

Abstract
Tuberous sclerosis complex (TSC) is an incurable multisystem disease characterized by mTORC1-hyperactive tumors. TSC1/2 mutations also occur in other neoplastic disorders, including lymphangioleiomyomatosis (LAM) and bladder cancer. Whether TSC-associated tumors will respond to immunotherapy is unknown. We report here that the programmed death 1 coinhibitory receptor (PD-1) is upregulated on T cells in renal angiomyolipomas (AML) and pulmonary lymphangioleiomyomatosis (LAM). In C57BL/6J mice injected with syngeneic TSC2-deficient cells, anti–PD-1 alone decreased 105K tumor growth by 67% (P < 0.0001); the combination of PD-1 and CTLA-4 blockade was even more effective in suppressing tumor growth. Anti–PD-1 induced complete rejection of TSC2-deficient 105K tumors in 37% of mice (P < 0.05). Double blockade of PD-1 and CTLA-4 induced rejection in 62% of mice (P < 0.01). TSC2 reexpression in TSC2-deficient TMKOC cells enhanced antitumor immunity by increasing T cell infiltration and production of IFN-γ/TNF-α by T cells, suggesting that TSC2 and mTORC1 play specific roles in the induction of antitumor immunity. Finally, 1 month of anti–PD-1 blockade reduced renal tumor burden by 53% (P < 0.01) in genetically engineered Tsc2+/– mice. Taken together, these data demonstrate for the first time to our knowledge that checkpoint blockade may have clinical efficacy for TSC and LAM, and possibly other benign tumor syndromes, potentially yielding complete and durable clinical responses.
Keywords: Oncology
Keywords: Cancer immunotherapy, Immunotherapy, T cells
TSC2-deficient tumors respond to anti-PD-1/anti-CTLA-4 immunotherapy and TSC2 promotes anti-tumor immunity.
Introduction
Tuberous sclerosis complex (TSC) is an incurable multisystem genetic disease affecting approximately 1 million people worldwide. TSC is characterized by hamartomatous benign tumors in multiple organs, including brain (subependymal giant cell astrocytoma; SEGA), skin (e.g., facial angiofibroma), kidney (angiomyolipomas; AML), and lung (lymphangioleiomyomatosis; LAM) (1, 2). AML is a frequent cause of TSC-associated death as a result of hemorrhage and/or renal insufficiency (3, 4). LAM, which affects almost exclusively women, is a progressive cystic lung disease characterized by abnormal proliferation of smooth muscle–like cells that can lead to dependence on supplemental oxygen and respiratory failure (5–7).
TSC is caused by inactivating mutations in 1 of 2 tumor suppressor genes, TSC1 or TSC2 (8–11). The TSC1 and TSC2 proteins (hamartin and tuberin, respectively) form a tertiary complex together with TBC1D7 to suppress mTOR complex 1 (mTORC1) signaling via the small GTPase Rheb (12). The TSC2 protein has a GTPase activating domain that stimulates the conversion of Rheb-GTP to Rheb-GDP (13–16). mTORC1 is a central regulator of cellular growth and metabolism (17). Hyperactivation of mTORC1 signaling can occur as a consequence of either TSC1/TSC2 inactivation or dysregulation of upstream signaling via PTEN and phosphoinositide 3-kinase (PI3K)/Akt. Such aberrant activation of mTORC1 is observed in many human cancers, as well as in TSC-associated tumors (18). mTOR inhibitors, such as rapamycin (sirolimus) and everolimus, are Food and Drug administration–approved (FDA-approved) therapies for specific TSC-associated manifestations, including AML, LAM, and SEGA (19–23). The response to therapy is incomplete, with a partial decrease in AML and SEGA tumor size and stabilization of lung function in women with LAM. Upon treatment cessation, however, AML and SEGA regrow and lung function declines in women with LAM (19–23). Therefore, continuous therapy with rapamycin is required, highlighting the need for other treatments that result in complete and durable clinical responses.
Alongside the intrinsic, cell-autonomous drivers of tumorigenesis, tumor growth is dependent on multiple extrinsic factors, including tumor endothelia, stromal components, and local immune cells (24). Tumor-infiltrating lymphocytes (TILs) play critical roles in tumorigenesis, particularly functional cytolytic T cells, which are crucial for mediating immune surveillance (25, 26). Tumors can escape immune surveillance via several mechanisms. One of these escape mechanisms is to modulate T cell responses by enhancing signaling through coinhibitory receptors or immune checkpoint proteins on T cells, including programmed cell death-1 (PD-1), cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motif (ITIM) domains (TIGIT) and T cell immunoglobulin and mucin-domain containing-3 (TIM-3) (27–32).
Understanding the interaction between T cells and tumor cells has led to the development of successful checkpoint blockade immunotherapy targeting PD-1 and CTLA-4 on T cells, as well as PD-ligand 1 (PD-L1), which is often expressed on tumor cells (33–35). These therapies have proven to be highly effective in treating multiple malignancies, including non–small cell lung cancer (NSCLC), renal cell carcinoma (RCC), melanoma, bladder cancer, and Hodgkin’s lymphoma (36).
It is currently unknown whether benign tumors, such as those arising in TSC, will also respond to checkpoint blockade therapy. In this study, we sought to examine the immune microenvironment of TSC-associated tumors. We found that T cells in AML have increased PD-1 expression compared with normal kidneys. Utilizing 2 TSC2-null cell lines, we found that s.c. tumor growth in immunocompetent mice was suppressed by anti–PD-1 or anti–CTLA-4 antibodies. Combination blockade of PD-1 and CTLA-4 demonstrated the most potent inhibition of tumor growth. Compellingly, PD-1 monotherapy led to complete tumor rejection in 37% of animals, and combination therapy led to complete tumor rejection in 62% of animals. Tumor clearance was associated with increased infiltration of T cells into the tumors. These T cells demonstrated enhanced proliferation and production of IFN-γ and TNF-α. Reexpression of TSC2 in TSC2-deficient cells increased tumor-infiltrating CD8+ and CD4+ T cells and IFN-γ/TNF-α–producing CD8+ and CD4+ T cells. Strikingly, the efficacy of dual PD-1 and CTLA-4 blockade was enhanced by TSC2 reexpression. This is the first evidence to our knowledge that TSC2 plays a specific role in antitumor immunity and the first preclinical evidence to our knowledge suggesting that benign tumor syndromes, such as those found in TSC, may respond to immune checkpoint blockade.
Results
PD-1 is highly expressed on T cells in AML and LAM.
To determine whether tumor-infiltrating T cells are present in the microenvironment of human AMLs, IHC staining for T lymphocytes using anti-CD3 antibody was performed on formalin-fixed and paraffin-embedded AML tissues. This revealed an 8-fold increase in the number of CD3+ T cells in AML tissues compared with normal kidney (Figure 1A).
Figure 1. Increased T cell infiltration and PD-1+ T cells in angiomyolipomas (AML) and lymphangioleiomyomatosis (LAM).

(A) Left, representative CD3 IHC in AML (n = 9) and normal kidney (n = 4). Scale bar: 50 μm. Right, quantification of CD3+ T cells in AML vs. normal kidney. Statistical significance was determined by Mann-Whitney’s U test. ***P < 0.001. (B) Multiparametric flow cytometry data of AML (n = 5) and normal kidney (n = 4) indicating the frequency (%) of CD3 T cells, CD19+ B cells, CD56+ NK cells, NKT cells, CD33+ myeloid cells, and CD15+ granulocytes. Data are presented as mean ± SD. Statistical significance determined by multiple 2-tailed t test followed by Holm-Sidak post-test for multiple comparisons. *P < 0.05. (C) Representative CD3 and HMB45 IHC in LAM (n = 10) and normal lung (n = 3). Scale bar: 50 μm. (D) Multicolor flow cytometry data for LAM (n = 4) and normal lung (n = 4) indicating the frequency (%) of CD3 T cells, CD19+ B cells, CD56+ NK cells, NKT cells, CD33+ myeloid cells, and CD15+ granulocytes. Data for bar graphs were calculated using multiple 2-tailed t test followed by Holm-Sidak post-test for multiple comparisons. (E) Representative IHC for CD3 and PD-1 in serial sections of AML (n = 10) and normal kidney (n = 4). Red arrows indicate PD-1+ T cells. Scale bar: 50 μm and 25 μm (inset).
In parallel, we analyzed 5 freshly resected AML vs. normal kidney by multiparametric flow cytometry. Tumor-infiltrating T cells were significantly enhanced (3-fold, P < 0.05) in AML tissues compared with normal kidney (Figure 1B). There was a trend toward increased CD33+ myeloid cells and CD15+ granulocytes in AMLs vs. normal, but no change in CD19+ B cells, CD56+ NK cells, or NKT cells (Figure 1B). We further examined the presence of myeloid cell populations in AMLs. In comparison with normal kidney, the percentages of CD33+CD15+CD14–HLA-DR– granulocyte-like myeloid derived suppressor cells (G-MDSCs) (mean 3.75 vs. 17.54; P = 0.039) and CD14+CD16– monocytes (mean 1.72 vs. 15.71; P = 0.036) were increased in the tumor tissues (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.98674DS1). However, the relative frequencies of CD14+CD163+HLA-DR– tumor-associated macrophages (TAMs) and CD14+CD163–HLA-DR– monocyte MDSCs (M-MDSCs) in AMLs were not significantly increased compared with normal kidney (Supplemental Figure 1A).
We also examined T cell infiltration in LAM by IHC. HMB45, an established marker of LAM cells, was used to identify LAM nodules. We found that LAM nodules showed an accumulation of T cells (Figure 1C), similar to AML. However, multiparametric flow cytometric analysis of fresh whole lung tissues from 4 LAM patients whose lungs were removed at the time of lung transplantation revealed only a small and insignificant increase in T cells compared with their 4 normal-lung controls (Figure 1D). This likely reflects the complexity of cellular pathology in the end-stage LAM, in which LAM nodules are a minority population. We did not observe a difference in G-MDSCs, monocytes, TAMs, or M-MDSCs in LAM vs. normal lung (Supplemental Figure 1B).
PD-1 expression on tumor-infiltrating T cells contributes to T cell dysfunction in the tumor microenvironment (36). We found more PD-1+ T cells in AML vs. normal kidney (Figure 1E) and in LAM nodules compared with normal lung (Supplemental Figure 1C).
PD-L1 is not expressed in AML and is rarely expressed in LAM, but it is highly expressed on tumor myeloid cell infiltrates.
PD-1 has 2 ligands, PD-L1 and PD-L2, which are both inhibitory. PD-L1 is widely expressed in the tumor microenvironment. It is frequently found on tumor cells, fibroblasts, endothelial cells, and hematopoietic cells. In contrast, PD-L2 is mainly expressed on myeloid cells and DCs, and also on some tumor cells (35). We used IHC to examine the expression of PD-L1 in fixed AML and LAM tissues. No PD-L1 positivity was observed in AML cells (n = 10), and only faint cytoplasmic PD-L1 staining was observed in LAM nodules, in a minority of cases (2/10 cases) (Supplemental Figures 2, A and B).
In contrast to this absent PD-L1 expression on AML and low expression on LAM cells, PD-L1 expression was detected on hematopoietic cells within AML and LAM nodules. In both tissue types, the relative frequencies of PD-L1–expressing G-MDSCs, M-MDSCs, TAMs, and monocytes were higher than PD-L2 (Supplemental Figure 2, C and D). The PD-L1–expressing G-MDSCs and monocytes were significantly upregulated in AML compared with normal kidney (Supplemental Figure 2E). The relative frequencies of PD-L1–expressing G-MDSCs, m-MDSCs, TAMs, and monocytes were not different in LAM vs. normal lung (Supplemental Figure 2F). Interestingly, PD-L2–expressing monocytes were also increased within AMLs when compared with normal kidney (Supplemental Figure 2G). We did not observe differences in the relative frequencies of G-MDSCs, m-MDSCs, TAMs, and monocytes in LAM compared with normal lung (Supplemental Figure 2H).
Combination of PD-1 and CTLA-4 blockade suppresses tumor growth and increases T cell infiltration in TSC2-deficient tumors.
Based on these findings that PD-1+ T cells infiltrate TSC2-deficient human tumors, we next examined the in vivo efficacy of PD-1–blocking monoclonal antibody alone or in combination with CTLA-4 blocking monoclonal antibody in preclinical s.c. models of TSC. We used 2 different TSC2-deficient cell lines, 105K and TMKOC, both of which were derived from renal tumors that developed in C57BL/6J Tsc2+/– mice (37, 38). The TMKOC cells, but not the 105K cells, were passaged through immunodeficient mice to enhance tumorigenicity (38). Injection (s.c.) of these cells into immunocompetent C57BL/6J mice led to tumor development. Once tumors became palpable, mice were then injected i.p. with isotype control, anti–PD-1, anti–CTLA-4, or combined anti–PD-1 and CTLA-4 antibodies, every 3 days for a total of 4 treatments (Figure 2A). In the 105K model, anti–PD-1 treatment suppressed tumor growth by 62% (P < 0.0001), whereas anti–CTLA-4 decreased tumor growth by 25% (P < 0.05). The combination of anti–PD-1 and –CTLA-4 inhibited tumor growth by 77% (P < 0.0001) in 105K tumors compared with isotype control (Figure 2B). In the TMKOC model, anti–CTLA-4 suppressed tumor growth by 57% (P < 0.0001), anti–PD-1 by 25% (P < 0.05), and the combination of both anti–PD-1 and anti–CTLA-4 by 50% (P < 0.001), compared with isotype control treatment (Figure 2B). In summary, in both the 105K and TMKOC models, individual PD-1 or CTLA-4 blockade inhibited tumor growth, while combined PD-1 and CTLA-4 blockade decreased tumor growth to a greater extent than single blockade.
Figure 2. PD-1 and CTLA-4 combination blockade suppresses TSC2-deficient tumor growth and increases the frequency of infiltrating CD8+ T cells.

(A) Experimental design for s.c. TSC2-deficient 105K tumors (left) and TSC2-deficient TMKOC tumors (right) treated with isotype control, αPD-1, αCTLA-4, or combined αPD-1 plus αCTLA-4 every 3 days for 4 treatments. (B) Growth of 105K tumors (left) and TMKOC tumors (right) treated as indicated (n = 8 per group). Data are presented as mean ± SD. Statistical significance was determined by nonparametric 1-way ANOVA followed by Holm-Sidak post-test between tumor sizes at day 27 for 105K tumors and day 15 for TMKOC tumors. (C) Flow cytometry quantification of tumor-infiltrating CD4+ and CD8+ T cells in TSC2-deficient 105K tumors (left) and TSC2-deficient TMKOC tumors (right) obtained 24 hours following the last treatment (n = 6–8 per group). Data are presented as mean ± SD. Statistical significance was determined by nonparametric 1-way ANOVA followed by Dunn’s multiple comparison test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Interestingly, the level of CD8+ T cell infiltration correlated with the degree of response to therapy. The combination of PD-1 and CTLA-4 antibody treatment increased CD8+ T cell infiltration in both models more than single-agent treatment (2-fold, P < 0.01, in 105K and 3-fold, P < 0.001, in TMKOC; Figure 2C and Supplemental Figure 3). Similarly, combined PD-1 and CTLA-4 antibody treatment increased total CD4+ T cell infiltration (Figure 2C). Taken together, these data demonstrate that combined anti–PD-1 and anti–CTLA-4 therapy enhances T cell infiltration and inhibits the growth of TSC2-deficient tumors.
To investigate whether proliferation of tumor-infiltrating T cells and/or cytokine production are altered as a result of combination coinhibitory blockade of PD-1 and CTLA-4, we isolated TILs from tumors 24 hours following the last treatment. These were stimulated ex vivo with PMA and ionomycin for 4 hours. Proliferation and intracellular cytokines were monitored by flow cytometry using Ki-67 and IFN-γ/TNF-α antibodies, respectively. Combined PD-1 and CTLA-4 blockade significantly increased Ki-67–expressing CD8+ T cells (Figure 3A). IFN-γ–producing CD8+ T cells were induced by PD-1 and CTLA-4 single blockade and also by the combination of the 2 antibodies in the 105K tumors (Figure 3B). Interestingly, in both models, combined PD-1 and CTLA-4 blockade enhanced the TNF-α–producing CD8 population of TILs (Supplemental Figure 4, A and B). In TMKOC tumors, TNF-α–producing CD8+ and CD4+ T cells were induced by single anti–CTLA-4 and combined anti–PD-1 and anti–CTLA-4 (Supplemental Figure 4B). We also assessed the double-positive IFN-γ and TNF-α cells and found that the dual blockade of PD-1 and CTLA-4 augmented this population within CD8+ and CD4+ T cells (Figure 3C). Thus, the combination of PD-1 and CTLA-4 antibody therapy more potently increased TIL proliferation and accumulation of intracellular cytokines when compared with monotherapy in both models.
Figure 3. PD-1 and CTLA-4 combination blockade increases proliferating and IFN-γ/TNF-α double-producing CD8+ T cells in TSC2-deficient tumors.

Tumor-infiltrating lymphocytes (TILs) were purified from s.c. 105K or TMKOC tumors using Ficoll gradient 24 hours following the last treatment, as illustrated in Figure 2A. TILs were stimulated with cell stimulation cocktail containing PMA and ionomycin in the presence of GolgiPlug for 4 hours. CD8+ and CD4+ T cells were analyzed by flow cytometry for lymphocyte markers and Ki-67 expression (A), intracellular IFN-γ production (B), and intracellular IFN-γ and TNF-α production (C) (n = 6–8 per group). Data are presented as mean ± SD. Statistical significance was determined by nonparametric 1-way ANOVA followed by Dunn’s multiple comparison test. *P < 0.05; **P < 0.01.
Combination blockade of CTLA-4 and PD-1 in TSC2-deficient tumors suppresses Tregs and increases CD8+ T Cell/Treg and CD4+ T cells/Treg ratios.
Tregs play critical roles in suppressing antitumor immune responses (39). Previous studies have shown that blocking PD-1 and/or CTLA-4 can enhance antitumor response by modulating Treg infiltration and/or function (40–42). The combined blockade of PD-1 and CTLA-4 reduced the frequency of Foxp3+CD4+ Tregs in both models (from 26% to 17% in the 105K, and from 42% to 30% in the TMKOC, P < 0.05, Figure 4A). In addition, the CD8/Treg and CD4/Treg ratios increased significantly following combination therapy in both models (CD8/Treg ratio: from 5 to 15 in the 105K and 5 to 12 in the TMKOC; CD4/Treg ratio: from 4.5 to 7 in the 105K and 2.2 to 3.9 in the TMKOC, P < 0.05, Figure 4B).
Figure 4. PD-1 and CTLA-4 combination blockade decreases the percentage of Foxp3+CD4+ Tregs and increases the CD8/Treg and CD4/Treg ratio in TSC2-deficient tumors.

(A) Flow cytometry quantification of tumor-infiltrating Foxp3+CD4+ Tregs in TSC2-deficient 105K tumors (left) and TSC2-deficient TMKOC tumors (right) obtained 24 hours following the last treatment, as illustrated in Figure 2A. (B) Ratios of CD8+ T cells to Tregs and CD4+ T cells to Tregs within 105K tumors (left) and TSC2-deficient TMKOC tumors (right) are shown (n = 6–8 per group). Data are presented as mean ± SD. Statistical significance was determined by nonparametric 1-way ANOVA followed by Dunn’s multiple comparison test. *P < 0.05.
PD-1 blockade alone or combined PD-1 and CTLA-4 blockade suppress PD-1+Tim-3+ and PD-1+TIGIT+ within the tumor-infiltrating CD8+ T cell population.
To further understand the activation state of the tumor-infiltrating T cells following blockade of PD-1 and/or CTLA-4, we assessed the expression of several coinhibitory receptors, focusing on PD-1 expression on CD8+ and CD4+ T cells alone or together with TIGIT, Tim-3, or CTLA-4. PD-1 single blockade led to a significant decrease in double-positive PD-1+TIGIT+ and PD-1+Tim-3+ CD8+ T cells by 30% (P < 0.01), which was also observed in the combined PD-1 and CTLA-4 treatment (Figure 5, A and B, and Supplemental Figure 5, A and B). CTLA-4 single blockade increased the frequency of PD-1+CTLA-4– in the CD8+ and CD4+ T cell populations in both models (Figure 5C and Supplemental Figure 5C). In the TMKOC model, monotherapeutic PD-1 blockade and dual PD-1 and CTLA-4 blockade resulted in a significant decrease in PD-1+CTLA4+ T cells (Supplemental Figure 5C).
Figure 5. PD-1 blockade alone or in combination with CTLA-4 suppresses PD-1+TIGIT+ and PD-1+Tim-3+ CD8+ T cells in 105K tumors.

Flow cytometry quantification of the tumor-infiltrating PD-1+ and/or TIGIT+ CD8+ or CD4+ T cells (A), PD-1+ and/or Tim-3+ CD8+ or CD4+ T cells (B), and PD-1+ and/or CTLA-4+ CD8+ or CD4+ T cells (C) in TSC2-deficient 105K tumors 24 hours following the last treatment, as illustrated in Figure 2A (n = 6–8 per group). Data are presented as mean ± SD. Statistical significance was determined by 2-way ANOVA followed by Holm-Sidak post-test for multiple comparison. *P < 0.05; **P < 0.01.
Combination blockade of PD-1 and CTLA-4 receptors significantly reduces F4/80+ macrophages, CD11b+Ly6G+Ly6Cmed G-MDSCs, and CD11b+ DCs in TSC2-deficient tumors.
Some myeloid populations, including MDSCs, have been shown to dampen T cell activity in tumors (38). Previous studies have shown that PD-1 blockade can decrease tumor-infiltrating MDSCs or TAMs in preclinical mouse models of melanoma and head and neck cancer (42–45). Therefore, we assessed the effects of immunotherapy on TAMs, MDSCs, and DCs. Combination PD-1 and CTLA-4 blockade reduced the relative frequencies of CD11b+F4/80+ macrophages and CD11b+Ly6G+Ly6Cmed G-MDSCs in 105K and TMKOC tumors (Figure 6, A and B, and Supplemental Figure 6, A and B). In addition, regulatory CD11c+CD11b+ DCs were reduced following combination therapy (Figure 6C and Supplemental Figure 6C). We also assessed activation of macrophages by examining the expression of CD80 and CD86 following the treatments in both 105K and TMKOC tumors. These populations were not affected (Figure 6A and supplemental Figure 6A). Tumor-infiltrating NKs were not altered in 105K tumors (Figure 6D), but single PD-1 blockade increased NKs in TMKOC tumors (Supplemental Figure 6D).
Figure 6. PD-1 and CTLA-4 combination blockade decreases CD11b+F4/80+ macrophages, CD11b+Ly6CmedLy6G+ G-MDSCs, and regulatory CD11b+ DCs in TSC2-deficient tumors.

Mice carrying TSC2-deficient 105K tumors were treated as illustrated in Figure 2A. Tumor-infiltrating lymphocytes (TILs) were isolated and stained for myeloid lineage and activation markers. Percentage of CD11b+F4/80+ macrophages and CD80+CD86+ macrophages (A), CD11b+Ly6CmedLy6G+ G-MDSCs and CD11b+Ly6C+Ly6G– M-MDSCs (B), CD11c+CD11b– DCs and CD11c+CD11b+ DCs (C), and NK cells (D) within CD45+ cells isolated from TSC2-deficient 105K tumors are shown (n = 6–8 per group). Data are presented as mean ± SD. Statistical significance was determined by nonparametric 1-way ANOVA followed by Dunn’s multiple comparison test. *P < 0.05.
PD-1 monotherapy or in combination with CTLA-4 promotes complete tumor rejection.
Blockade of PD-1 and/or CTLA-4 has been shown to enhance effector T cell responses and induce T cell–mediated tumor rejection in some models (41, 42, 46, 47). To evaluate whether checkpoint inhibition of PD-1 and CTLA-4 leads to long-term tumor regression, we treated mice carrying 105K tumors with anti–PD-1, anti–CTLA-4, or the combination (Figure 7A) and followed them for up to 100 days. Consistent with prior results (Figure 2B), single PD-1, CTLA-4, or combination blockade inhibited tumor growth (Figure 7B). Mice treated with anti–CTLA-4 alone had a median survival of 48 days, with no tumor-free mice (Figure 7C and Table 1). In contrast, 3 of 8 mice treated with anti–PD-1 were tumor free at 100 days (Figure 7C and Table 1), and 5 of 8 mice that received combined inhibition of PD-1 and CTLA-4 were tumor free at 100 days (Figure 7C and Table 1). These data demonstrate, for the first time to our knowledge, that checkpoint blockade of TSC2-deficient tumors can lead to complete and sustained antitumor responses.
Figure 7. PD-1 blockade alone or in combination with CTLA-4 blockade increases complete tumor rejection at 100 days.

(A) Experimental design. Mice were followed up to 100 days. (B) Tumor growth analyses following the first treatment. (C) Tumor-free survival analysis of mice bearing TSC2-deficient 105K tumors treated as indicated (n = 6–8 per group). Statistical significance was determined by Log-rank test. *P < 0.05; **P < 0.01. (D) Percentage of mice bearing s.c. 105K tumors treated with the indicated monoclonal antibody blockade therapy that completely rejected their tumors.
Table 1. PD-1 blockade alone or in combination with CTLA-4 blockade increases complete tumor rejection at 100 days.

Sequential rapamycin treatment followed by PD-1 blockade delays TSC2-deficient tumor regrowth.
Rapamycin and its analogues (rapalogs) clinically suppress tumors in TSC patients. Rapalogs have a cytostatic effect with tumor regrowth after treatment discontinuation (19–23). In mice, this tumor regrowth begins within days of rapamycin discontinuation. Rapamycin has been shown to inhibit T cell activation (48), suggesting a complex interplay between suppression of the immune system and suppression of tumorigenesis. We assessed a sequential treatment regimen of rapamycin followed by PD-1 blockade (Figure 8A) to determine how rapamycin treatment prior to PD-1 blockade impacts response. Mice with palpable TSC2-deficient 105K tumors were injected i.p. with vehicle or rapamycin for 6 treatments over 2 weeks. The rapamycin-treated mice then received 4 treatments of anti–PD-1 or isotype control antibodies, beginning 24 hours after the final rapamycin treatment. As expected, rapamycin inhibited tumor growth (Figure 8B), and anti–PD-1 sequential therapy had antitumor activity (Figure 8C), consistent with our earlier data (Figure 2B). Tumor progression after completion of anti–PD-1 therapy was significantly inhibited compared with isotype control antibody, with a net decrease of 67% in tumor volume at 15 days following the last treatment (P < 0.001, Figure 8D). PD-1 inhibition lengthened the survival of mice (Figure 8E); however, we noted that only 9% of them had a complete response. These data suggest that the addition of PD-1 blockage therapy can enhance the therapeutic efficacy of rapamycin.
Figure 8. Sequential treatment of rapamycin followed by PD-1 blockade slows tumor regrowth.

(A) Schematic representation of treatment schedule. Mice carrying s.c. TSC2-deficient 105K tumors were treated with rapamycin (3 mg/kg) or vehicle every 2 days for 6 treatments in total. Twenty-four hours following the last rapamycin treatment, rapamycin-treated mice received isotype control or αPD-1 every 3 days for 4 treatments in total and were followed up to 36 days. (B) Tumor growth analysis of 105K tumor-bearing mice receiving vehicle (n = 8) or rapamycin treatment (n = 14). Data are presented as mean ± SD. Statistical significance was determined by Mann-Whitney U test, ****P < 0.0001. (C) Tumor growth analysis of 105K tumor–bearing mice pretreated with rapamycin and then receiving isotype control (n = 7) or αPD-1 antibody (n = 11). (D) The average tumor volume difference of the isotype control or αPD-1 antibody–treated group after the last antibody treatment (day 22) and the final measurement (day 36) is shown. Data are presented as mean ± SD. Statistical significance was determined by Mann-Whitney U test. (E) Tumor-free survival analysis of mice bearing TSC2-deficient 105K tumors treated as indicated (n = 7–11 per group). Statistical significance was determined by Log-rank test. ***P < 0.001; ****P < 0.0001.
TSC2 reexpression promotes antitumor responses and enhances the potency of dual PD-1 and CTLA-4 blockade.
We sought to elucidate whether loss of TSC2 is specifically involved in the generation of an immunosuppressive tumor microenvironment by profiling immune cell infiltration in TSC2-deficient and TSC2 reexpressing tumors in syngeneic C57BL/6J mice. Expression of TSC2 was assessed by immunoblotting in TSC2-null cells expressing control vector (referred to as TSC2-null) or TSC2 reexpressing cells (referred to as TSC2 add-back) (Figure 9A). We found that TSC2 add-back cells grew 10% faster than TSC2-null cells in vitro (P < 0.001, Supplemental Figure 7A). However, when the 2 cell lines were injected s.c. into Cd8–/– mice, TSC2 add-back tumors proliferated much slower than TSC2-null tumors (Supplemental Figure 7B). Similarly, when TSC2-null and TSC2 add-back cells were injected s.c. into immunocompetent C57BL/6 mice, TSC2 reexpression suppressed tumor growth by 68% at 28 days after injection (P < 0.001) and increased tumor-free survival when compared with TSC2-null cells by a median of 14 days (Figure 9, B and C).
Figure 9. Reexpression of TSC2 in TSC2-deficient tumors enhances T cell–mediated antitumor immunity and promotes dual PD-1 and CTLA-4 blockade.

(A) Whole cell lysates from TSC2-null TMKOC cells and TSC2 add-back TMKOC cells were analyzed by immunoblotting with antibodies against TSC2 and β-actin. (B) Tumor growth curve of mice bearing TSC2-null TMKOC tumors or TSC2 add-back TMKOC tumors (n = 6 per group). Data are presented as mean ± SD. Statistical significance was determined by Mann-Whitney U test. (C) Tumor-free survival analysis of mice bearing TSC2-null TMKOC or TSC2 add-back TMKOC tumors. (D–F) Tumors from mice injected with TSC2-null TMKOC or TSC2 add-back TMKOC cells were harvested and analyzed for CD45+ hematopoietic lineage cells with lymphocytes and myeloid markers by flow cytometry: (D) CD45+ cells and CD11b+ myeloid cells, (E) CD8+ and CD4+ T cells, and (F) CD8+ and CD4+ T cell intracellular IFN-γ and TNF-α production. (G) Ki-67 expression in CD8+ and CD4+ T cells. Data are presented as mean ± SD. Statistical significance was determined by Mann-Whitney U test. (H) Tumor growth curve of mice bearing TSC2-null TMKOC tumors or TSC2 add-back TMKOC tumors treated with isotype control or combined anti–PD-1 and anti–CTLA-4 antibodies (n = 10 per group). Data are presented as mean ± SD. Statistical significance was determined by nonparametric 1-way ANOVA followed by Dunn’s multiple comparison test. (I) Overall survival analysis of mice bearing TSC2-null TMKOC or TSC2 add-back TMKOC tumors treated as described in H (n = 14 per group). Statistical significance was determined by Log-rank test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (J) Percentage of mice bearing s.c. TSC2-null or TSC2 add-back TMKOC tumors treated as described in H that completely rejected their tumors.
In order to assess whether TSC2 is a bona fide regulator of tumor immunity, we generated single-cell suspensions from tumors that had been grown to the same size (800 mm3) and analyzed single cells by multi-color flow cytometry. The frequency of tumor-infiltrating CD45+ hematopoietic cells was similar in the TSC2-null vs. TSC2 add-back tumors (Figure 9D). The percentage of CD11b+ myeloid cells was not changed in the TSC2 add-back tumors (Figure 9D). There was a 3-fold increase in the percentage of infiltrating CD8+ T cells (P < 0.0001) and a 2-fold increase in the percentage of CD4+ T cells (P < 0.01, Figure 9E). We also observed a significant increase in IFN-γ/TNF-α–producing CD8+ and CD4+ T cells (Figure 9F) in the tumors with TSC2 reexpression. There was a trend toward increased percentage of Ki-67+CD8+ and Ki-67+CD4+ T cells (Figure 9G). There was also a significant increase in M-MDSCs (Supplemental Figure 8A), but there was no change in TAMs, G-MDSCs, or DCs (Supplemental Figure 8, A and B).
Given that TSC2 plays a critical role in antitumor immunity, we then examined whether TSC2 can enhance checkpoint blockade therapy. Mice with palpable TSC2-null and TSC2 add-back TMKOC tumors were treated with control isotype or combined anti–PD-1 and anti–CTLA-4 every 3 days for 4 treatments in total. Consistent with Figure 9B, TSC2 reexpression reduced tumor growth (Figure 9H), and as expected, dual PD-1 and CTLA-4 blockade suppressed the proliferation in TSC2-null tumors (Figure 9H). Remarkably, TSC2 add-back tumors had a greater response to combined anti–PD-1 and anti–CTLA-4 treatment compared with TSC2-null tumors (P < 0.05, Figure 9H). Importantly, TSC2 add-back tumors treated with dual anti–PD-1 and anti–CTLA-4 exhibited more infiltration of CD8+ CD4+ T cells, enhanced production of IFN-γ and TNF-α, and increased expression of proliferative marker Ki-67 compared with TSC2-null tumors treated with isotype control antibodies (Supplemental Figure 9, A–C).
To investigate whether TSC2 is beneficial for long-term tumor regression following checkpoint blockade therapy, we treated TSC2-null and TSC2 add-back TMKOC tumors with isotype control or combined anti–PD-1 and anti–CTLA-4 and followed them up to 40 days. Consistent with prior results (Figure 9C), TSC2 add-back prolonged median survival by 10 days, with no tumor-free mice (Figure 9I and Table 2). Strikingly, mice carrying TSC2 add-back tumor treated with combined anti–PD-1 and anti–CTLA-4 had improved overall survival (P < 0.0001) and more tumor-free survivors (5/14 vs. 1/14) compared with TSC2-null tumors treated with combined anti–PD-1 and anti–CTLA-4 (Figure 9I and Table 2).
Table 2. Tumor-free mice 30 days off treatment.
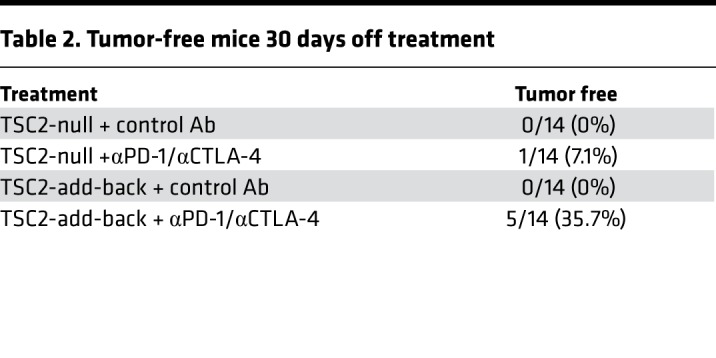
Cumulatively, these data indicate that the absence of TSC2 in tumors plays a specific role in generating an immunosuppressive tumor microenvironment and that TSC2 reexpression elicits T cell–mediated antitumor immunity that is enhanced by PD-1 and CTLA-4 blockade.
Anti–PD-1 inhibits renal tumorigenesis in Tsc2+/– mice.
Given that anti–PD-1 antibody therapy has a substantial effect on tumor growth in the s.c. models, we tested the efficacy of PD-1 blockade in a genetically engineered mouse model of TSC. Tsc2+/– mice on A/J strain background develop benign kidney tumors, known as cystadenomas (49). Tsc2+/– mice (6-and-a-half months old) were treated with isotype control or anti–PD-1 antibody every 3 days for a total of 8 treatments (Figure 10A). The mice were harvested 24 hours after the last treatment. The effect of anti–PD-1 antibody on gross and microscopic kidney tumor burden was analyzed as described previously (49, 50). The gross kidney tumor score, based on cyst size and number, was decreased by 41% in anti–PD-1–treated mice (score = 17.3) compared with isotype control treated mice (score = 29.3) (P < 0.01, Figure 10B). The microscopic kidney tumor score was also reduced by 53% in anti–PD-1–treated mice (score = 1.4) compared with isotype control–treated mice (score = 3.0) (P < 0.01, Figure 10C). These data further confirm that anti–PD-1 blocking antibody has efficacy in treating TSC tumors.
Figure 10. PD-1 blockade suppresses kidney tumor burden in Tsc2+/– mice.

(A) Experimental design. (B) Gross kidney tumor score was determined for each kidney using an established method based on number and size. Data are presented as mean ± SD. Statistical significance was determined by Mann-Whitney U test. (C) Microscopic kidney tumor score was assessed for each kidney by an established method of determining tumor size and filling from H&E sections (n = 14–16 per group). Data are presented as mean ± SD. Statistical significance was determined by Mann-Whitney U test. **P < 0.01
Discussion
TSC is an autosomal dominant disorder with diverse manifestations, including seizures, autism, cognitive disability, and tumors of the brain, heart, kidney, skin, and lung (1, 2). Tumors often develop during childhood. Treatment of TSC-associated tumors, including AML, SEGA, and LAM, with mTORC1 inhibitors leads to partial responses, with tumor regrowth upon treatment discontinuation, consistent with a cytostatic cellular response (19–23). Novel therapeutic strategies that induce complete clinical responses, thereby reducing the need for lifelong mTORC1 inhibitor therapy, could have a major clinical impact for both adults and children with TSC. Immunotherapy approaches have yielded substantial clinical benefits in selected patients with melanoma, RCC, NSCLC, bladder cancer, and other malignancies, but their potential efficacy in benign tumors, such as those that occur in TSC, is largely unexplored. We report here that single-agent PD-1 and CTLA-4 blockade or their combination inhibits the growth of TSC2-deficient tumors in mice, associated with increasing tumor-infiltrating CD8+ and CD4+ T cells and decreasing Tregs, G-MDSCs, and regulatory CD11b+ DCs. Importantly, complete regression of established tumors can be achieved in preclinical mouse models of TSC tumorigenesis using anti–PD-1 monotherapy. The combination of anti–PD-1 and anti–CTLA-4 nearly doubles the number of tumor-free survivors. Our studies provide the first preclinical evidence to our knowledge that TSC-associated tumors and LAM can be treated with checkpoint blockade immunotherapy, with the potential for long-term disease-free responses that cannot currently be achieved with mTORC1 inhibitors.
Human AML tissues showed an 8-fold accumulation of T cells compared with normal kidney. The presence of preexisting intratumoral lymphocytes has been found to correlate with the success of immunotherapy (51). We hypothesized that similar results would be seen in LAM, since LAM cells have biallelic TSC2 mutations and express melanoma and smooth muscle markers, making them essentially indistinguishable from the smooth muscle cell component of AML. While we observed T cells within LAM nodules, this was not significantly higher, quantitatively, when compared with normal lung — consistent with previous studies (52). We suspect that this reflects the substantial cellular heterogeneity within the lungs of women with end-stage LAM, since the majority of the samples we studied were from lungs removed at the time of transplantation for LAM.
PD-1+ T cells were present in both AML and LAM nodules, but we did not find extensive expression of PD-L1 in either AML or LAM. This was consistent with a prior study in which 7 cases of AML (53) were stained using a PD-L1 antibody that recognizes a C-terminal epitope of the protein, which was developed recently by Mahoney et al. (54). An earlier study used a different PD-L1 antibody and reported PD-L1 expression in 3 of 110 AMLs (55). Similarly in LAM, we observed only faint cytoplasmic staining in 2 of 10 cases. In contrast to the absent or low expression of PD-L1 on AML and LAM cells, respectively, PD-L1 expression was increased on G-MDSCs and monocytes in AML relative to normal kidney. These data suggest that the upregulated PD-1 on intratumoral T cells in TSC may be engaged by the expression of PD-L1 on tumor-infiltrating immunosuppressive immune cells, rather than on tumor cells themselves. The absence of PD-L1 expression in AML and LAM (with hyperactivation of mTORC1 signaling) is intriguing, as multiple studies have demonstrated that mTORC1 activation tightly regulates PD-L1 expression in several different human tumors (56–59). Our data suggest that loss of TSC2 and activation of mTORC1 are insufficient to induce PD-L1 expression and that other genetic and/or epigenetic events in malignant tumors may be required.
We observed substantial efficacy of anti–PD-1 antibodies, alone or in combination with anti–CTLA-4 antibodies, in preclinical models of TSC. We used 2 different cell lines derived from renal tumors in Tsc2+/– mice, allowing us to work with syngeneic mice with intact immune systems. While both of these lines were derived from similar tumors, these lines differ in that TMKOC cells were passaged through immunodeficient mice to develop a more tumorigenic cell line (38). In both models, single-agent PD-1 and CTLA-4 antibody therapy was effective, while combination blockade of PD-1 and CTLA-4 was even more effective in reducing tumor growth. Our data is consistent with previous reports in other cancer murine models (41, 42, 60, 61) that combination blockade therapy resulted in enhanced tumor-infiltrating CD4+ and CD8+ T cells, increased CD8/Treg and CD4/Treg ratios, and reduced Tregs, TAMs, and CD11b+ DCs. We used anti–PD-1 (RMP1-30) for detection, which has been previously shown to recognize a distinct epitope from the therapeutic anti–PD-1 (RMP1-14) (62). The anti–CTLA-4 antibody (UC10-4B9) we used for detection was also previously used in studies of therapeutic anti–CTLA-4 (9D9) treatment (63). Surprisingly, we found that PD-1 blockade alone or together with CTLA-4 reduced double-positive PD-1+Tim3+ and PD-1+TIGIT+ CD8+ T cells but not the PD-1+CD8+ T cells. In contrast, other studies have demonstrated that PD-1 blockade resulted in increased intratumoral PD-1+CD8+ T cells (42, 64). However, one recent report showed that PD-1 monoclonal antibody does not affect PD-1 expression on CD8+ T cells (65). The underlying reasons for these differential responses to anti–PD-1 therapy are unclear at this time.
Sirolimus (rapamycin) and everolimus are the mainstays of TSC and LAM therapy. Continuous therapy is required, since tumors regrow when treatment is stopped. To elucidate the translational potential of anti–PD-1 in the setting of mTORC1 inhibitory therapy, we tested sequential therapy by administering rapamycin followed by anti–PD-1 treatment. This regimen resulted in only 9% of the mice with complete tumor rejection, which is much lower than single-agent anti–PD-1 (37% complete tumor rejection) and combination anti–PD-1 and anti–CTLA-4 therapy (67% complete tumor rejection). Rapamycin is well known for its immunosuppressive properties, but its effects on T cell responses are not yet completely understood. One study has demonstrated that rapamycin weakens antitumor CD8+ T cell response in tumor-bearing mice vaccinated with CyaA-E7, regardless of the dosage and treatment length (66). On the other hand, there are studies supporting the notion that rapamycin treatment boosts immunity by generating CD8+ T cell memory responses in vaccine-induced antitumoral and antiviral response in mice (67–69). Furthermore, Berezhnoy et al. showed that rapamycin enhances the formation of CD8+ memory T cells, but these reactivated memory cells have reduced cytotoxic effector functions (70). This may explain the reduced complete regression rate in our model when anti–PD-1 was preceded by rapamcyin. Importantly, however, anti–PD-1 therapy delayed the regrowth of tumors when rapamcyin was discontinued, compared with tumors that did not receive anti–PD-1 blockade. These data suggest that there is a potential for co-utilization of mTORC1 inhibitors and immunotherapy, although the timing of the 2 agents will require further optimization.
The Tsc2+/– develops renal cysts and cystadenomas (71), providing a model of spontaneous tumorigenesis analogous to humans carrying germline TSC2 mutations. To investigate whether checkpoint inhibition has efficacy in Tsc2+/– mice, we treated Tsc2+/– mice with anti–PD-1 antibody or isotype control for 24 days (treated every 3 days for a total of 8 doses), beginning at 6.5 months of age, when a significant disease burden is already present. The mice were harvested at day 25. Both macroscopic cysts and tumors (evident by visual inspection of the intact kidney) and microscopic lesions (quantitated on 5 H&E-stained sections per kidney using an established metric that incorporated lesion number, size, and luminal filling) were quantitated. We found that PD-1 blockade resulted in a 53% reduction in microscopic tumor score (P < 0.01) and a 41% reduction in macroscopic score (P < 0.01). Together with the data from our s.c. mouse models of TSC, there findings further support the preclinical efficacy of immune checkpoint inhibition for the treatment of TSC2-deficient tumors.
Our analysis of infiltrating immune cells in TSC2-null and TSC2 add-back tumors showed that TSC2 plays a role in regulating the tumor immune microenvironment by enhancing the accumulation of double-positive IFN-γ/TNF-α–producing CD4+ and CD8+ T cells at tumor sites. This appears to be a highly specific response, since the number of CD45+ cells is the same in the TSC2-null and TSC2 add-back tumors. Importantly, we found that the antitumor effect of TSC2 reexpression is potentiated by dual PD-1 and CTLA-4 blockade, suggesting that TSC2 is not merely an intrinsic tumor suppressor gene, but is also involved in modulating tumor immunity and enhancing the potency of checkpoint blockade immunotherapy.
In summary, our data demonstrate that blocking PD-1 or CTLA-4 alone is sufficient to delay tumor growth in preclinical models of TSC and that anti–PD-1 can yield complete tumor-free responses. Dual blockade of PD-1 and CTLA-4 is more effective than single-agent therapy in achieving complete tumor rejection and improving long-term survival. This is associated with enhanced accumulation of proliferative and cytokine-producing T cells and decreased infiltration of immunosuppressive Tregs, G-MDSCs, and regulatory CD11b+ DCs. Blocking the PD-1 pathway in mice following rapamycin treatment also prolonged short-term survival. We conclude that single-agent or combination therapy utilizing PD-1 and CTLA-4 blockade may represent a promising approach for TSC-associated tumors and the sporadic form of LAM. Our data may also have relevance for many sporadic human tumors with activation of mTORC1 (estimated to represent approximately 50% of all human malignancies) (72), as well as other genetic syndromes associated with multiple benign tumors, such as neurofibromatosis (NFI) and familial adenomatous polyposis (FAP).
Methods
Cell lines and transfection.
The 105K cells, derived from a Tsc2+/– C57BL/6 mouse renal tumor, were cultured in DMEM with 10% FBS as described previously (37). TMKOC cells, originally derived from a renal tubular cystadenoma of a C57BL/6 Tsc2+/– mouse, then passaged through immunodeficient mice as described previously (38). TMKOC cells were maintained in DMEM with 10% FBS. For analysis of tumor-infiltrating immune cells in TSC2-null and TSC2 add-back experiments, TMKOC cells were cotransfected with pRP-mCherry-hyPBase plasmid and pPB-empty vector or pPB-TSC2 vector (VectorBuilder) using Lipofectamine 3000 in culture. Forty-eight hours after transfection, cells were selected using puromycin (1 μg/ml) for 7 days to allow establishment of stable cell lines. Protein expression was detected by immunoblot analysis using antibodies specific for TSC2 (dilution 1:1,000; Cell Signaling Technology; 4308) or β-actin (dilution 1:1,000; Cell Signaling Technology; 3700).
S.c. cell injection.
Six-week-old male C57BL/6J and Cd8–/– mice were purchased from The Jackson Laboratory. The 105K tumor cells, TMKOC tumor cells, or TMKOC stably expressing pPB-control or pPB-TSC2 vectors (2.5 × 106 cells) were resuspended in 75 μl of PBS, and then 75 μl of growth factor reduced Matrigel (Corning, 356237) was added to the cell suspension. The cell/Matrigel suspensions were injected s.c. into the left flanks of male C57BL/6J mice.
In vivo PD-1/CTLA-4 blockade therapy and rapamycin treatment.
TMKOC or 105K tumor cells injected s.c. were allowed to grow to 150–200 mm3 before initiation of PD-1/CTLA-4 blockade therapy. Mice were then treated with 4 i.p. injections (once every 3 days) of rat IgG2a (2A3, BioXcell)/mouse IgG2b (MPC-11, BioXcell) isotype control antibodies, anti–PD-1 blocking antibody (200 μg/mouse, RMP1-14, BioXCell), anti–CTLA-4 blocking antibody (100 μg/mouse, 9D9, BioXCell), or combined anti–PD-1 (200 μg/mouse) and CTLA-4 (100 μg/mouse) blocking antibodies. For the TMKOC tumor cells expressing control or TSC2 vector, tumors were established and treated as described above. For sequential rapamycin and PD-1 blockade experiments, 105K tumor cells injected s.c. were allowed to grow to 150–200mm3. Rapamycin (LC Laboratories) was dissolved in 100% ethanol and then diluted in a vehicle solution containing 5% Tween 80 (MilliporeSigma) and 5% PEG400 (MilliporeSigma) before injection. Mice received 6 i.p. injections of vehicle or rapamycin (3 mg/kg) every 2 days for 2 weeks. Twenty-four hours after cessation of the last rapamycin treatment, mice were treated with rat IgG2a isotype control or anti–PD-1 (200 μg/mouse) blocking antibodies every 3 days for a total of 4 treatments.
Regrowth experiments.
Mice were s.c. injected with 105K tumor cells and treated with rat IgG2a isotype control, anti–PD-1 (200 μg/mouse), anti–CTLA-4 (100 μg/mouse), or combination anti–PD-1 (200 μg/mouse) and anti–CTLA-4 (100 μg/mouse) blocking antibodies as described above. Tumor regrowth following the last treatment was monitored longitudinally using a caliper for 100 days. Mice were s.c. injected with TSC2-null or TSC2 add-back TMKOC tumor cells and treated with rat IgG2a isotype control or combination anti–PD-1 (200 μg/mouse) and anti–CTLA-4 (100 μg/mouse) blocking antibodies as described above. Tumor regrowth following the last treatment was monitored longitudinally using a caliper for 40 days.
Isolation of TILs from mouse tumors.
S.c. solid tumors were disrupted mechanically with a surgical scalpel and then enzymatically digested in HBSS (Invitrogen) containing 400 U/ml collagenase IV and 20U/ml DNase I, with constant rocking at 37oC for 45 minutes. The resulting cell suspension was passed through a 70-μm cell strainer and washed once with 1× PBS containing 0.4% FBS and 2.5 mM EDTA. Tumor-infiltrating immune cells were separated from other cells by centrifugation with the deceleration brake set at 1 at 1,260 g at 4oC for 25 minutes in a Ficoll gradient (17-1440-03, GE Healthcare). After centrifugation, TILs were collected for staining for flow cytometry.
Flow cytometry staining of mouse tumors.
For intracellular cytokine staining, isolated TILs were first incubated in cell stimulation cocktail containing PMA and ionomycin (1:500 dilution, 00-4975-93, eBioscience) for 4 hours at 37oC in RPMI media containing 10% heat-inactivated FBS and 0.05 mM 2-mercaptoethanol; they were then stained with Zombie NIR (423105, BioLegend) for 10 minutes at room temperature. Cells were surfaced stained with the following directly conjugated antibodies: CD44-FITC (clone XMG1.2), Tim-3–APC (clone RMT3-23), CD62L-BV510 (clone MEL-14), CD45-BV605 (clone 30-F11), CD4-BV650 (clone RM4-5), CD8-BV711 (clone 53-6.7), CD3-BV785 (clone 17A2), NK1.1-FITC (clone PK136), Ly6G-PerCP Cy5.5 (clone 1A8), CD86–PE-Cy7 (clone GL-1), CD11c-BV510 (clone N418), Ly6C-BV605 (clone HK1.4), CD80-BV650 (clone 16-10A1), CD45-BV711 (clone 30-F11), and CD11b-BV785 (clone M1/70) (all from BioLegend), as well as PD-1–eFluor710 (RMP1-30) and TIGIT–PE-Cy7 (clone GIGD7) (both from eBioscience). Cell suspensions were washed, fixed, permeabilized, and stained with conjugated antibodies: IFN-γ–FITC (clone XMG1.2), Ki-67–PE (clone 16A8), TNF-α–APC (clone MP6-XT22), and CTLA-4–BV421 (clone UC10-4B9) (all from BioLegend), as well as Foxp3-eFluor450 (clone FJK-16s, eBioscience). Flow cytometry was performed on a BD LSRFortessa SORP equipped to detect 17 fluorescent parameters. Compensation and data analysis were carried out using FACSDiva and FlowJo software.
Tissue preparation and flow cytometry staining of human samples.
Fresh AML, LAM, normal kidney, and normal lung tissues were disrupted mechanically with a surgical scalpel and then enzymatically digested in RPMI (Invitrogen) containing 10% FBS, 100 U/ml collagenase IV (Invitrogen), and 50 μg/ml DNase I (MilliporeSigma). Staining procedures and antibodies were described previously (73).
IHC.
Tissue sections were dewaxed in 3 changes of xylene followed by 3 changes of 100% ethanol (Thermo Fisher Scientific). Heat-induced antigen retrieval was performed in a pressure cooker (Russel) for 10 minutes in sodium citrate or EDTA antigen retrieval solutions. Sections were blocked in 5% normal goat serum (MilliporeSigma) for 60 minutes, followed by incubation with PD-1 (dilution 1:200, 86163, Cell Signaling Technology), PD-L1 (dilution 1:100, 13684, Cell Signaling Technology), CD3 (dilution 1:1,000, ab5690, Abcam), or HMB-45 (dilution 1:200, M0634, Dako) antibodies at 4oC overnight. Sections were incubated with mouse or rabbit HRP for 30 minutes.
Renal kidney tumor assessment in Tsc2+/– mice.
Tsc2+/– mice on the A/J background were generated in house as described previously (49, 50).The severity of renal kidney tumors was scored using previously established gross and microscopic kidney tumor scoring methods (49, 50). The gross kidney tumor score was determined as a summed score for all tumors in a kidney according to size: <1 mm, score 1; 1–1.5 mm, score 2; 1.5–2 mm, score 5; >2 mm, score 10. For microscopic kidney tumor score, kidneys were first cut into 1 mm–thick pieces and then fixed in 10% formalin. Each tumor or cyst was measured (length, width), and the percent of the lumen filled by tumor was determined (0% for a simple cyst and 100% for a completely filled, solid tumor) from 5-μm H&E sections for each of the 1 mm–thick pieces for each kidney. These measurements were converted into a score as previously established (50).
Statistics.
Nonparametric 1-way ANOVA followed by post Dunn’s, 2-way ANOVA or multiple 2-tailed t tests followed by Holm-Sidak post-test were used to determine the statistical significance of flow cytometry data. Nonparametric 1-way ANOVA or Mann-Whitney’s U tests were used to determine the statistical significance of tumor volume. Survival curves were assessed by the log rank (Mantel-Cox). A P value of 0.05 or less was considered significant. Statistical calculations were performed with Prism 6.0 (GraphPad Software Inc.).
Study approval.
All animal studies were reviewed and approved by IACUC of Brigham and Women’s Hospital. Fresh tissues from patients were obtained with IRB approval of Partners Human Research Committee of Brigham and Women’s Hospital. Written informed consent was obtained from participants prior to inclusion in this study.
Author contributions
HJL designed and performed experiments, analyzed data, and wrote/revised the manuscript. PHL performed experiments and analyzed data. HD, MCS, SV, and HCL performed experiments. NA recruited the study participants. KKW, GJF, and AHS provided intellectual input and edited the manuscript. EPH conceived the project, supervised experiments, and edited/revised the manuscript.
Supplementary Material
Acknowledgments
We thank Vera Krymskaya for providing us the TMKOC cell line. We thank Julie Nijmeh for discussion and revision of the manuscript, as well as Harry Filippakis and John Daley for technical advice. This work was supported by the Department of Defense Tuberous Sclerosis Medical Research Program (grant TS160057)and the Engles Program for TSC and LAM Research. HJL was supported by a Tuberous Sclerosis Alliance postdoctoral fellowship (grant 480241).
Version 1. 04/19/2018
Electronic publication
Footnotes
Conflict of interest: GJF has patents/pending royalties on the PD-1 pathway from Roche, Merck, Bristol-Myers-Squibb, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Dako, and Novartis.
Reference information: JCI Insight. 2018;3(8):e98674. https://doi.org/10.1172/jci.insight.98674.
Contributor Information
Heng-Jia Liu, Email: hliu1@bwh.harvard.edu.
Heng Du, Email: HDU2@bwh.harvard.edu.
Spencer Vaughan, Email: spencervaughan.w@gmail.com.
Nicola Alesi, Email: nalesi@bwh.harvard.edu.
Kwok-Kin Wong, Email: Kwok-Kin.Wong@nyumc.org.
References
- 1.Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- 2.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 3.Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66(3):924–934. doi: 10.1111/j.1523-1755.2004.00838.x. [DOI] [PubMed] [Google Scholar]
- 4.Byard RW, Blumbergs PC, James RA. Mechanisms of unexpected death in tuberous sclerosis. J Forensic Sci. 2003;48(1):172–176. [PubMed] [Google Scholar]
- 5.Cudzilo CJ, et al. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest. 2013;144(2):578–585. doi: 10.1378/chest.12-2813. [DOI] [PubMed] [Google Scholar]
- 6.Taveira-DaSilva AM, Moss J. Clinical features, epidemiology, and therapy of lymphangioleiomyomatosis. Clin Epidemiol. 2015;7:249–257. doi: 10.2147/CLEP.S50780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taveira-DaSilva AM, Steagall WK, Moss J. Lymphangioleiomyomatosis. Cancer Control. 2006;13(4):276–285. doi: 10.1177/107327480601300405. [DOI] [PubMed] [Google Scholar]
- 8.Henske EP, Jóźwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- 9.Cheadle JP, Reeve MP, Sampson JR, Kwiatkowski DJ. Molecular genetic advances in tuberous sclerosis. Hum Genet. 2000;107(2):97–114. doi: 10.1007/s004390000348. [DOI] [PubMed] [Google Scholar]
- 10.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2000;97(11):6085–6090. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosset C, Netto CBO, Ashton-Prolla P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet Mol Biol. 2017;40(1):69–79. doi: 10.1590/1678-4685-gmb-2015-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dibble CC, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47(4):535–546. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garami A, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11(6):1457–1466. doi: 10.1016/S1097-2765(03)00220-X. [DOI] [PubMed] [Google Scholar]
- 14.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13(15):1259–1268. doi: 10.1016/S0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5(6):578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 17.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169(2):361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 18.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bissler JJ, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 20.Bissler JJ, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franz DN, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 22.Krueger DA, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 23.Young L, et al. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med. 2013;1(6):445–452. doi: 10.1016/S2213-2600(13)70090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Lin PC. Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin Cancer Biol. 2017;47:185–195. doi: 10.1016/j.semcancer.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–146. doi: 10.1038/nrc3670. [DOI] [PubMed] [Google Scholar]
- 26.Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35(2):161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmadzadeh M, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114(8):1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fourcade J, et al. PD-1 is a regulator of NY-ESO-1-specific CD8+ T cell expansion in melanoma patients. J Immunol. 2009;182(9):5240–5249. doi: 10.4049/jimmunol.0803245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston RJ, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26(6):923–937. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 31.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walunas TL, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–413. doi: 10.1016/1074-7613(94)90071-X. [DOI] [PubMed] [Google Scholar]
- 33.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 34.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14(8):561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 35.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alsaab HO, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parkhitko AA, et al. Autophagy-dependent metabolic reprogramming sensitizes TSC2-deficient cells to the antimetabolite 6-aminonicotinamide. Mol Cancer Res. 2014;12(1):48–57. doi: 10.1158/1541-7786.MCR-13-0258-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atochina-Vasserman EN, et al. Pharmacological targeting of VEGFR signaling with axitinib inhibits Tsc2-null lesion growth in the mouse model of lymphangioleiomyomatosis. Am J Physiol Lung Cell Mol Physiol. 2015;309(12):L1447–L1454. doi: 10.1152/ajplung.00262.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016;28(8):401–409. doi: 10.1093/intimm/dxw025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dyck L, Wilk MM, Raverdeau M, Misiak A, Boon L, Mills KH. Anti-PD-1 inhibits Foxp3+Treg cell conversion and unleashes intratumoural effector T cells thereby enhancing the efficacy of a cancer vaccine in a mouse model. Cancer Immunol Immunother. 2016;65(12):1491–1498. doi: 10.1007/s00262-016-1906-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duraiswamy J, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors--response. Cancer Res. 2014;74(2):633–634. doi: 10.1158/0008-5472.CAN-13-2752. [DOI] [PubMed] [Google Scholar]
- 42.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107(9):4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon SR, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–499. doi: 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krempski J, et al. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol. 2011;186(12):6905–6913. doi: 10.4049/jimmunol.1100274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu GT, et al. PD-1 blockade attenuates immunosuppressive myeloid cells due to inhibition of CD47/SIRPα axis in HPV negative head and neck squamous cell carcinoma. Oncotarget. 2015;6(39):42067–42080. doi: 10.18632/oncotarget.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Tötterman TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother. 2010;33(3):225–235. doi: 10.1097/CJI.0b013e3181c01fcb. [DOI] [PubMed] [Google Scholar]
- 47.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 48.Dumont FJ, Su Q. Mechanism of action of the immunosuppressant rapamycin. Life Sci. 1996;58(5):373–395. doi: 10.1016/0024-3205(95)02233-3. [DOI] [PubMed] [Google Scholar]
- 49.Pollizzi K, Malinowska-Kolodziej I, Stumm M, Lane H, Kwiatkowski D. Equivalent benefit of mTORC1 blockade and combined PI3K-mTOR blockade in a mouse model of tuberous sclerosis. Mol Cancer. 2009;8:38. doi: 10.1186/1476-4598-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee L, et al. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42(3):213–227. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- 51.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 52.Klarquist J, et al. Melanoma-associated antigen expression in lymphangioleiomyomatosis renders tumor cells susceptible to cytotoxic T cells. Am J Pathol. 2009;175(6):2463–2472. doi: 10.2353/ajpath.2009.090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choueiri TK, et al. PD-L1 expression in nonclear-cell renal cell carcinoma. Ann Oncol. 2014;25(11):2178–2184. doi: 10.1093/annonc/mdu445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahoney KM, et al. PD-L1 Antibodies to Its Cytoplasmic Domain Most Clearly Delineate Cell Membranes in Immunohistochemical Staining of Tumor Cells. Cancer Immunol Res. 2015;3(12):1308–1315. doi: 10.1158/2326-6066.CIR-15-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osterburg AR, et al. NK cell activating receptor ligand expression in lymphangioleiomyomatosis is associated with lung function decline. JCI Insight. 2016;1(16):e87270. doi: 10.1172/jci.insight.87270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 57.Crane CA, et al. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene. 2009;28(2):306–312. doi: 10.1038/onc.2008.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu C, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25(5):590–604. doi: 10.1016/j.ccr.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lastwika KJ, et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016;76(2):227–238. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 60.van Hooren L, et al. Local checkpoint inhibition of CTLA-4 as a monotherapy or in combination with anti-PD1 prevents the growth of murine bladder cancer. Eur J Immunol. 2017;47(2):385–393. doi: 10.1002/eji.201646583. [DOI] [PubMed] [Google Scholar]
- 61.Liu Y, et al. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol Immunother. 2009;58(5):687–697. doi: 10.1007/s00262-008-0591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lázár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci USA. 2008;105(30):10483–10488. doi: 10.1073/pnas.0804453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berrien-Elliott MM, et al. Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T-cell tolerance. Cancer Res. 2013;73(2):605–616. doi: 10.1158/0008-5472.CAN-12-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen S, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3(2):149–160. doi: 10.1158/2326-6066.CIR-14-0118. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, et al. Enhancing CD8+T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017;32(3):377–391.e9. doi: 10.1016/j.ccell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chaoul N, et al. Rapamycin Impairs Antitumor CD8+ T-cell Responses and Vaccine-Induced Tumor Eradication. Cancer Res. 2015;75(16):3279–3291. doi: 10.1158/0008-5472.CAN-15-0454. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y, Sparwasser T, Figlin R, Kim HL. Foxp3+ T cells inhibit antitumor immune memory modulated by mTOR inhibition. Cancer Res. 2014;74(8):2217–2228. doi: 10.1158/0008-5472.CAN-13-2928. [DOI] [PubMed] [Google Scholar]
- 68.Li Q, et al. Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. J Immunol. 2012;188(7):3080–3087. doi: 10.4049/jimmunol.1103365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turner AP, et al. Sirolimus enhances the magnitude and quality of viral-specific CD8+ T-cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant. 2011;11(3):613–618. doi: 10.1111/j.1600-6143.2010.03407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest. 2014;124(1):188–197. doi: 10.1172/JCI69856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(+/–) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest. 1999;104(6):687–695. doi: 10.1172/JCI7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ilagan E, Manning BD. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer. 2016;2(5):241–251. doi: 10.1016/j.trecan.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lizotte PH, et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight. 2016;1(14):e89014. doi: 10.1172/jci.insight.89014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.