO4)2 was found to be the optimal Lewis acid, which enabled the right balance between substrate activation for the SET step and an undesired reductive decomposition of the cycloaddition product. Symmetrical aryl enones bearing electron-rich and electron-poor substituents and heteroaryl enones were well tolerated under the optimized conditions. The reaction was also found to be applicable to unsymmetrical aryl-alkyl enones, although slightly diminished yields were observed with these substrates.
Scheme 39.

Photocatalytic [4+2] cyloaddition.50
The photocatalytic [4+2] hetero Diels-Alder cycloaddition reaction has been proposed to proceed as depicted in Scheme 40. Reduction of *RuII(bpy)32+ by Hünig's base generates RuI(bpy)3+, a reductant which engages in a SET with the Lewis-acid activated aryl enone 40.2 (the rate of a one-electron reduction of methyl enone was found to be much slower than the rate of the reduction of aryl enone). Intramolecular cyclization of anion radical 40.3 affords trans-substituted cyclohexane intermediate 40.4, which isomerizes to the more stable aryl ketyl radical 40.5. The key step is the selective formation of the C–O bond from 40.5. Subsequently, loss of an electron either to the photogenerated amine cation radical or another equivalent of enone affords the [4+2] hetero Diels-Alder cycloaddition product 40.7.
Scheme 40.
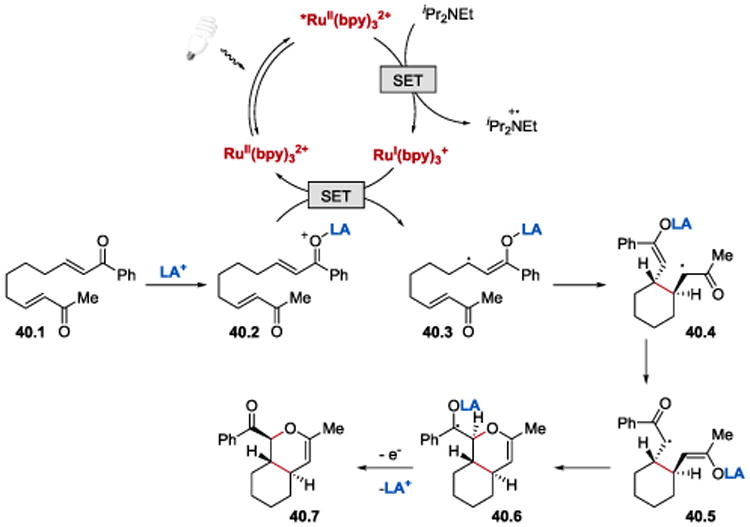
Proposed mechanism of the regioselective [4+2] hetero Diels-Alder cycloaddition reaction.50
Yoon et al. also reported that aryl cyclopropyl ketones react with olefins to afford highly substituted cyclopentane rings under similar photoredox conditions (Scheme 41).102 In contrast to the [2+2] cycloaddition reaction, this [3+2] process was found to be applicable to substrates containing not only pendant enone acceptors, but also styrenes, cyclic aliphatic olefins, as well as aryl and aliphatic alkynes. In addition, it was empirically determined that LiBF4 was not sufficiently Lewis acidic to activate cyclopropyl ketones towards the single-electron reduction; a more strongly acidic Lewis acid additive, La(OTf)3, was required to facilitate the reaction.
Scheme 41.

Photocatalytic [3+2] cycloaddition of aryl cyclopropyl ketones.102
More recently, the Yoon group reported an asymmetric intermolecular version of the [3+2] cycloaddition process (Scheme 42).57 The enantioselectivity of the reaction was controlled by the use of a catalytic amount of Gd(III) pybox complex. It was demonstrated that a wide range of aryl and heteroaryl cyclopropyl ketones could undergo the coupling reaction with styrenes, N-vinylcarbazole, and conjugated dienes to afford densely substituted cyclopentanes in excellent yields with high enantioselectivities and moderate diastereoselectivities.
Scheme 42.

Photocatalytic enantioselective intermolecular [3+2] cycloaddition. aReaction conducted using 20 mol% Gd(OTf)3 and 30 mol% of the ligand at –20 °C.57
Scheme 43 depicts the working hypothesis of the mechanism of the [3+2] cycloaddition process. In analogy to the previously discussed examples, this transformation is proposed to proceed via reductive quenching cycle of *RuII(bpy)32+. RuI(bpy)3+ transfers an electron to phenyl ketone activated by the chiral Gd(III) Lewis acid complex 43.1. The SET yields a ketyl radical 43.2, which undergoes a cyclopropyl ring opening followed by alkene addition, cyclization and reoxidation. The formation of a neutral product 43.7 can occur either by chain-terminating reduction of the photogenerated amine cation radical or by chain-propagating electron-transfer to another equivalent of Gd(III)-activated substrate 43.1.
Scheme 43.
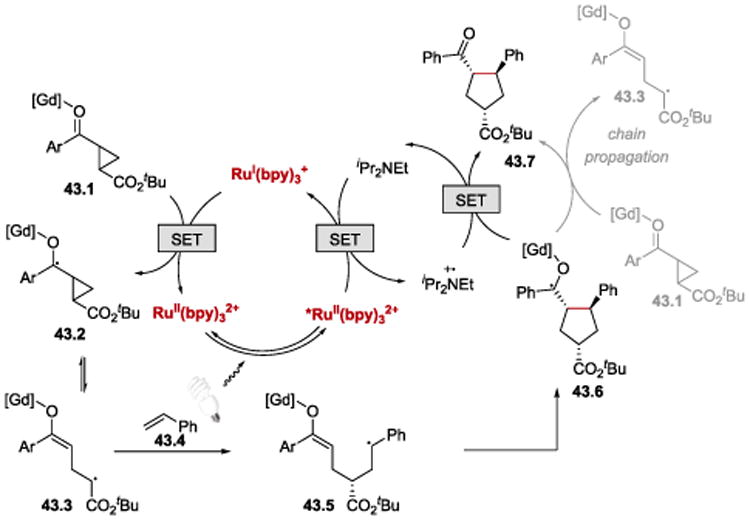
Proposed mechanism for enantioselective [3+2] cycloaddition.57
Interestingly, Yoon et. al. observed that the outcome of the photocatalytic reaction of enones strongly depends on the nature of the acid co-catalyst employed for the substrate activation.47, 49 While Lewis acids, for instance LiBF4, promote the [2+2] cycloaddition, Brønsted acids, such as HCO2H, have been reported to favor the reductive coupling reaction (Scheme 44). Notably, these two reactions differ by their overall redox balance: the [2+2] cycloaddition is net redox-neutral, whereas the reductive cyclization constitutes a two-electron reduction of the enone substrate. It was argued that the reactivity of the neutral radical intermediate formed in the presence of a Brønsted acid is very distinct from the chemistry of the anion radical generated under Lewis acidic conditions: the neutral radical preferentially undergoes 5-exo-trig cyclization rather than the [2+2] cycloaddition. Thus, the acid co-catalyst determines the nature of the reactive intermediate, which, in turn, has a profound influence on the overall transformation, its stereoselectivity, and oxidation state of the products.
Scheme 44.

Divergent reactivity of photoredox-generated radical and anion radical intermediates.49
It is worth noting that the photoreductive cyclization of enones, in contrast to the [2+2] cycloaddition reaction, is applicable to aliphatic enones, activated alkynes and styrenes, and is generally trans selective (Scheme 45).49
Scheme 45.

Photocatalytic reductive cyclization of enones. aReaction was conducted with 2.5 mol% of Ir(ppy)2(dtbbpy)(PF6).49
Another net reductive coupling reaction under photoredox conditions involving conjugated ketyl radical intermediates was reported by Xia and co-workers (Scheme 46).103 Chalcones we shown to undergo reductive dimerization when treated with RuII(bpy)3(PF6)2 photoredox catalyst, Sm(OTf)3 Lewis acid and Hünig's base as the terminal reductant. Sm(III)-stabilized anion radical 46.2 was proposed to dimerize to generate dienolate 46.3. Sequential protonation and intramolecular aldol reaction provided an access to polysubstituted cyclopentanol derivatives 46.5. Nine examples were reported, and the reaction was shown to tolerate neither substitution at β and β-position nor ortho-substituted Ar1 groups.
Scheme 46.
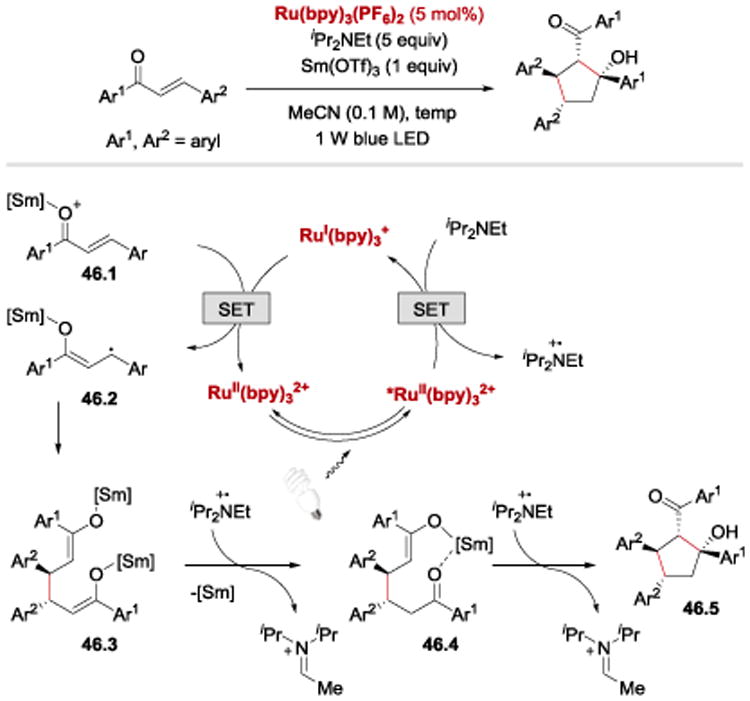
Reductive chalcone dimerization under photoredox conditions.103
In 2017, Gong and Meggers demonstrated that conjugated ketyl radicals undergo intermolecular enantioselective radical-radical coupling reaction with N-centered radicals derived from N-aryl carbamates (Scheme 47).104 The conjugate amination of α,β-unsaturated 2-acyl imidazoles proceeds with very high yields and excellent enantioselectivities in the presence of a chiral-at-rhodium Lewis acid catalyst Δ-RhO, a weak phosphate base, and an iridium-based photoredox catalyst IrIII[dF(CF3)ppy]2(5,5′-dCF3bpy)PF6.
Scheme 47.

Enantioselective β-amination of enones.104
It was proposed that the reaction proceeds as depicted in Scheme 48. 104 A PCET to the photoexcited *Ir(III) catalyst from the Brønsted-base-activated carbamate 48.1 generates a N-centered radical 48.2 and Ir(II) species. A single-electron reduction of the rhodium-coordinated substrate 48.3 by Ir(II) leads to the formation of rhodium enolate radical intermediate 48.4, which subsequently undergoes recombines with the carbamoyl N-radical 48.2 to afford 48.5. Protonation of 48.5 followed by product release from Scheme 48.6 and coordination of a new substrate to the chiral rhodium catalyst closes the catalytic cycle. The asymmetric induction providing S-configured products is governed by the Δ-configured rhodium catalyst.
Scheme 48.
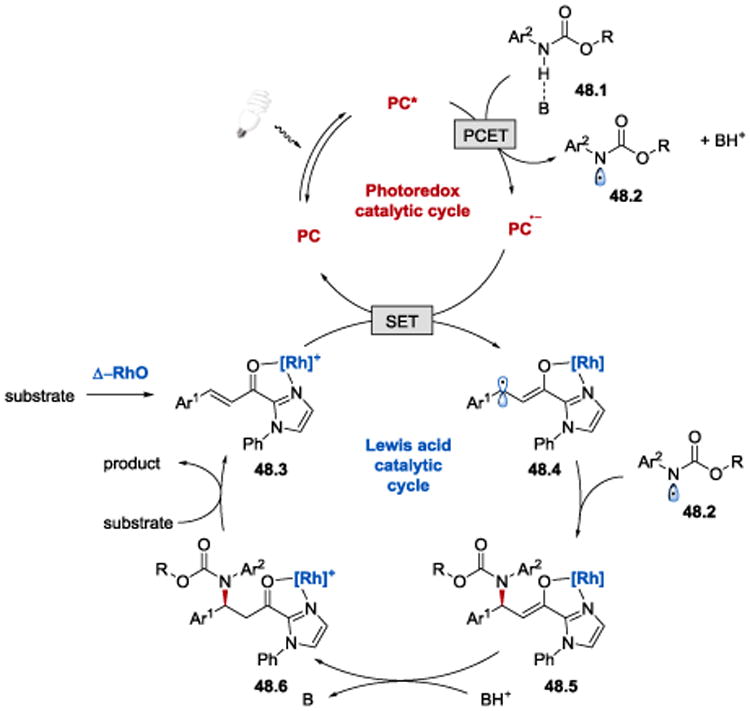
Proposed mechanism of enantioselective β-amination of enones.104
Gong and Meggers also reported that under slightly modified reaction conditions 2-acyl imidazoles react with N-alkyl amides to furnish δ-alkylation products (Scheme 49).105 This asymmetric remote C(sp3)–H functionalization tolerated different β- and N-substituents on 2-acyl imidazole substrates; with regard to the amide coupling partner, the reaction was shown to work best with PMP-containing substrates, presumably due to the more facile oxidation of electron-rich amides under PCET conditions.
Scheme 49.

Asymmetric δ-functionalization of N-alkyl amides under photoredox conditions.105
The alkylation reaction was proposed to proceed as depicted in Scheme 50 and follow the mechanism previously discussed for the β-amination of enones (Scheme 48).104, 105 The amidyl radical 50.2 generated from 50.1 under PCET conditions was proposed to undergo an intramolecular 1,5-hydrogen atom transfer (1,5-HAT) to form a carbon-centered radical 50.3. Radical-radical coupling between 50.3 and 50.5 furnishes the C–C bond in an enantioselective fashion.
Scheme 50.
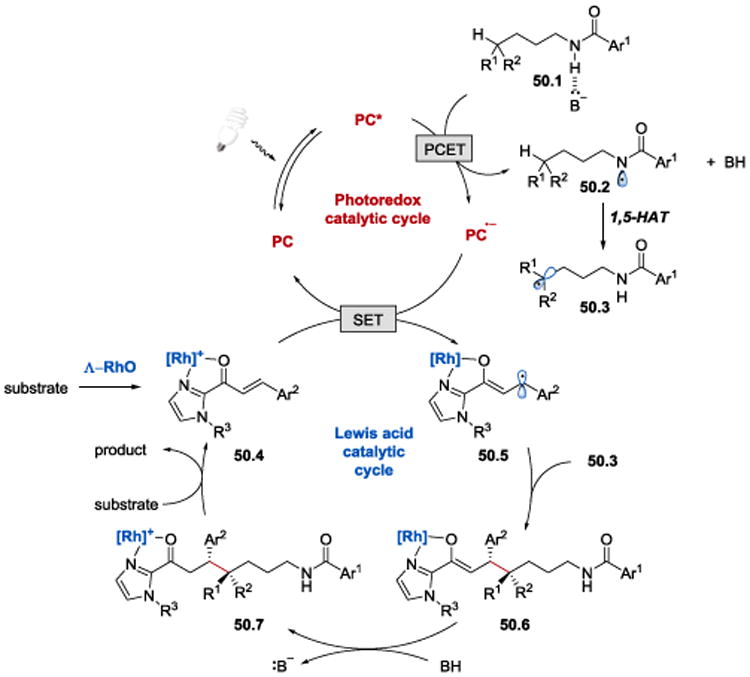
Proposed mechanism of asymmetric δ-functionalization of N-alkyl amides under photoredox conditions.104
Addition of ketyl and α-aminyl anion radicals to π-systems
Another important class of transformations characteristic of ketyl and α-aminyl anion radicals are their additions to activated π-bonds. In 2013, Knowles and co-workers developed a new photocatalytic protocol for an intramolecular ketyl-olefin coupling reaction (Scheme 51) using the concept of proton-coupled electron transfer (PCET).54 The cyclization was applicable to aryl ketones with pendant acrylate esters, acrylonitrile, and styrenyl acceptors.
Scheme 51.

Photoredox-catalysed intramolecular ketyl-olefin coupling reaction.54
Knowles et al. postulated that the reaction proceeds as depicted in Scheme 52. Photoexcitation of RuII(bpy)32+ generates *RuII(bpy)32+, which is proposed to be reduced to RuI(bpy)3+ by HEH. At the same time, the Brønsted acid catalyst can engage in reversible hydrogen-bonding interactions with the ketone substrate 52.1 to form a hydrogen-bonded Brønsted acid-ketone complex, which can participate in a concerted PCET. In this step, an electron transfer from the strongly reducing RuI(bpy)3+ occurs concomitantly with the proton transfer to the ketone oxygen to afford a neutral ketyl intermediate 52.2. The ketyl radical 52.2 can then undergo an intramolecular Michael addition to form a cyclopentane ring and an α-carbonyl radical 52.3. Hydrogen atom abstraction from HEH by the α-carbonyl radical 52.3 generates the final product 52.4 and HEH•. HEH•, being a strong reductant, can reduce *RuII(bpy)32+ and close the catalytic cycle upon proton transfer.
Scheme 52.
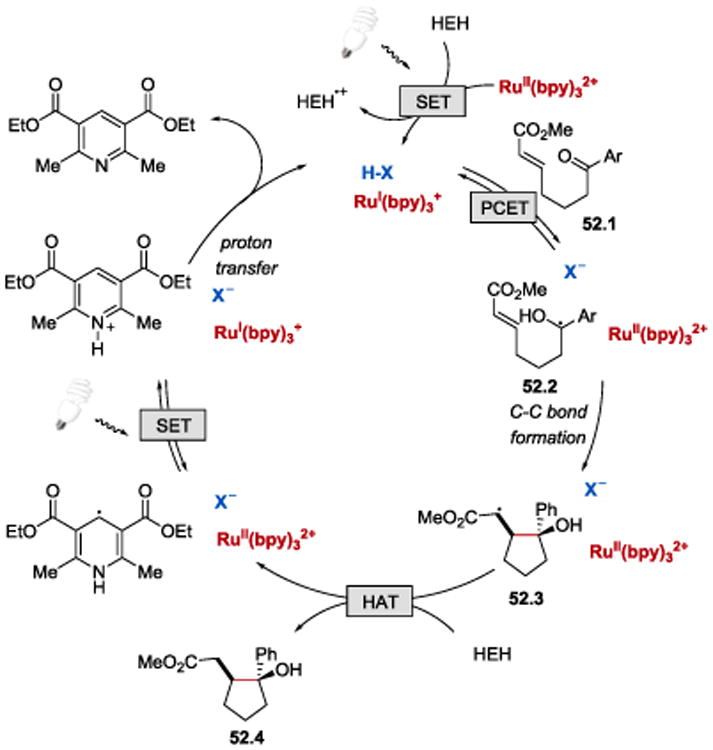
Proposed mechanism for photoredox-catalysed intramolecular ketyl-olefin coupling reaction.54
Also in 2013, Knowles group applied the concept of PCET into development of a highly enantioselective intramolecular azapinacol coupling reaction (Scheme 53).53 The enantioselectivity of the reductive coupling of ketones and hydrazones was realized by the use of a chiral triphenylsilyl-substituted phosphoric acid. The coupling reaction proceeded with good yields and high enantioselectivities from both electron-rich and electron-poor acetophenone derivatives. Heterocyclic ketones were also tolerated, although an erosion in both yield and ee was observed in these cases.
Scheme 53.

Photoredox-catalysed asymmetric intramolecular aza-pinacol coupling reaction. aReaction run at 0 °C in THF.53
In analogy to their previous work,53, 54 the authors proposed that the reaction is initiated by an off-cycle excitation of the Ir(III) photocatalyst, followed by reduction of *Ir(III) by HEH (Scheme 54). Concerted PCET from Ir(II) to a hydrogen-bonded complex between the ketone 54.1 and the phosphoric acid furnishes a neutral ketyl radical as a H–bonded adduct to the chiral phosphate 54.2. The association between of the ketyl radical and the phosphate controls the enantioselectivity of the C–C bond forming step. The final product 54.4 is generated upon hydrogen atom abstraction from HEH by the hydrazyl radical 54.3. Reduction of *Ir(III) by HEH• affords pyridinium ion, whose subsequent deprotonation by the phosphate anion regenerates the active catalyst and completes the catalytic cycle.
Scheme 54.
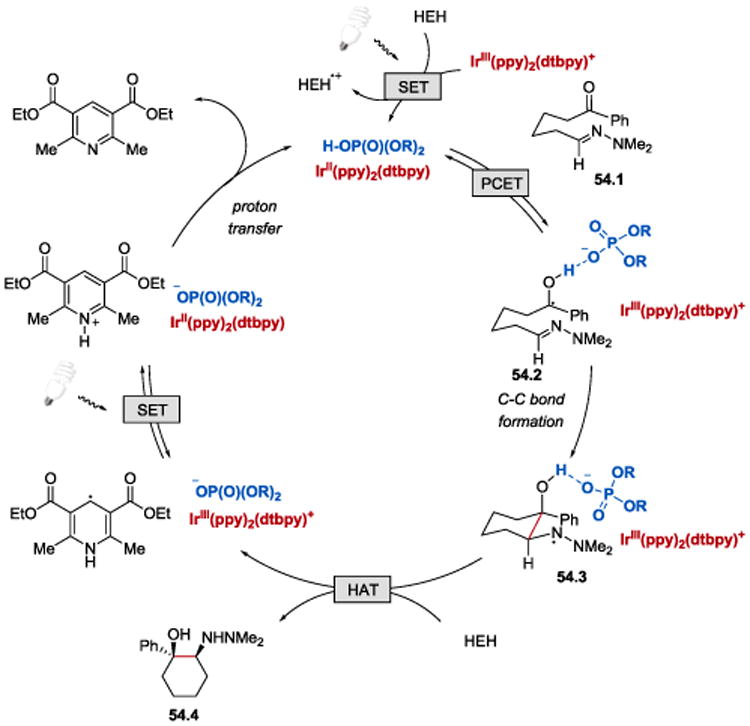
Proposed mechanism for the photoredox-catalysed asymmetric intramolecular aza-pinacol coupling reaction.53
In 2016, Rueping group reported that ketyl radicals generated from aromatic aldehydes under photoredox conditions can undergo an intramolecular addition to alkenes and alkynes to yield chromanol derivatives (Scheme 55).59 Aldehydes containing electron-withdrawing and electron-donating substituents were well tolerated under the reaction conditions and afforded the cyclized products in modest d.r.
Scheme 55.

Intramolecular photoredox-catalysed ketyl-alkene/alkyne coupling. aReaction performed with 2.5 mol% of photocatalyst.59
The authors proposed that the reaction proceeds via photoexcitation of IrIII(ppy)2(dtbbpy)+ to *IrIII(ppy)2(dtbbpy)+, followed by reduction of *IrIII(ppy)2(dtbbpy)+ by Hünig's base (Scheme 56). The Hünig's base cation radical 56.2 undergoes a [1,2]-H shift to form 56.3, which is deprotonated by another molecule of 56.1 followed by oxidation to afford ammonium derivative 56.5 and iminium ion 56.6. Either 56.5 or 56.3 can engage in a PCET with the aldehyde substrate 56.7, facilitating its reduction by IrII(ppy)2(dtbbpy)and formation of a neutral ketyl radical 56.8. The ketyl radical addition to the multiple bond (6-exo-dig or 6-exo-trig cyclization) forms the chromane ring and a benzyl radical 56.9, which abstracts a hydrogen atom to deliver the final product 56.10.
Scheme 56.
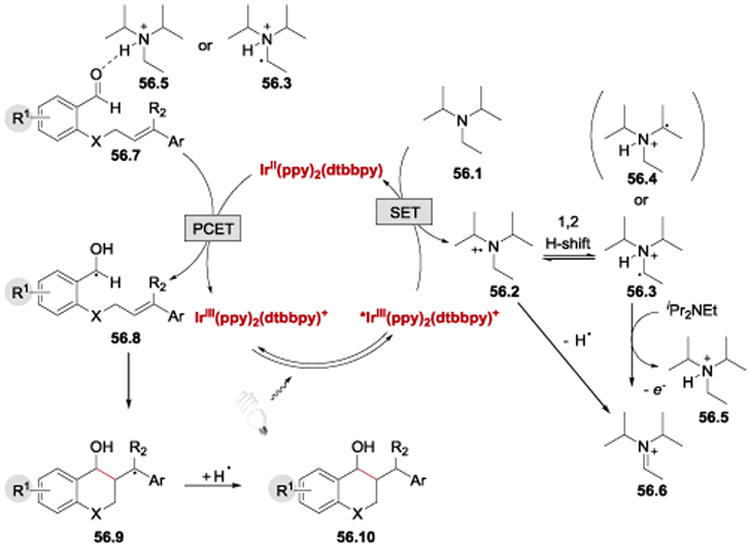
Proposed reaction mechanism of the photoredox-catalysed intramolecular ketyl-olefin/alkyne coupling.59
In addition to intramolecular coupling reactions, ketyl and α-aminoalkyl anion radicals generated upon a single-electron reduction of carbonyl derivatives under photoredox conditions have been reported to engage in intermolecular reactions with activated olefins. In 2016, Chen et al. reported visible-light-induced allylation and intermolecular Michael addition of aldehydes and ketones (Scheme 57).60 The formation of homoallylic alcohols was achieved with iridium-based photocatalyst, IrIII(ppy)2(dtbbpy)(PF6), and when Hantzsch ester (HEH) was employed as a stoichiometric reductant. The allylation reaction proceeded efficiently with (hetero)aryl aldehydes, aryl-, ester-, and amide-substituted ketones. Michael addition of ketyl radical to (vinylsulfonyl)benzene also afforded the desired product in high yield. Aryl imines with various substitution patterns were also shown to readily participate in this polarity reversal reaction. In addition, it was demonstrated that they can undergo coupling reactions with other acceptors such as vinyl ketones, esters, and nitriles. Most remarkably, the reaction was also applicable to alkyl imines, which were previously elusive coupling partners due to their highly negative reduction potential.106 Owing to their inherent instability, alkyl imines were prepared in situ under the photoredox reaction conditions.
Scheme 57.

Allylation reaction of aldehydes, ketones and imines under photoredox conditions.60
It has been proposed that the reaction proceeds as depicted in Scheme 58. IrIII(ppy)2(dtbbpy)+ undergoes photoexcitation with blue light to form *IrIII(ppy)2(dtbbpy)+. The resulting *IrIII(ppy)2(dtbbpy)+ (E1/2*III/II = +0.66 V vs SCE)93 is then reduced by Hantzsch ester to IrII(ppy)2(dtbbpy). The highly reducing IrII(ppy)2(dtbbpy) (E1/2III/II = −1.51 V vs SCE)93 transfers a single-electron to the aldehyde or imine activated by hydrogen bonding with Hantzsch ester cation radical (HEH•+). Following the PCET process, hydroxymethyl or α-aminyl radical 58.3 undergoes addition to either the allyl sulfone 58.4 to afford the homoallylic alcohol 58.5, or to the Michael acceptor 58.6, to furnish the product of the Michael addition 58.7. In this photoredox transformation, Hantzsch ester plays a dual role: it serves as electron/proton donor, and activates the aldehydes for the PCET step.
Scheme 58.
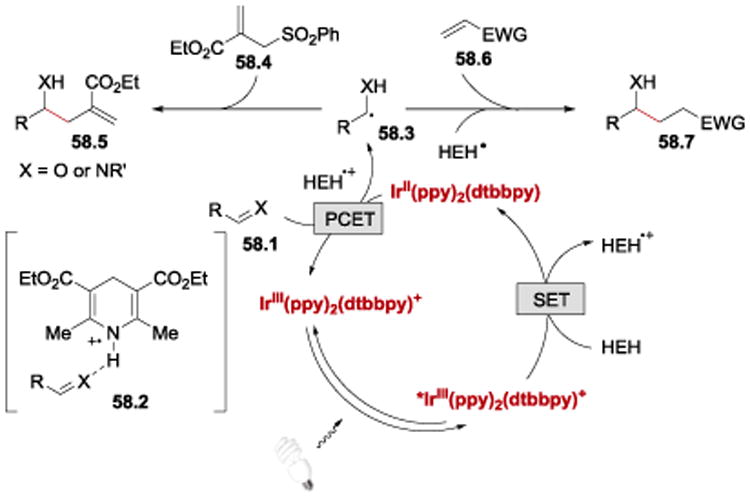
Proposed mechanism of the allylation and Michael addition reactions.60
Recently, Ngai et al. disclosed a reductive coupling reaction between aldehydes and alkenylpyridnes under dual Lewis acid/photoredox catalysis (Scheme 59).107 The C–C bond forming event involving a ketyl radical addition to activated vinylpyridines was accomplished with RuII(bpy)3(PF6)2 as a photoredox catalyst, Hantzsch ester as a terminal reductant, and La(OTf)3 as the Lewis acid activator. Under these conditions, a broad range of aryl and heteroaryl aldehydes, including complex substrates, were readily converted into secondary alcohols in high yields. With respect to the alkenylpyridine coupling partners the reaction tolerated a wide scope of substituents at the α-position of alkenylpyridines, including hydrogen atom, alkyl group, as well as aromatic and heteroaromatic moieties. The corresponding coupling products were obtained in good to excellent yields and with modest diastereoselectivities. In addition to ketyl radicals, α-aminoalkyl anion radicals generated from aromatic imines upon a SET were reported to undergo the coupling reaction with 4-vinylpyridine. The reaction was observed to proceed well regardless of the electronic structure of the iminyl aromatic rings.
Scheme 59.

Photoredox-catalysed coupling reaction between aldehydes/imines and and 4-vinylpyridine. 1H NMR yields are provided in parenthesis.107
The proposed mechanistic details of the coupling reaction are depicted in Scheme 60. Irradiation of Ru(bpy)32+ with visible light produces a long-lived (1.1 μs)108 photoexcited state, *RuII(bpy)32+, which is then reduced by the catalytically generated intermediate Hantzsch ester radical (HEH•) to form a strongly reducing ruthenium (I) species, RuI(bpy)3+, and pyridinium ion PyH+. Ru(bpy)3+ (E1/2red= −1.33 V vs SCE)66 subsequently participates in a PCET with the aldehyde activated by hydrogen bonding with PyH+ (60.1) to afford ketyl radical intermediate 60.2 and regenerate the RuI(bpy)32+ catalyst. Radical 60.2 adds to Lewis acid-activated 4-vinylpyridine 60.4 to form radical 60.5, which abstracts a hydrogen atom from HEH, generating the Lewis acid-bounded product 60.6 and another molecule of a reductant (HEH•) capable of reducing *RuII(bpy)32+. Replacement of the product by new 4-vinylpyridine 60.3 substrate liberates the desired coupling product 60.7 and completes the catalytic cycle.
Scheme 60.
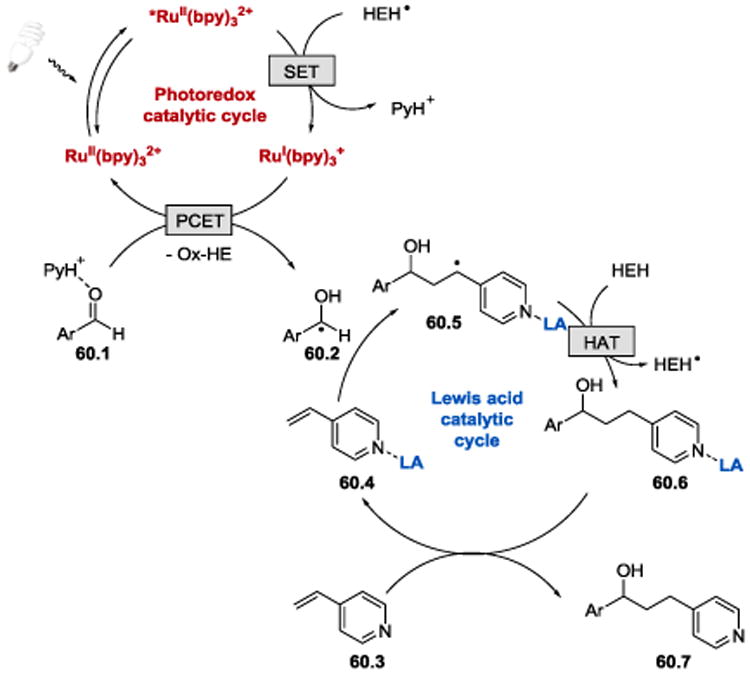
Proposed reaction mechanism of the photoredox coupling reaction between aldehydes and alkenylpyridines.107
Conclusions and Future Outlook
Recent years have witnessed a tremendous progress in reactions of carbonyl derivatives initiated by a SET from organometallic photoredox catalysts. Generation of ketyl and α-aminyl anion radicals from aldehydes, ketones, and imines under photochemical conditions has obviated the need for strong stoichiometric reductants, and provided an access to important synthons under mild reaction conditions using simple reagents and catalysts. Moreover, it has enabled a variety of transformations that would otherwise be difficult or even impossible to achieve. A number of these photoredox reactions tolerate a wide array of functional groups, facilitate novel reactivities (e.g. β-functionalization of ketones), and allow the synthesis of medicinally-relevant compounds and structures of unprecedented molecular architectures. Recently described catalytic enantioselective reactions that rely on the use of a chiral catalyst have provided means for precise stereocontrol over the reactivity of ion-radicals and significantly advanced the field of asymmetric radical chemistry. With the continuing interest of the scientific community in photoredox catalysis, one might expect to see further applications of the photocatalytically-generatedketyl and α-aminyl anion radicalsin synthesis.
Scheme 9.
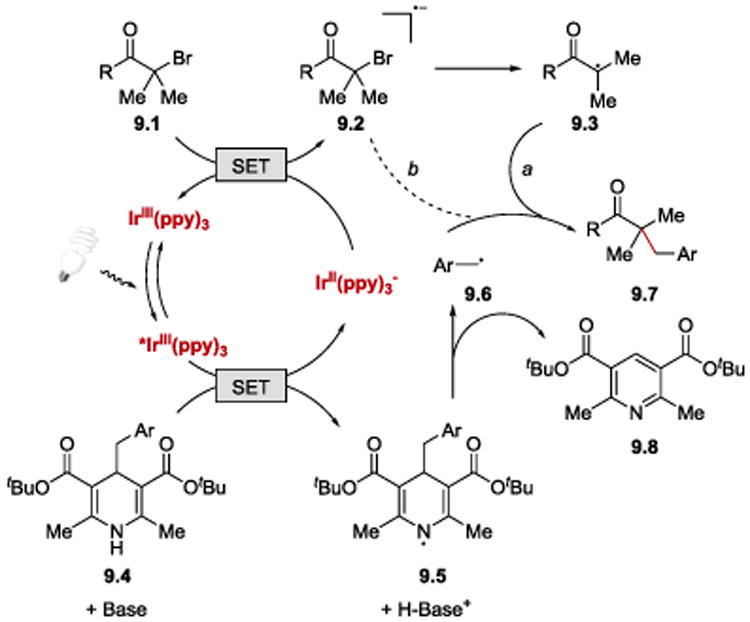
Proposed mechanism of the visible-light photoredox transfer alkylation reaction.82
Scheme 19.

Coupling of benzylic ethers with imines: ether substrate scope.95
Scheme 32.
Proposed mechanism of the visible-light activated Rh/Ru dual catalysis.97
Acknowledgments
We thank National Institute of General Medical Sciences (R35GM119652) and start-up funds from Stony Brook University for supporting our research. K.N.L. received the graduate fellowship from the NIH Chemical-Biology training grant (T32GM092714). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Footnotes relating to the main text should appear here. These might include comments relevant to but not central to the matter under discussion, limited experimental and spectral data, and crystallographic data.
References
- 1.Corey EJ. Pure Appl Chem. 1967;14:19–37. [Google Scholar]
- 2.Seebach D. Angew Chem Int Ed. 1979;18:239–258. [Google Scholar]
- 3.Nicolaou KC, Ellery SP, Chen JS. Angew Chem Int Ed. 2009;48:7140–7165. doi: 10.1002/anie.200902151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reduction potentials provided in this Review were reported against SCE in MeCN unless otherwise stated. When a reduction potential was reported against a different electrode in the original reference, the reduction potential against SCE was calculated by using the conversion factor described in the following reference: Pavlishchuk VV, Addison AW. Inorg Chim Acta. 2000;298:97–102.
- 5.Roth HG, Romero NA, Nicewicz DA. Synlett. 2016;27:714–723. [Google Scholar]
- 6.Pradhan SK, Kadam SR, Kolhe JN, Radhakrishnan TV, Sohani SV, Thaker VB. J Org Chem. 1981;46:2622–2633. [Google Scholar]
- 7.Ikeda T, Yue S, Hutchinson CR. J Org Chem. 1985;50:5193–5199. [Google Scholar]
- 8.Beckwith ALJ, Roberts DH. J Am Chem Soc. 1986;108:5893–5901. doi: 10.1021/ja00279a039. [DOI] [PubMed] [Google Scholar]
- 9.Ardisson J, Férézou JP, Julia M, Pancrazi A. Tetrahedron Lett. 1987;28:2001–2003. [Google Scholar]
- 10.Sugawara T, Otter BA, Ueda T. Tetrahedron Lett. 1988;29:75–78. [Google Scholar]
- 11.Enholm EJ, Prasad G. Tetrahedron Lett. 1989;30:4939–4942. [Google Scholar]
- 12.Enholm EJ, Burroff JA. Tetrahedron Lett. 1992;33:1835–1838. [Google Scholar]
- 13.Hays DS, Fu GC. J Org Chem. 1996;61:4–5. doi: 10.1021/jo960846e. [DOI] [PubMed] [Google Scholar]
- 14.Corey EJ, Pyne SG. Tetrahedron Lett. 1983;24:2821–2824. [Google Scholar]
- 15.Yeh CH, Korivi RP, Cheng CH. Adv Synth Catal. 2013;355:1338–1344. [Google Scholar]
- 16.Estévez RE, Oller-López JL, Robles R, Melgarejo CR, Gansäuer A, Cuerva JM, Oltra JE. Org Lett. 2006;8:5433–5436. doi: 10.1021/ol0620390. [DOI] [PubMed] [Google Scholar]
- 17.Elango TP, Ramakrishnan V, Vancheesan S, Kuriacose JC. Tetrahedron. 1985;41:3837–3843. [Google Scholar]
- 18.Fukuzawa S, Nakanishi A, Fujinami T, Sakai S. J Chem Soc Chem Comm. 1986;0:624–625. [Google Scholar]
- 19.Otsubo K, Inanaga J, Yamaguchi M. Tetrahedron Lett. 1986;27:5763–5764. [Google Scholar]
- 20.Molander GA, Kenny C. Tetrahedron Lett. 1987;28:4367–4370. [Google Scholar]
- 21.Molander GA, Kenny C. J Am Chem Soc. 1989;111:8236–8246. [Google Scholar]
- 22.Mikami K, Yamaoka M. Tetrahedron Lett. 1998;39:4501–4504. [Google Scholar]
- 23.Froling A. Recl Trav Chim Pays Bas. 1974;93:47–51. [Google Scholar]
- 24.Shono T, Nishiguchi I, Omizu H. Chem Lett. 1976;5:1233–1236. [Google Scholar]
- 25.Shono T, Ohmizu H, Kawakami S, Sugiyama H. Tetrahedron Lett. 1980;21:5029–5032. [Google Scholar]
- 26.Shono T, Kashimura S, Mori Y, Hayashi T, Soejima T, Yamaguchi Y. J Org Chem. 1989;54:6001–6003. [Google Scholar]
- 27.Belotti D, Cossy J, Pete JP, Portella C. Tetrahedron Lett. 1985;26:4591–4594. [Google Scholar]
- 28.Belotti D, Cossy J, Pete JP, Portella C. J Org Chem. 1986;51:4196–4200. [Google Scholar]
- 29.Cossy J, Belotti D, Pete JP. Tetrahedron Lett. 1987;28:4547–4550. [Google Scholar]
- 30.Cossy J, Pete JP, Portella C. Tetrahedron Lett. 1989;30:7361–7364. [Google Scholar]
- 31.Cossy J, Belotti D, Cuong NK, Chassagnard C. Tetrahedron. 1993;49:7691–7700. [Google Scholar]
- 32.Cossy J, Belotti D. Tetrahedron. 2006;62:6459–6470. [Google Scholar]
- 33.Hirao T. Top Curr Chem. 2007;279:53–75. [Google Scholar]
- 34.Streuff J. Synthesis-Stuttgart. 2013;45:281–307. [Google Scholar]
- 35.Castro Rodríguez M, Rodríguez García I, Rodríguez Maecker RN, Pozo Morales L, Oltra JE, Rosales Martínez A. Org Process Res Dev. 2017;21:911–923. [Google Scholar]
- 36.Frey G, Hausmann JN, Streuff J. Chem Eur J. 2015;21:5693–5696. doi: 10.1002/chem.201500102. [DOI] [PubMed] [Google Scholar]
- 37.Saito T, Nishiyama H, Tanahashi H, Kawakita K, Tsurugi H, Mashima K. J Am Chem Soc. 2014;136:5161–5170. doi: 10.1021/ja501313s. [DOI] [PubMed] [Google Scholar]
- 38.Morcillo SP, Miguel D, Campana AG, Alvarez de Cienfuegos L, Justicia J, Cuerva JM. Org Chem Front. 2014;1:15–33. [Google Scholar]
- 39.Skubi KL, Blum TR, Yoon TP. Chem Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- 42.Kärkäs MD, Porco JA, Stephenson CRJ. Chem Rev. 2016;116:9683–9747. doi: 10.1021/acs.chemrev.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hedstrand DM, Kruizinga WH, Kellogg RM. Tetrahedron Lett. 1978;19:1255–1258. [Google Scholar]
- 45.Fukuzumi S, Ishikawa K, Hironaka K, Tanaka T. J Chem Soc Perk T 2. 1987;0:751–760. [Google Scholar]
- 46.Shibata T, Kabumoto A, Shiragami T, Ishitani O, Pac C, Yanagida S. J Phys Chem. 1990;94:2068–2076. [Google Scholar]
- 47.Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]
- 48.Du J, Yoon TP. J Am Chem Soc. 2009;131:14604–14605. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du JN, Espelt LR, Guzei IA, Yoon TP. Chem Sci. 2011;2:2115–2119. doi: 10.1039/C1SC00357G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hurtley AE, Cismesia MA, Ischay MA, Yoon TR. Tetrahedron. 2011;67:4442–4448. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang M, Rouch WD, McCulla RD. Eur J Org Chem. 2012:6187–6196. [Google Scholar]
- 52.Petronijevic FR, Nappi M, MacMillan DWC. J Am Chem Soc. 2013;135:18323–18326. doi: 10.1021/ja410478a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rono LJ, Yayla HG, Wang DY, Armstrong MF, Knowles RR. J Am Chem Soc. 2013;135:17735–17738. doi: 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]
- 54.Tarantino KT, Liu P, Knowles RR. J Am Chem Soc. 2013;135:10022–10025. doi: 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]
- 55.Du JN, Skubi KL, Schultz DM, Yoon TP. Science. 2014;344:392–396. doi: 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakajima M, Fava E, Loescher S, Jiang Z, Rueping M. Angew Chem Int Ed. 2015;54:8828–8832. doi: 10.1002/anie.201501556. [DOI] [PubMed] [Google Scholar]
- 57.Amador AG, Sherbrook EM, Yoon TP. J Am Chem Soc. 2016;138:4722–4725. doi: 10.1021/jacs.6b01728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fava E, Millet A, Nakajima M, Loescher S, Rueping M. Angew Chem Int Ed Engl. 2016;55:6776–6779. doi: 10.1002/anie.201511235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fava E, Nakajima M, Nguyen ALP, Rueping M. J Org Chem. 2016;81:6959–6964. doi: 10.1021/acs.joc.6b01006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qi L, Chen YY. Angew Chem Int Ed. 2016;55:13312–13315. doi: 10.1002/anie.201607813. [DOI] [PubMed] [Google Scholar]
- 61.Larraufie MH, Pellet R, Fensterbank L, Goddard JP, Lacote E, Malacria M, Ollivier C. Angew Chem Int Ed. 2011;50:4463–4466. doi: 10.1002/anie.201007571. [DOI] [PubMed] [Google Scholar]
- 62.Wang C, Qin J, Shen X, Riedel R, Harms K, Meggers E. Angew Chem Int Ed. 2016;55:685–688. doi: 10.1002/anie.201509524. [DOI] [PubMed] [Google Scholar]
- 63.Jeffrey JL, Petronijević FR, MacMillan DWC. J Am Chem Soc. 2015;137:8404–8407. doi: 10.1021/jacs.5b05376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uraguchi D, Kinoshita N, Kizu T, Ooi T. J Am Chem Soc. 2015;137:13768–13771. doi: 10.1021/jacs.5b09329. [DOI] [PubMed] [Google Scholar]
- 65.Fuentes de Arriba AL, Urbitsch F, Dixon DJ. Chem Commun. 2016;52:14434–14437. doi: 10.1039/c6cc09172e. [DOI] [PubMed] [Google Scholar]
- 66.Kalyanasundaram K. Coord Chem Rev. 1982;46:159–244. [Google Scholar]
- 67.Huynh MHV, Meyer TJ. Chem Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reduction potentials of the compounds cited in the schemes are obtained from the following references: Cyclopropylketone: Mandell L, Johnston JC, Day RA. J Org Chem. 1978;43:1616–1618.; Enone: Roh Y, Jang HY, Lynch V, Bauld NL, Krische MJ. Org Lett. 2002;4:611–613. doi: 10.1021/ol0172065.; Carbonyl compounds: reference #5. N-Benzylidenemethanesulfonamide: reference #64. Reduction potentials of photoredox catalyst were taken from the references cited throughout the Review.
- 69.Mayer JM. Annu Rev Phys Chem. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- 70.Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Kent CA, Westlake BC, Paul A, Ess DH, McCafferty DG, Meyer TJ. Chem Rev. 2012;112:4016–4093. doi: 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- 71.Yayla HG, Knowles RR. Synlett. 2014;25:2819–2826. [Google Scholar]
- 72.Roughley SD, Jordan AM. J Med Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 73.Van Bergen TJ, Hedstrand DM, Kruizinga WH, Kellogg RM. J Org Chem. 1979;44:4953–4962. [Google Scholar]
- 74.Ono N, Tamura R, Kaji A. J Am Chem Soc. 1980;102:2851–2852. [Google Scholar]
- 75.Nakamura K, Fujii M, Mekata H, Oka S, Ohno A. Chem Lett. 1986;15:87–88. [Google Scholar]
- 76.Tanner DD, Singh HK, Kharrat A, Stein AR. J Org Chem. 1987;52:2142–2146. [Google Scholar]
- 77.Fukuzumi S, Mochizuki S, Tanaka T. J Phys Chem. 1990;94:722–726. [Google Scholar]
- 78.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- 79.Fukuzumi S, Koumitsu S, Hironaka K, Tanaka T. J Am Chem Soc. 1987;109:305–316. [Google Scholar]
- 80.Boujlel K, Martigny P, Simonet J. J Electroanal Chem Interfacial Electrochem. 1983;144:437–442. [Google Scholar]
- 81.Chen WX, Liu Z, Tian JQ, Li J, Ma J, Cheng X, Li GG. J Am Chem Soc. 2016;138:12312–12315. doi: 10.1021/jacs.6b06379. [DOI] [PubMed] [Google Scholar]
- 82.Zhu XQ, Li HR, Li Q, Ai T, Lu JY, Yang Y, Cheng JP. Chem Eur J. 2003;9:871–880. doi: 10.1002/chem.200390108. [DOI] [PubMed] [Google Scholar]
- 83.Sim BA, Griller D, Wayner DDM. J Am Chem Soc. 1989;111:754–755. [Google Scholar]
- 84.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. In: Photochemistry and Photophysics of Coordination Compounds II. Balzani V, Campagna S, editors. Vol. 281. Springer Berlin Heidelberg; Berlin, Heidelberg: 2007. pp. 143–203. [Google Scholar]
- 85.Dange NS, Hussain Jatoi A, Robert F, Landais Y. Org Lett. 2017;19:3652–3655. doi: 10.1021/acs.orglett.7b01651. [DOI] [PubMed] [Google Scholar]
- 86.Dixon IM, Collin JP, Sauvage JP, Flamigni L, Encinas S, Barigelletti F. Chem Soc Rev. 2000;29:385–391. [Google Scholar]
- 87.Ishitani O, Pac C, Sakurai H. J Org Chem. 1983;48:2941–2942. [Google Scholar]
- 88.Ishitani O, Yanagida S, Takamuku S, Pac C. J Org Chem. 1987;52:2790–2796. [Google Scholar]
- 89.Ishitani O, Pac C, Sakurai H. J Org Chem. 1983;48:2941–2942. [Google Scholar]
- 90.Wagner PJ, Truman RJ, Puchalski AE, Wake R. J Am Chem Soc. 1986;108:7727–7738. doi: 10.1021/ja00284a041. [DOI] [PubMed] [Google Scholar]
- 91.Schoeller WW, Niemann J, Rademacher P. J Chem Soc Perk T 2. 1988;0:369–373. [Google Scholar]
- 92.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712–5719. [Google Scholar]
- 93.Pischel U, Zhang X, Hellrung B, Haselbach E, Muller PA, Nau WM. J Am Chem Soc. 2000;122:2027–2034. [Google Scholar]
- 94.Hager D, MacMillan DWC. J Am Chem Soc. 2014;136:16986–16989. doi: 10.1021/ja5102695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Slinker JD, Gorodetsky AA, Lowry MS, Wang J, Parker S, Rohl R, Bernhard S, Malliaras GG. J Am Chem Soc. 2004;126:2763–2767. doi: 10.1021/ja0345221. [DOI] [PubMed] [Google Scholar]
- 96.Ma JJ, Harms K, Meggers E. Chem Commun. 2016;52:10183–10186. doi: 10.1039/c6cc04397f. [DOI] [PubMed] [Google Scholar]
- 97.Cooper BE, Owen WJ. J Organomet Chem. 1971;29:33–40. [Google Scholar]
- 98.Chen M, Zhao X, Yang C, Xia W. Org Lett. 2017;19:3807–3810. doi: 10.1021/acs.orglett.7b01677. [DOI] [PubMed] [Google Scholar]
- 99.Tyson EL, Farney EP, Yoon TP. Org Lett. 2012;14:1110–1113. doi: 10.1021/ol3000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.House HO, Huber LE, Umen MJ. J Am Chem Soc. 1972;94:8471–8475. [Google Scholar]
- 101.Lu Z, Shen M, Yoon TP. J Am Chem Soc. 2011;133:1162–1164. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao G, Yang C, Guo L, Sun H, Lin R, Xia W. J Org Chem. 2012;77:6302–6306. doi: 10.1021/jo300796j. [DOI] [PubMed] [Google Scholar]
- 103.Zhou Z, Li Y, Han B, Gong L, Meggers E. Chem Sci. 2017;8:5757–5763. doi: 10.1039/c7sc02031g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yuan W, Zhou Z, Gong L, Meggers E. Chem Commun. 2017;53:8964–8967. doi: 10.1039/c7cc04941b. [DOI] [PubMed] [Google Scholar]
- 105.Kunai A, Harada J, Nishihara M, Yanagi Y, Sasaki K. Bull Chem Soc Jpn. 1983;56:2442–2446. [Google Scholar]
- 106.Lee KN, Lei Z, Ngai MY. J Am Chem Soc. 2017;139:5003–5006. doi: 10.1021/jacs.7b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Juris A, Balzani V, Belser P, von Zelewsky A. Helv Chim Acta. 1981;64:2175–2182. [Google Scholar]