

Abstract
Background
Multiple myeloma (MM) is the second most common hematologic malignancy. Aberrant epigenetic modifications have been reported in MM and could be promising therapeutic targets. As response rates are overall limited but deep responses occur, it is important to identify those patients who could indeed benefit from epigenetic-targeted therapy.
Methods
Since HDACi and DNMTi combination have potential therapeutic value in MM, we aimed to build a GEP-based score that could be useful to design future epigenetic-targeted combination trials. In addition, we investigated the changes in GEP upon HDACi/DNMTi treatment.
Results
We report a new gene expression-based score to predict MM cell sensitivity to the combination of DNMTi/HDACi. A high Combo score in MM patients identified a group with a worse overall survival but a higher sensitivity of their MM cells to DNMTi/HDACi therapy compared to a low Combo score. In addition, treatment with DNMTi/HDACi downregulated IRF4 and MYC expression and appeared to induce a mature BMPC plasma cell gene expression profile in myeloma cell lines.
Conclusion
In conclusion, we developed a score for the prediction of primary MM cell sensitivity to DNMTi/HDACi and found that this combination could be beneficial in high-risk patients by targeting proliferation and inducing maturation.
Subject terms: Myeloma, Targeted therapies
Introduction
Multiple myeloma (MM) is a most often fatal neoplasia characterized by the accumulation of malignant plasma cells (MMCs) in the bone marrow (BM). The profile of DNA methylation in MM comprises genomic global hypomethylation and simultaneous promoter hypermethylation of known or potential tumor-suppressor genes1, 2. Recently, hypermethylation of several potential suppressor genes was demonstrated to be associated with significantly shorter overall survival (OS)1.
Decitabine (5-aza-2’-deoxycytidine) and 5-azacytidine are both clinically used DNA methyltransferase (DNMT) inhibitors (DNMTi) for the treatment of myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML)3. In MM, clinical trials are ongoing with DNMTi as monotherapy or combined with lenalidomide or dexamethasone4. Histone deacetylases (HDACs) also represent promising molecular targets for the treatment of different cancers, including MM5–15. Romidepsin and Vorinostat (SAHA) are two HDAC inhibitors (HDACi) that have been approved by the Food and Drug Administration (FDA) for the treatment of cutaneous T-cell lymphoma16 and several pan-HDACi are currently evaluated in clinical trials in MM4, 14. Combination of panobinostat/bortezomib/dexamethasone (PANORAMA) and of vorinostat/bortezomib (VANTAGE 088) have been tested in two large phase III clinical trials17, 18. Results from the VANTAGE 088 trial showed that the association of vorinostat and bortezomib significantly prolonged progression-free survival (PFS) in patients with relapsed or refractory MM17. For the PANORAMA trial, re-evaluation of the results recently showed a significant improvement of the PFS when the pan-HDACi panobinostat was combined with bortezomib and dexamethasone in a prespecified subgroup of patients previously exposed to with both bortezomib and an immunomodulatory agent (IMiD) with relapsed MM resulted in a significant PFS improvement. In addition, the overall response rate was also higher: 59 vs 41%. The FDA and European Medicines Agency pproved panobinostat only very recently in patients who have received at least two prior lines of therapy, including bortezomib and an IMiD19–21. However, this combination is associated with high toxicity, including thrombocytopenia (67%), lymphopenia (53%), diarrhoea (26%), and asthenia or fatigue (24%). Several other ongoing trials are evaluating panobinostat in combination with other partners (both standard-of-care agents and targeted therapies) for newly diagnosed or relapsing/refractory MM patients19.
Recently, Matthews et al. investigated the potential of combining panobinostat with a BH3-only mimetic (ABT-737), recombinant human tumor necrosis factor-related apoptosis-inducing ligand (rhTRAIL), or 5-azacitidine, in vivo, using the Vk*MYC transgenic MM mouse model22. HDACi/rhTRAIL or HDACi/ABT-737 combinations are associated with important drug-induced toxicity in vivo. In contrast, HDACi and DNMTi demonstrated a significant reduction of tumor load and prolonged survival of mice without observing major toxicity22, 23. In patients with solid cancers or advanced hematological malignancies, HDACi and DNMTi combination was well tolerated24 and suggested promising activity in MDS, AML16, 24, 25, and refractory advanced non-small cell lung cancer26. Together, these observations suggest that targeting the aberrant tumor-specific epigenetic program simultaneously with DNMTi and HDACi treatment could have therapeutic interest in MM. However, identification of biomarkers predictive for sensitivity of MMCs to epigenetic therapies remains an important objective to improve clinical trials. We recently reported gene expression (gene expression profiling (GEP))-based risk scores to predict the sensitivity of MMCs to DNMTi27, 28 and HDACi28. Since HDACi and DNMTi combination have potential therapeutic value in MM, we aimed to build a GEP-based score that could be useful to design future epigenetic-targeted combination trials. In addition, we investigated the changes in GEP upon HDACi/DNMTi treatment in order to identify mechanisms underlying the enhanced anti-MM activity using human MM cell lines and the 5T33MM model.
Materials and methods
Human myeloma cell lines (HMCLs) and primary MM cells of patients
HMCLs (N = 40) were obtained as previously described29–34 or purchased from DSMZ and American Type Culture Collection. Microarray data are deposited in the ArrayExpress public database (accession numbers E-TABM-937 and E-TABM-1088). Patients presenting with previously untreated MM (N = 206) or monoclonal gammopathy of undetermined significance (MGUS; N = 5) at the university hospitals of Heidelberg and Montpellier, as well as 7 healthy donors have been included in the study approved by the ethics committee of Montpellier and Heidelberg after written informed consent in accordance with the Declaration of Helsinki. Clinical parameters and treatment regimens of the MM patients included in the Heidelberg–Montpellier (HM) cohort were previously described35. GEP of purified MMCs was assayed using Affymetrix U133 2.0 plus microarrays (Affymetrix, Santa Clara, CA, USA) as described36 and data normalized using the MAS5 Affymetrix algorithm. The .CEL and MAS5 files are deposited in the ArrayExpress public database (http://www.ebi.ac.uk/arrayexpress/), under accession number E-MTAB-362. We also used publicly available MAS5 normalized GEP data (GEO, http://www.ncbi.nlm.nih.gov/geo/, accession number GSE2658) from purified MMCs of a cohort of 345 patients treated with total therapy 2 protocol (UAMS-TT2 cohort) at the University of Arkansas for Medical Sciences (UAMS, Little Rock, USA)37. t(4;14) translocation was evaluated using MMSET spike expression38 and del17p13 surrogated by TP53 probe set signal39 for UAMS-TT2 patients. We also used Affymetrix data of 152 relapsed MM patients subsequently treated with bortezomib (GSE9782) from the study of Mulligan et al.40 Gene expression data of normal memory B cells (MB), preplasmablasts, plasmablasts, and early plasma cells41, 42 are deposited in the ArrayExpress databases under accession numbers E-MEXP-2360 and E-MEXP-3034.
Identification of genes deregulated by the HDACi/DNMTi combination
HMCLs (XG-5, XG-6, XG-7, XG-12, XG-19, XG-20, OPM2, RPMI8226, and LP1) were treated with 0.5 μmol/L decitabine (Sigma, St Louis, MO) for 7 days in RPMI 1640, 10% fetal bovine serum supplemented with interleukin (IL)-6 for IL-6-dependent HMCLs. During the last 24 h, 0.33 μmol/L trichostatin A (TSA; Sigma) was added as described by Heller et al.43Whole-genome GEP was assayed with Affymetrix U133 2.0 plus microarrays (Affymetrix).
C57BL/KaLwRij mice were purchased from Harlan CPB (Horst, The Netherlands). Mice were housed according to the conditions approved by the Ethical Committee for Animal Experiments of the Vrije Universiteit Brussel (license no. LA1230281). At day 0, naive C57BL/KaLwRij mice were injected with 5 × 105 5T33MM cells. After establishment of disease (day 16), mice were treated with the combination of decitabine (0.2 mg/kg) (intraperitoneal injection, daily) and quisinostat (1.5 mg/kg) (subcutaneous injection, once every other day) or vehicle. Compounds were kindly provided by Johnson & Johnson (Beerse, Belgium) and used as a filter-sterilized 10% hydroxypropyl-cyclodextran suspension. After 5 days, mice were sacrified and the bone marrow was isolated from hind legs. For mRNA analysis, tumor cells were purified by depletion of CD11b+ contaminating cells. Cytospins were made before and after depletion to count the percentage of plasma cells as described previously23. Samples with >95% plasma cells (N = 4 in each group) were used for RNA isolation using the RNeasy Kit (Qiagen, Venlo, The Netherlands). RNA was further processed and hybridized to the Mouse Genome 430 2.0 Array (Affymetrix, Santa Clara, CA, USA) as previously described23, 27. Microarray data are available at ArrayExpress database (Accession number: E-MTAB-3178).
Sensitivity of primary myeloma cells to HDACi/DNMTi combination
Primary myeloma cells of 10 patients were cultured with or without graded concentrations of decitabine and TSA. Primary myeloma cells of 15 patients were cultured with or without graded concentrations of 5-azacitidine (Sigma) and vorinostat (SAHA) (Sigma). MMC cytotoxicity was evaluated using anti-CD138-PE monoclonal antibody (mAb; Immunotech, Marseille, France) as described27, 44. Results were analyzed using GraphPad Prism (http://www.graphpad.com/scientific-software/prism/).
Statistical analysis
Gene expression data were analyzed using the SAM (Significance Analysis of Microarrays) software45 as published38. The statistical significance of differences in OS between groups of patients was calculated by log-rank test. Multivariate analysis was performed using the Cox proportional hazards model. Survival curves were plotted using the Kaplan–Meier method. All these analyses have been done with R.2.10.1 (http://www.r-project.org/), bioconductor version 2.5 and Genomicscape (http://www.genomicscape.com)46. A histone acetylation/DNA methylation risk score (termed Combo score) was built using our previously published methodology with the decitabine/HDACi combination deregulated prognostic genes27, 28. Briefly, the Combo score was constructed as the sum of the Cox model beta coefficients of each of the decitabine/HDACi combination deregulated genes with a prognostic value, weighted by ±1 according to the patient MMC signal above or below the probeset maxstat value27, 28, 38. Significantly enriched pathways were identified using Reactome functional interaction map. Murine orthologs of HMCL genes were identified using OrthoRetriever tool (https://lighthouse.ucsf.edu/orthoretriever/). Gene set enrichment analysis was carried out by computing overlaps with canonical pathways and gene ontology gene sets obtained from the Broad Institute47. Clustering was performed and visualized with Cluster and TreeView48.
Real-time reverse transcriptase PCR (RT-PCR)
RNA was converted to cDNA using the Qiagen’s QuantiTect Rev. Transcription Kit (Qiagen, Hilden, Germany). The assays-on-demand primers and probes and the TaqMan Universal Master Mix were used according to the manufacturer’s instructions (Applied Biosystems, Courtaboeuf, France). The measurement of gene expression was performed using the Roche LC480 Sequence Detection System. For each primer, serial dilutions of standard cDNA were amplified to create a standard curve, and values of unknown samples were estimated relative to this standard curve in order to assess PCR efficiency. Ct values were obtained for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and the respective genes of interest during the log phase of the cycle. Gene expression was normalized to that of GAPDH (ΔCt = Ct gene of interest−Ct GAPDH) and compared with the values obtained for a known positive control using the following formula: 100/2ΔΔCt where ΔΔCt = ΔCt unknown −ΔCt positive control.
Immunophenotypic analyses
For immunophenotypic analysis, the Cytofix/Cytoperm Kit (BD Biosciences) was used for intracellular staining of IRF-4 (clone IRF4.3E4 from BioLegend (San Diego, CA, USA)) or MYC (clone #12189, Cell Signaling). Flow cytometric analysis was performed with a FORTESSA cytometer using FACSDiva 6.1 (Becton Dickinson, San Jose, CA, USA). The Kaluza software (Beckman Coulter) was used for data analysis. The fluorescence intensity of the cell populations was quantified using the staining index formula: [mean fluorescence intensity (MFI) obtained for a given mAb minus MFI obtained with a control mAb]/[2 times the standard deviation of the MFI obtained with the same control mAb]49.
Results
Identification of genes with deregulated expression by decitabine and HDACi combination and associated with a prognostic value in MM
Using gene expression microarrays, we analyzed gene expression changes in HMCLs after (Supplementary Table S1) combination treatment with sub-lethal doses of decitabine and TSA43. HDACi/DNMTi treatment resulted in a significant upregulation of 375 genes (SAM supervised paired analysis, false discovery rate (FDR) < 5%; Supplementary Table S2) without significant downregulated genes. Reactome analysis revealed a significant enrichment of genes involved in immunological and inflammatory diseases, p53 and TAp63 anti-oncogenic signaling pathways, and cell-to-cell communication signals (Table 1). We also used the syngeneic, immunocompetent 5T33MM to study the in vivo transcriptional response of MM cells to HDACi/DNMTi combination as previously described23. Similar to the HMCL, the sub-lethal doses showing minimal effects on BM plasmacytosis ensuring the yield of good-quality RNA were used (data not shown). In vivo HDACi/DNMTi treatment resulted in a significant deregulation of 415 genes (SAM supervised paired analysis, FDR < 5%; Supplementary Table S3). To identify genes commonly deregulated by the combination in vitro and in vivo, we compared results obtained with the HMCLs and the 5T33MM model. A list of 52 common HDACi/DNMTi deregulated genes was identified (Supplementary Table S4). Using Maxstat R function and Benjamini–Hochberg multiple testing correction38, we then investigated the association between the expression levels of these 52 genes and patient prognosis in order to find genes regulated by HDAC and DNMT that could have an important function in MM pathophysiology. Among the 52 genes, 4 genes had a bad prognostic value and 11 a good one in our cohort of 206 newly diagnosed patients (HM cohort) (Supplementary Table S5). The prognostic information of HDACi/DNMTi combination deregulated genes was gathered in a Combo score as described in the Materials and methods section (Fig. 1a). Combo score values in normal, premalignant, or malignant plasma cells are displayed in Fig. 1b. The Combo score value was not significantly different in plasma cells from MGUS patients compared to normal BM plasma cells (BMPCs). However, MMCs of patients have a significantly higher Combo score than plasma cells from MGUS patients (P < 0.001) and HMCLs had the highest score (P < 0.001) (Fig. 1b). Investigating the Combo score in the eight groups of the molecular classification of MM, the Combo score was significantly higher in the proliferation and hyperdiploid subgroups (P < 0.001, P = 0.001, and P < 0.001, respectively) and significantly lower in the low bone disease, CD2, and MY subgroups (P = 0.02, P = 0.003, and P = 0.002, respectively)50 (Supplementary Figure S1).
Table 1.
Pathway enrichment analysis using Reactome of genes significantly overexpressed in decitabine/TSA-treated HMCLs
| Gene set name | k/K | # Genes in Gene Set (K) | # Genes in Overlap (k) | P-value | FDR q-value | Genes |
|---|---|---|---|---|---|---|
| Interferon alpha/beta signaling (R) | 0.0069 | 67 | 18 | 0.0000 | 5.98E-13 | IFITM1, IFITM2, IFIT1, IFIT3, IFIT2, RSAD2, IFI27, EGR1, STAT1, XAF1, OASL, OAS1, OAS2, IRF7, IRF6, USP18, GBP2, MX1 |
| Interferon gamma signaling (R) | 0.0074 | 72 | 17 | 0.0000 | 1.52E-11 | TRIM22, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, CD44, HLA-DQB1, STAT1, HLA-DPB1, HLA-DRA, OASL, OAS1, OAS2, IRF7, IRF6, GBP2, GBP1 |
| Inflammatory bowel disease (IBD) (K) | 0.0068 | 66 | 13 | 0.0000 | 6.70E-08 | GATA3, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, STAT4, STAT1, HLA-DPB1, IL4R, HLA-DRA |
| Allograft rejection (K) | 0.0039 | 38 | 10 | 0.0000 | 2.37E-07 | PRF1, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HLA-DRA |
| Graft-versus-host disease (K) | 0.0043 | 42 | 10 | 0.0000 | 5.03E-07 | PRF1, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HLA-DRA |
| Asthma (K) | 0.0032 | 31 | 9 | 0.0000 | 5.03E-07 | HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HLA-DRA |
| Cell adhesion molecules (CAMs) (K) | 0.0148 | 144 | 16 | 0.0000 | 8.00E-07 | ALCAM, SDC4, SDC2, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, CDH1, NRCAM, SELL, HLA-DRA, CD6 |
| Antigen processing and presentation (K) | 0.0080 | 78 | 11 | 0.0000 | 1.02E-05 | HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HSPA1A, HSPA2, HLA-DRA |
| Intestinal immune network for IgA production (K) | 0,0049 | 48 | 9 | 0.0000 | 1.15E-05 | HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HLA-DRA |
| Systemic lupus erythematosus (K) | 0.0138 | 135 | 13 | 0.0000 | 5.28E-05 | C3, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HIST1H2AE, HIST1H2BG, HIST1H2BD, HLA-DRA |
| TCR signaling (R) | 0.0067 | 65 | 8 | 0.0000 | 8.54E-04 | PAG1, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DPB1, HLA-DRA |
| MHC class II antigen presentation (R) | 0.0091 | 89 | 9 | 0.0001 | 0.001 | HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HLA-DRA |
| Rheumatoid arthritis (K) | 0.0092 | 90 | 9 | 0.0001 | 0.001 | HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, HLA-DRA |
| Phagosome (K) | 0.0158 | 154 | 11 | 0.0002 | 0.003 | C3, HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DMA, HLA-DMB, HLA-DPB1, CLEC7A, HLA-DRA |
| Costimulation by the CD28 family (R) | 0.0064 | 62 | 7 | 0.0002 | 0.003 | HLA-DQA1, HLA-DRB5, HLA-DRB1, HLA-DPA1, HLA-DQB1, HLA-DPB1, HLA-DRA |
| IL12 signaling mediated by STAT4 (N) | 0.0031 | 30 | 5 | 0.0003 | 0.005 | PRF1, HLA-DRB1, STAT4, HLA-DRA, ETV5 |
| Validated transcriptional targets of TAp63 isoforms (N) | 0.0050 | 49 | 6 | 0.0003 | 0.006 | EGR2, CDKN1A, MFGE8, NQO1, IGFBP3, PMAIP1 |
| T-cell activation (P) | 0.0083 | 81 | 7 | 0.0009 | 0.01 | HLA-DQA1, HLA-DPA1, HLA-DMA, HLA-DMB, ITPR1, HLA-DRA, PIK3CD |
| Direct p53 effectors (N) | 0.0135 | 132 | 9 | 0.0009 | 0.01 | ATF3, TGFA, SFN, CDKN1A, DUSP1, HSPA1A, CD82, IGFBP3, PMAIP1 |
| ECM–receptor interaction (K) | 0.0089 | 87 | 7 | 0.0013 | 0.01 | SDC4, FN1, COL1A1, COL1A2, CD44, ITGA7, LAMB3 |
| IL4-mediated signaling events (N) | 0.0066 | 64 | 6 | 0.0013 | 0.02 | EGR2, PARP14, COL1A1, COL1A2, IL13RA2, IL4R |
| Proteoglycans in cancer (K) | 0.0209 | 204 | 11 | 0.0016 | 0.02 | CBLB, ANK3, SDC4, SDC2, FN1, CDKN1A, CD44, FLNB, ITPR1, HPSE, PIK3CD |
| p53 signaling pathway (K) | 0.0070 | 68 | 6 | 0.0018 | 0.02 | SFN, CDKN1A, CD82, SESN3, IGFBP3, PMAIP1 |
| Calcineurin-regulated NFAT-dependent transcription in lymphocytes (N) | 0.0048 | 47 | 5 | 0.0020 | 0.02 | CBLB, GATA3, EGR1, EGR2, GBP3 |
| Apoptotic execution phase (R) | 0.0048 | 47 | 5 | 0.0020 | 0.02 | GSN, TJP2, CDH1, H1F0, DSP |
| Proteoglycan syndecan-mediated signaling events (N) | 0.0004 | 4 | 2 | 0.0026 | 0.03 | SDC4, SDC2 |
| ErbB1 downstream signaling (N) | 0.0103 | 100 | 7 | 0.0028 | 0.03 | NCKAP1, EPS8, SFN, EGR1, DUSP1, CHN2, STAT1 |
Gene annotation and networks were generated with the Reactome Functional Interaction Cytoscape plugin (http://www.cytoscape.org/)
Fig. 1.

Clustergram of the signals of the 15 genes used to build the Combo score in myeloma cells of 206 previously untreated patients. a The signals of the 15 probe sets in MMCs of 206 patients, ordered by increasing Combo score, are displayed from low (deep blue) to high (deep red) expression. Among the 15 probe sets, the genes associated with a poor outcome are identified by a red frame. b Combo score in normal bone marrow plasma cells (n = 7), in premalignant plasma cells of patients with monoclonal gammopathy of undetermined significance (MGUS, n = 5), in multiple myeloma cells of patients with intramedullary MM (n = 206), and in human myeloma cell lines (n = 40)
Evaluation of the prognostic significance of the Combo score
Using a maxstat analysis for OS51, the Combo score was significantly associated with high-risk myeloma in the two independent cohorts of newly diagnosed patients, namely, HM and UAMS-TT2 (Fig. 2a, b). Maxstat statistic test split the HM patient cohort into two groups: a high-risk group of 48.6 % patients (Combo score > −6.55) with a 54.9 months median OS and a low-risk group of 51.4% patients (Combo score ≤ −6.55) with not reached median survival (P = 7E−9; Fig. 2a). In the UAMS-TT2 cohort, a Combo score >−6.55 is also associated with a high risk (P = 0.002; Fig. 2b) in 47.2% of the patients. Next, we investigated whether the Combo score could also have a prognostic value in relapsed patients. Using the Mulligan cohort, including patients treated with bortezomib monotherapy after relapse, the Combo score kept prognostic value (P = 0.003; Fig. 2c).
Fig. 2.

Prognostic value of Combo score in multiple myeloma. a Patients of the HM cohort were ranked according to increased Combo score and a maximum difference in OS was obtained with Combo score = −6.55 splitting patients into high-risk (48.6%) and low-risk (51.4%) groups. b The prognostic value of Combo score was validated using an independent cohort of 345 patients from UAMS treated with TT2 therapy (UAMS-TT2 cohort). The parameters to compute the Combo score of patients of UAMS-TT2 cohort and the proportions delineating the two prognostic groups were those defined in the HM cohort. c The prognostic value of Combo score was validated in a cohort of 188 patients at relapse treated with bortezomib monotherapy (Mulligan cohort)40
The prognostic value of the Combo score was then compared to usual prognostic factors (β2M, ISS, t(4;14), and del17p) and the published GEP-based risk scores, i.e., UAMS-HRS52, IFM score53, RS score54, proliferation scores, GPI35, and scores surrogating response to treatment DM score27 and HA score28. In univariate COX analysis, all of these factors had prognostic value (Supplementary Table S5). Compared two by two in multivariate COX analysis, Combo score, ISS, IFM score, t(4;14), GPI, DM score, HA score, and β2M remained independent in the HM cohort. In the UAMS-TT2 cohort, when compared two by two, Combo score tested with HRS, IFM score, t(4;14), del17p, RS, GPI, HA score, and DM score remained independent prognostic factors. When tested all together, Combo score, β2M, and RS remained independent in the HM cohort, whereas UAMS-HRS, t(4;14), and del17p were independent in the UAMS-TT2 cohort (Supplementary Table S6).
The Combo score is predictive for myeloma cell sensitivity to DNMTi and HDACi combination
The efficacy of the Combo score to predict sensitivity of myeloma cells to DNMTi/HDACi treatment was investigated using primary MMC of patients co-cultured with their bone marrow microenvironment in vitro27, 28. MMC of patients with a high Combo score (n = 5) were significantly more sensitive to decitabine and TSA combination than MMC of patients with a low Combo score (n = 5) (Fig. 3a). We confirmed these results using another DNMTi and HDACi association. Primary MMCs of patients with a high Combo score (n = 7) exhibited a significant higher sensitivity to the clinical grade inhibitors 5-azacitidine/SAHA combination than MMC of patients with a low Combo score (n = 8) (Fig. 3b). Altogether, these data indicated that patients with high risk identified using the Combo score could benefit from HDACi/DNMTi treatment.
Fig. 3.

Combo score predicts for sensitivity of primary myeloma cells of patients to HDACi/DNMTi combined treatment. a Mononuclear cells from tumor samples of 10 patients with MM were cultured for 4 days in the presence of IL-6 (2 ng/ml) with or without graded decitabine and TSA concentrations. At day 4 of culture, the count of viable CD138+ MMCs was determined using flow cytometry. The gray columns represent the mean ± SD of primary myeloma cell counts (expressed as the percentage of the count without adding drugs) of the five patients with a low Combo score and the white columns that of the five patients with a high Combo score. b 5-Azacitidine and SAHA combination was also investigated using samples of 15 myeloma patients. The gray columns represent the mean ± SD of primary myeloma cell counts (expressed as the percentage of the count without adding drugs) of the 8 patients with a low Combo score and the white columns that of the 7 patients with a high Combo score
MMC of patients with a low Combo score value are characterized by a mature BMPC gene signature, whereas patients with high Combo score have a proliferating and MYC-associated gene signatures
In order to evaluate whether different gene signatures could be identified comparing the high and low Combo score groups, we performed a gene set enrichment analysis (GSEA). MMC of patients with a low Combo score displayed a significant enrichment in genes associated with normal mature BMPCs (gene set: MOREAUX MULTIPLE MYELOMA BY TACI UP, P < 0.0001, Supplementary Figure S2 and Supplementary Table S7) and bone microenvironment dependence (gene sets: KEGG CYTOKINE CYTOKINE RECEPTOR INTERACTION, P < 0.0001, Supplementary Figure S2 and Supplementary Tables S8). In contrast, MMCs of patients with a high Combo score exhibited a significant enrichment in genes associated with proliferating plasmablastic progenitors (gene sets: WHITFIELD CELL CYCLE S, REACTOME CELL CYCLE P < 0.0001 and P = 0.01, Supplementary Figure S3 and Supplementary Tables S9 and S10), MYC deregulation (gene sets: ODONNEL TARGETS OF MYC, DANG MYC TARGETS UP, P < 0.0001 and 0.002, Supplementary Figure S3 and Supplementary Table S11 and S12), and transcription (gene set: REACTOME TRANSCRIPTION, P < 0.0001, Supplementary Figure S3 and supplementary Table S13). Investigating the Combo score in normal plasma cell differentiation, the Combo score value was significantly higher in preplasmablasts (PrePB, P = 0.03) compared to MB cells and in plasmablasts (PB, P = 0.04) compared to preplasmablasts (Fig. 4). Early plasma cells were found to have the highest score (P < 0.001) and the Combo score decreased drastically to the lowest value in mature BMPC (P < 0.001) (Fig. 4).
Fig. 4.
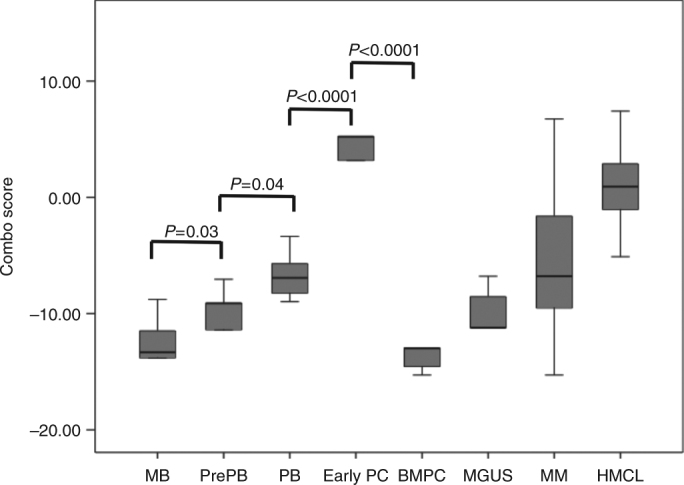
Combo score in normal plasma cell differentiation. Combo score in normal memory B cells (MB, n = 5), normal preplasmablasts (PrePB, n = 5), normal plasmablasts (PB, n = 5), normal early plasma cells (Early PC, n = 5), normal bone marrow plasma cells (n = 5), in premalignant plasma cells of patients with monoclonal gammopathy of undetermined significance (MGUS, n = 5), multiple myeloma cells of patients with intramedullary MM (n = 206), and human myeloma cell lines (n = 40)
HDACi/DNMTi combination results in significant downregulation of IRF4 and MYC expression
Since MM patients with a high Combo score are characterized by deregulation of MYC target genes, we next investigated the link between DNMTi/HDACi combined treatment and MYC deregulation using nine different HMCLs. Interestingly, GSEA analysis underlined that DNMTi/HDACi treatment results in a significant downregulation of genes silenced by H3K27 and DNA methylation, HDAC, EZH2, IRF4, and MYC target genes, suggesting that the effects of the combination could be mediated by IRF4 and MYC downregulation (Table 2). IRF4 and MYC downregulation after HDACi/DNMTi treatment was validated at the mRNA level by using QPCR (Fig. 5a) and at the protein level (Fig. 5b). Of note, this strong common downregulation of MYC and IRF4 expression was higher compared to DNMTi or HDACi treatment alone (Supplementary Figure S4A-B).
Table 2.
Top gene set significantly enriched in untreated compared to MMC (n=9) treated by DNMTi/HDACi combination
| Gene set name | Genes in gene set (K) | Description | Genes in overlap (k) | k/K | P-value | FDR q-value |
|---|---|---|---|---|---|---|
| HELLER_HDAC_TARGETS_SILENCED_BY_METHYLATION_UP | 461 | Genes upregulated in multiple myeloma (MM) cell lines treated with both decitabine [PubChem=451668] TSA [PubChem=5562] | 49 | 0.1063 | 8.57E-31 | 4.06E-27 |
| SENESE_HDAC3_TARGETS_DN | 536 | Genes downregulated in U2OS cells (osteosarcoma) upon knockdown of HDAC3 [GeneID=8841] by RNAi | 41 | 0.0765 | 1.58E-20 | 1.07E-17 |
| PILON_KLF1_TARGETS_DN | 1972 | Genes downregulated in erythroid progenitor cells from fetal livers of E13.5 embryos with KLF1 [GeneID=10661] knockout compared to those from the wild-type embryos | 78 | 0.0396 | 7.34E-20 | 4.35E-17 |
| HELLER_SILENCED_BY_METHYLATION_DN | 105 | Genes downregulated in at least one of the three multiple myeloma (MM) cell lines treated with the DNA hypomethylating agent decitabine (5-aza-2'-deoxycytidine) [PubChem=451668] | 20 | 0.1905 | 2.33E-18 | 1.03E-15 |
| HELLER_HDAC_TARGETS_SILENCED_BY_METHYLATION_DN | 281 | Genes downregulated in multiple myeloma (MM) cell lines treated with both decitabine [PubChem=451668] TSA [PubChem=5562] | 29 | 0.1032 | 2.39E-18 | 1.03E-15 |
| MEISSNER_BRAIN_HCP_WITH_H3K4ME3_AND_H3K27ME3 | 1069 | Genes with high-CpG-density promoters (HCP) bearing histone H3 dimethylation at K4 (H3K4me2) and trimethylation at K27 (H3K27me3) in the brain | 52 | 0.0486 | 4.33E-17 | 1.47E-14 |
| HELLER_HDAC_TARGETS_UP | 317 | Genes upregulated in at least one of the three multiple myeloma (MM) cell lines by TSA [PubChem=5562] | 29 | 0.0915 | 6.26E-17 | 1.98E-14 |
| NUYTTEN_EZH2_TARGETS_UP | 1037 | Genes upregulated in PC3 cells (prostate cancer) after knockdown of EZH2 [GeneID=2146] by RNAi | 49 | 0.0473 | 1.13E-15 | 2.97E-13 |
| NUYTTEN_EZH2_TARGETS_DN | 1024 | Genes downregulated in PC3 cells (prostate cancer) after knockdown of EZH2 [GeneID=2146] by RNAi | 46 | 0.0449 | 5.41E-14 | 1.03E-11 |
| HELLER_SILENCED_BY_METHYLATION_UP | 282 | Genes upregulated in at least one of the three multiple myeloma (MM) cell lines treated with the DNA hypomethylating agent decitabine (5-aza-2'-deoxycytidine) [PubChem=451668] | 24 | 0.0851 | 1.50E-13 | 2.49E-11 |
| CHICAS_RB1_TARGETS_CONFLUENT | 567 | Genes upregulated in confluent IMR90 cells (fibroblast) after knockdown of RB1 [GeneID=5925] by RNAi | 33 | 0.0582 | 2.26E-13 | 3.45E-11 |
| HELLER_HDAC_TARGETS_DN | 292 | Genes downregulated in at least one of the three multiple myeloma (MM) cell lines by TSA [PubChem=5562] | 24 | 0.0822 | 3.20E-13 | 4.74E-11 |
| DANG_BOUND_BY_MYC | 1103 | Genes whose promoters are bound by MYC [GeneID=4609], according to MYC Target Gene Database | 46 | 0.0417 | 7.35E-13 | 9.56E-11 |
| ZHAN_MULTIPLE_MYELOMA_CD1_VS_CD2_UP | 66 | Genes upregulated in CD-1 compared to CD-2 cluster of multiple myeloma samples | 13 | 0.197 | 1.32E-12 | 1.53E-10 |
| WEI_MYCN_TARGETS_WITH_E_BOX | 795 | Genes whose promoters contain E-box motifs and whose expression changed in MYCN-3 cells (neuroblastoma) upon induction of MYCN [GeneID=4613] | 37 | 0.0465 | 6.23E-12 | 6.71E-10 |
| MARTINEZ_TP53_TARGETS_DN | 593 | Genes downregulated in mice with skin-specific knockout of TP53 [GeneID=7157] | 31 | 0.0523 | 1.89E-11 | 1.79E-09 |
| MARTINEZ_RB1_TARGETS_UP | 673 | Genes upregulated in mice with skin-specific knockout of RB1 [GeneID=5925] by Cre-lox | 32 | 0.0475 | 1.00E-10 | 7.29E-09 |
| PUJANA_ATM_PCC_NETWORK | 1442 | Genes constituting the ATM-PCC network of transcripts whose expression positively correlated (Pearson correlation coefficient, PCC≥0.4) with that of ATM [GeneID=472] across a compendium of normal tissues | 49 | 0.034 | 1.75E-10 | 1.23E-08 |
| SHAFFER_IRF4_TARGETS_IN_MYELOMA_VS_MATURE_B_LYMPHOCYTE | 101 | IRF4 [GeneID=3662] target genes up-regulated in primary myeloma vs. mature B lymphocytes | 13 | 0.1287 | 3.57E-10 | 2.32E-08 |
| MARTINEZ_RB1_AND_TP53_TARGETS_DN | 591 | Genes downregulated in mice with skin-specific double knockout of both RB1 and TP53 [GeneID=5925;7157] by Cre-lox | 29 | 0.0491 | 3.71E-10 | 2.37E-08 |
| SENESE_HDAC1_TARGETS_UP | 457 | Genes upregulated in U2OS cells (osteosarcoma) upon knockdown of HDAC1 [GeneID=3065] by RNAi | 25 | 0.0547 | 6.79E-10 | 3.92E-08 |
| BILD_MYC_ONCOGENIC_SIGNATURE | 206 | Genes selected in supervised analyses to discriminate cells expressing c-Myc [GeneID=4609] from control cells expressing GFP | 17 | 0.0825 | 8.63E-10 | 4.92E-08 |
| CHICAS_RB1_TARGETS_GROWING | 243 | Genes upregulated in growing IMR90 cells (fibroblast) after knockdown of RB1 [GeneID=5925] by RNAi | 18 | 0.0741 | 1.57E-09 | 8.46E-08 |
| BENPORATH_CYCLING_GENES | 648 | Genes showing cell-cycle stage-specific expression [PMID=12058064] | 29 | 0.0448 | 2.99E-09 | 1.48E-07 |
| REACTOME_CELL_CYCLE | 421 | Genes involved in cell cycle | 23 | 0.0546 | 3.36E-09 | 1.61E-07 |
| CHICAS_RB1_TARGETS_SENESCENT | 572 | Genes upregulated in senescent IMR90 cells (fibroblast) after knockdown of RB1 [GeneID=5925] by RNAi | 27 | 0.0472 | 3.44E-09 | 1.63E-07 |
Fig. 5.

HDACi/DNMTi induces a significant downregulation of IRF4 and MYC expression. a Seven HMCLs were treated for 7 days with decitabine (DNMTi) and TSA during the last 24 h. Data are the MYC and IRF4 gene expression in treated HMCLs compared to the control (arbitrary signal = 100) assayed using real-time PCR. b Protein expression of MYC and IRF4 was assayed using flow cytometry in three HMCLs, namely XG7, XG12, and XG19
HDACi/DNMTi combination results in epigenetic reprogramming of MMCs
As we identified HDACi/DNMTi combined treatment is associated with MYC/IRF4 axis dysregulation thought to play a role in MM pathophysiology, we sought to determine whether HDACi/DNMTi treatment is associated with an epigenetic reprogramming of MMCs. Cluster analysis of the top 100 genes deregulated by HDACi/DNMTi (GSEA analysis) revealed that treated HMCLs cluster with normal BMPC, suggesting that HDACi/DNMTi combined treatment induced a normal BMPC gene program (Fig. 6). Altogether, these data indicate an epigenetic reprogramming of MMCs by HDACi/DNMTi combination, associated with MYC/IRF4 axis targeting and normal BMPC gene expression pattern induction.
Fig. 6.

HDACi/DNMTi induces a normal BMPC gene expression program in MM cells. The signals of the top 100 genes deregulated by HDACi/DNMTi in myeloma cell lines (n = 49), myeloma cell lines treated by DNMTi/HDACi (n = 9), normal memory B cells (MB, n = 5), normal plasmablasts (PB, n = 5), and normal bone marrow plasma cells (n = 5) are displayed from low (deep blue) to high (deep red) expression
Discussion
Clinical trials suggested promising activity of HDACi/DNMTi combination in MDS, AML16, 24, 25, and refractory advanced non-small cell lung cancer26. In MM, the oral HDACi panobinostat was recently approved by the US Food and Drug Administration for use in combination with bortezomib and dexamethasone in patients with relapsed MM19. In addition, ongoing trials are investigating the therapeutic interest of panobinostat with other agents including next-generation proteasome inhibitors (carfilzomib and ixazomib), IMiD and dexamethasone, bortezomib and IMiD, or monoclonal antibodies in relapsed/refractory MM patients19. Moreover, DNMTi and HDACi combination resulted in a significant anti-myeloma activity in the Vk*MYC transgenic MM and 5T33MM mouse models22, 23. Importantly, cooperation between histone modifications and DNA methylation is known to be important for the establishment of global epigenetic patterns as well as loci-specific gene regulation55. This crosstalk can be mediated by biochemical interactions between SET domain histone methyltransferases and DNMTs55. Only upregulated genes were identified in the HMCLs treated with DNMTi/HDACi compared with untreated HMCLs with a significant enrichment in genes involved in immunological and inflammatory diseases, p53 and TAp63 signaling pathways, and intercellular communication signals. Significant induction of TP53 and TP53 homolog TAp63 target genes known to exert anti-oncogenic roles in cancers56 and could participate in the MMC toxicity of HDACi/DNMTi combination.
In this study, we constructed a GEP-based Combo score that allows identification of high-risk patients associated with MMC’s higher sensitivity to HDACi/DNMTi combination in vitro. Since HDACi/DNMTi combination are well tolerated24 showing promising activity in cancers (including hematological malignancies16, 24–26) and have potential therapeutic value in MM22, 23, the Combo score could enable the identification of MM patients who could benefit from this treatment.
The 15 genes used to build the Combo score, included 4 genes associated with a bad prognosis and 11 associated with a good prognosis (Fig. 1). Among these genes, some of them could highlight pathways involved in MM biology and sensitivity to DNMTi/HDACi combination. Patients with high Combo score, and poor survival, are characterized by a higher expression of the 4 bad prognostic genes and a lower expression of the 11 good ones in MMCs. Primary MMCs of patients with high Combo score can be efficiently targeted by the upregulation of gene products encoded by genes upregulated by HDACi/DNMTi, in particular the 11 good prognostic genes. A full understanding of the reason why the Combo score could predict for HDACi/DNMTi sensitivity will be provided by an extensive study of the function of the products encoded by HDACi/DNMTi-regulated genes in MMC survival and/or proliferation. Among them, NFKBIZ was identified. NFKBIZ codes the inhibitor of nuclear factor-κB (NFκB) (IκB) protein IκBζ. IκBζ is an atypical and mainly nuclear IκB protein working as a co-transcription factor of NFκB to modulate the expression of NFκB target genes57. NFκB is a major pathway involved in MMC survival and is activated by various gene mutations58–60. NFKBIZ overexpression was reported to induce cell death61 and inhibit transcriptional activity of STAT3 with Mcl1 downregulation and apoptosis induction62. Mcl1 is known to be the major antiapoptotic protein involved in MMC survival63, 64. This significant overexpression of NFKBIZ after DNMTi/HDACi treatment was validated by quantitative RT-PCR in seven HMCLs (Supplementary Figure S5). As a significant enrichment in genes associated with proliferation was identified in MMC of patients with a high Combo score value, the higher sensitivity of high Combo score patients to DNMTi/HDACi combination could be explained by the fact that incorporation of DNMTi into DNA is restricted to cell cycling cells3. Furthermore, HDACi have been shown to induce G1 cell cycle arrest through dephosphorylation of retinoblastoma protein and increase expression of p53 and p219, 12, 14. Using methylation-specific PCR, several studies have identified hypermethylation of tumor-suppressor genes including cyclin-dependent kinase inhibitors (CDKI, p15, and p16) and p1465–67. DNMTi/HDACi treatment induced p21 and p57 CDKI expression in MMC (Supplementary Table S2). Thus DNMTi/HDACi combination could be useful to induce the expression of major tumor-suppressor genes in MMC.
Furthermore, core histone proteins must be synthetized rapidly during the brief S-phase when a cell is dividing68. As a result, the histone mRNAs are highly cell cycle regulated, increasing 35-fold as cells enter S-phase and decreasing again at the end of S-phase68. Altogether, these data could clarify why MMC of high Combo score patients, distinguished by an active growth, can be efficiently targeted by HDACi/DNMTi treatment.
Most interestingly, HDACi/DNMTi treatment induced reprogramming of MMC through IRF4 and MYC axis targeting and induction of a normal BMPC gene expression program. IRF4 is a transcription factor critical for normal plasma cell development69. Abnormal activity of IRF4 linked to a translocation or the presence of superenhancers plays a key role in MM development70, 71. IRF4 is overexpressed in a subset a patients harboring t(6;14)(p25;q32) translocation70. MYC is an IRF4 target presenting prominent role in MM pathogenesis69. We report here that HDACi/DNMTi combination triggers a significant downregulation of MYC, IRF4, and several IRF4-MYC target genes. Interestingly, this strong common downregulation of MYC and IRF4 expression was not identified after DNMTi or HDACi treatment alone27, 28. It was demonstrated that IMiDs could target IRF4 and MYC expression through binding to cereblon E3 ubiquitin ligase that promote proteasomal degradation of IKZF1 and IKZF3 transcriptional factors72, 73.
These data suggest that DNMTi/HDACi could be of therapeutic interest in combination with IMiDs in high-risk MM with elevated Combo score value.
Electronic supplementary material
Acknowledgements
We thank the Microarray Core Facility of IRMB (http://irb.montp.inserm.fr/en/index.php?page = Plateau&IdEquipe = 6). This work was supported by grants from French INCA (Institut National du Cancer) Institute (2012-109/087437 and PLBIO15-256), Languedoc Roussillon CRLR (R14026FF), LR-FEDER Hemodiag, Fondation de France (201400047510), ITMO Cancer (MM&TT), SIRIC Montpellier (INCa-DGOS-Inserm 6045), Kom Op tegen Kanker, Stichting tegen Kanker and Fonds voor Wetenschappelijk Onderzoek, Belgium, the German Federal Ministry of Education (BMBF) “CAMPSIMM” (01ES1103) and within the framework of the e:Med research and funding concept “CLIOMMICS” (01ZX1309), and the Deutsche Forschungsgemeinschaft (SFB/TRR79; subproject B1). L.H. is supported by a grant from Labex EpiGenMed.
Author contributions
A.B. and K.M. designed the research and wrote the paper. H.D., A.S., C.M.-T., L.V., S.B., G.C., and G.H. collected bone marrow samples and clinical data and participated in the writing of the paper. A.K., R.T., and J.-l.V. participated in the bioinformatics studies. N.R., G.R., and S.B. provided with technical assistance. K.V., L.H., and B.K. participated in the research and wrote the paper. E.D.B. and J.M. designed and supervised the research and wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors share first authorship: Angelique Bruyer, Ken Maes.
These authors contributed equally: Elke De Bruyne, Jerome Moreaux.
Electronic supplementary material
Supplementary information is available for this paper at 10.1038/s41416-018-0025-x.
References
- 1.Heuck CJ, et al. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J. Immunol. 2013;190:2966–2975. doi: 10.4049/jimmunol.1202493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker BA, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2010;117:553–562. doi: 10.1182/blood-2010-04-279539. [DOI] [PubMed] [Google Scholar]
- 3.Hollenbach PW, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE. 2010;5:e9001. doi: 10.1371/journal.pone.0009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maes K, et al. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers. 2013;5:430–461. doi: 10.3390/cancers5020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng R, et al. KD5170, a novel mercaptoketone-based histone deacetylase inhibitor, exerts antimyeloma effects by DNA damage and mitochondrial signaling. Mol. Cancer Ther. 2008;7:1494–1505. doi: 10.1158/1535-7163.MCT-08-0183. [DOI] [PubMed] [Google Scholar]
- 6.Hideshima T, et al. Induction of differential apoptotic pathways in multiple myeloma cells by class-selective histone deacetylase inhibitors. Leukemia. 2013;28:457–460. doi: 10.1038/leu.2013.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaiser M, et al. The effects of the histone deacetylase inhibitor valproic acid on cell cycle, growth suppression and apoptosis in multiple myeloma. Haematologica. 2006;91:248–251. [PubMed] [Google Scholar]
- 8.Khan SB, Maududi T, Barton K, Ayers J, Alkan S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br. J. Haematol. 2004;125:156–161. doi: 10.1111/j.1365-2141.2004.04882.x. [DOI] [PubMed] [Google Scholar]
- 9.Lavelle D, Chen YH, Hankewych M, DeSimone J. Histone deacetylase inhibitors increasep21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am. J. Hematol. 2001;68:170–178. doi: 10.1002/ajh.1174. [DOI] [PubMed] [Google Scholar]
- 10.Minami J, et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia. 2013;28:680–689. doi: 10.1038/leu.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitsiades CS, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc. Natl. Acad. Sci. USA. 2004;101:540–545. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitsiades N, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–4062. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- 13.Catley L, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–2622. doi: 10.1182/blood-2003-01-0233. [DOI] [PubMed] [Google Scholar]
- 14.Neri P, Bahlis NJ, Lonial S. Panobinostat for the treatment of multiple myeloma. Expert Opin. Investig. Drugs. 2012;21:733–747. doi: 10.1517/13543784.2012.668883. [DOI] [PubMed] [Google Scholar]
- 15.Neri P, et al. In vivo anti-myeloma activity and modulation of gene expression profile induced by valproic acid, a histone deacetylase inhibitor. Br. J. Haematol. 2008;143:520–531. doi: 10.1111/j.1365-2141.2008.07387.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang QL, et al. The proteasome inhibitor bortezomib interacts synergistically with the histone deacetylase inhibitor suberoylanilide hydroxamic acid to induce T-leukemia/lymphoma cells apoptosis. Leukemia. 2009;23:1507–1514. doi: 10.1038/leu.2009.41. [DOI] [PubMed] [Google Scholar]
- 17.Dimopoulos M, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 2013;14:1129–1140. doi: 10.1016/S1470-2045(13)70398-X. [DOI] [PubMed] [Google Scholar]
- 18.Richardson PG, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood. 2013;122:2331–2337. doi: 10.1182/blood-2013-01-481325. [DOI] [PubMed] [Google Scholar]
- 19.Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015;21:4767–4773. doi: 10.1158/1078-0432.CCR-15-0530. [DOI] [PubMed] [Google Scholar]
- 20.Richardson PG, et al. Pomalidomide, bortezomib and low-dose dexamethasone in lenalidomide-refractory and proteasome inhibitor-exposed myeloma. Leukemia. 2017;31:2695–2701. doi: 10.1038/leu.2017.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.San-Miguel JF, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15:1195–1206. doi: 10.1016/S1470-2045(14)70440-1. [DOI] [PubMed] [Google Scholar]
- 22.Matthews GM, et al. Preclinical screening of histone deacetylase inhibitors combined with ABT-737, rhTRAIL/MD5-1 or 5-azacytidine using syngeneic Vk*MYC multiple myeloma. Cell Death Dis. 2013;4:e798. doi: 10.1038/cddis.2013.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maes K, et al. In vivo treatment with epigenetic modulating agents induces transcriptional alterations associated with prognosis and immunomodulation in multiple myeloma. Oncotarget. 2015;6:3319–3334. doi: 10.18632/oncotarget.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin. Cancer Res. 2009;15:3970–3977. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
- 25.Fandy TE, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114:2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juergens RA, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreaux J, et al. Development of gene expression-based score to predict sensitivity of multiple myeloma cells to DNA methylation inhibitors. Mol. Cancer Ther. 2012;11:2685–2692. doi: 10.1158/1535-7163.MCT-12-0721. [DOI] [PubMed] [Google Scholar]
- 28.Moreaux J, et al. Gene expression-based prediction of myeloma cell sensitivity to histone deacetylase inhibitors. Br. J. Cancer. 2013;109:676–685. doi: 10.1038/bjc.2013.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu ZJ, et al. Agonist anti-gp130 transducer monoclonal antibodies are human myeloma cell survival and growth factors. Leukemia. 2000;14:188–197. doi: 10.1038/sj.leu.2401632. [DOI] [PubMed] [Google Scholar]
- 30.Moreaux J, et al. TACI expression is associated with a mature bone marrow plasma cell signature and C-MAF overexpression in human myeloma cell lines. Haematologica. 2007;92:803–811. doi: 10.3324/haematol.10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreaux J, et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica. 2011;96:574–582. doi: 10.3324/haematol.2010.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebouissou C, et al. A gp130 interleukin-6 transducer-dependent SCID model of human multiple myeloma. Blood. 1998;91:4727–4737. [PubMed] [Google Scholar]
- 33.Tarte K, et al. Induced expression of B7-1 on myeloma cells following retroviral gene transfer results in tumor-specific recognition by cytotoxic T cells. J. Immunol. 1999;163:514–524. [PubMed] [Google Scholar]
- 34.Zhang XG, et al. Reproducible obtaining of human myeloma cell lines as a model for tumor stem cell study in human multiple myeloma. Blood. 1994;83:3654–3663. [PubMed] [Google Scholar]
- 35.Hose D, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica. 2011;96:87–95. doi: 10.3324/haematol.2010.030296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Vos J, et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene. 2002;21:6848–6857. doi: 10.1038/sj.onc.1205868. [DOI] [PubMed] [Google Scholar]
- 37.Barlogie B, et al. Total therapy 2 without thalidomide in comparison with total therapy 1: role of intensified induction and posttransplantation consolidation therapies. Blood. 2006;107:2633–2638. doi: 10.1182/blood-2005-10-4084. [DOI] [PubMed] [Google Scholar]
- 38.Kassambara A, et al. Genes with a spike expression are clustered in chromosome (sub)bands and spike (sub)bands have a powerful prognostic value in patients with multiple myeloma. Haematologica. 2012;97:622–630. doi: 10.3324/haematol.2011.046821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiong W, et al. An analysis of the clinical and biologic significance of TP53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood. 2008;112:4235–4246. doi: 10.1182/blood-2007-10-119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulligan G, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109:3177–3188. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- 41.Jourdan M, et al. Characterization of a transitional preplasmablast population in the process of human B cell to plasma cell differentiation. J. Immunol. 2011;187:3931–3941. doi: 10.4049/jimmunol.1101230. [DOI] [PubMed] [Google Scholar]
- 42.Jourdan M, et al. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood. 2009;114:5173–5181. doi: 10.1182/blood-2009-07-235960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heller G, et al. Genome-wide transcriptional response to 5-aza-2’-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008;68:44–54. doi: 10.1158/0008-5472.CAN-07-2531. [DOI] [PubMed] [Google Scholar]
- 44.Mahtouk K, et al. An inhibitor of the EGF receptor family blocks myeloma cell growth factor activity of HB-EGF and potentiates dexamethasone or anti-IL-6 antibody-induced apoptosis. Blood. 2004;103:1829–1837. doi: 10.1182/blood-2003-05-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kassambara A, et al. GenomicScape: an easy-to-use web tool for gene expression data analysis. Application to investigate the molecular events in the differentiation of B cells into plasma cells. PLoS Comput. Biol. 2015;11:e1004077. doi: 10.1371/journal.pcbi.1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jourdan M, et al. Differential effects of lenalidomide during plasma cell differentiation. Oncotarget. 2016;7:28096–28111. doi: 10.18632/oncotarget.8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhan F, et al. The molecular classification of multiple myeloma. Blood. 2006;108:2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hothorn T, Lausen B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 2003;43:121–137. doi: 10.1016/S0167-9473(02)00225-6. [DOI] [Google Scholar]
- 52.Shaughnessy JD, Jr., et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 53.Decaux O, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myelome. J. Clin. Oncol. 2008;26:4798–4805. doi: 10.1200/JCO.2007.13.8545. [DOI] [PubMed] [Google Scholar]
- 54.Reme T, Hose D, Theillet C, Klein B. Modeling risk stratification in human cancer. Bioinformatics. 2013;29:1149–1157. doi: 10.1093/bioinformatics/btt124. [DOI] [PubMed] [Google Scholar]
- 55.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 56.Humphries LA, et al. Pro-apoptotic TP53 homolog TAp63 is repressed via epigenetic silencing and B-cell receptor signalling in chronic lymphocytic leukaemia. Br. J. Haematol. 2013;163:590–602. doi: 10.1111/bjh.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hildebrand DG, et al. IkappaBzeta is a transcriptional key regulator of CCL2/MCP-1. J. Immunol. 2013;190:4812–4820. doi: 10.4049/jimmunol.1300089. [DOI] [PubMed] [Google Scholar]
- 58.Bolli N, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lohr JG, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walker BA, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015;33:3911–3920. doi: 10.1200/JCO.2014.59.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Totzke G, et al. A novel member of the IkappaB family, human IkappaB-zeta, inhibits transactivation of p65 and its DNA binding. J. Biol. Chem. 2006;281:12645–12654. doi: 10.1074/jbc.M511956200. [DOI] [PubMed] [Google Scholar]
- 62.Wu Z, et al. Nuclear protein IkappaB-zeta inhibits the activity of STAT3. Biochem Biophys. Res Commun. 2009;387:348–352. doi: 10.1016/j.bbrc.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 63.Derenne S, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100:194–199. doi: 10.1182/blood.V100.1.194. [DOI] [PubMed] [Google Scholar]
- 64.Jourdan M, et al. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene. 2003;22:2950–2959. doi: 10.1038/sj.onc.1206423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braggio E, et al. Methylation status of nine tumor suppressor genes in multiple myeloma. Int J. Hematol. 2010;91:87–96. doi: 10.1007/s12185-009-0459-2. [DOI] [PubMed] [Google Scholar]
- 66.Mateos MV, et al. Methylation is an inactivating mechanism of the p16 gene in multiple myeloma associated with high plasma cell proliferation and short survival. Br. J. Haematol. 2002;118:1034–1040. doi: 10.1046/j.1365-2141.2002.03749.x. [DOI] [PubMed] [Google Scholar]
- 67.Takada S, et al. Methylation status of fragile histidine triad (FHIT) gene and its clinical impact on prognosis of patients with multiple myeloma. Eur. J. Haematol. 2005;75:505–510. doi: 10.1111/j.1600-0609.2005.00560.x. [DOI] [PubMed] [Google Scholar]
- 68.Harris ME, et al. Regulation of histone mRNA in the unperturbed cell cycle: evidence suggesting control at two posttranscriptional steps. Mol. Cell Biol. 1991;11:2416–2424. doi: 10.1128/MCB.11.5.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaffer AL, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454:226–231. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iida S, et al. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 1997;17:226–230. doi: 10.1038/ng1097-226. [DOI] [PubMed] [Google Scholar]
- 71.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kronke J, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu G, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.