

The recombinant production and crystal structure of chorismate mutase from Burkholderi thailandensis, which is a proxy for more virulent Burkholderia species, are presented. The enzyme shares conserved binding-cavity residues with other chorismate mutases, including those with which it has no appreciable sequence identity.
Keywords: structural genomics, Seattle Structural Genomics Center for Infectious Disease, Burkholderia thailandensis, chorismate mutase, isomerases
Abstract
Burkholderia thailandensis is often used as a model for more virulent members of this genus of proteobacteria that are highly antibiotic-resistant and are potential agents of biological warfare that are infective by inhalation. As part of ongoing efforts to identify potential targets for the development of rational therapeutics, the structures of enzymes that are absent in humans, including that of chorismate mutase from B. thailandensis, have been determined by the Seattle Structural Genomics Center for Infectious Disease. The high-resolution structure of chorismate mutase from B. thailandensis was determined in the monoclinic space group P21 with three homodimers per asymmetric unit. The overall structure of each protomer has the prototypical AroQγ topology and shares conserved binding-cavity residues with other chorismate mutases, including those with which it has no appreciable sequence identity.
1. Introduction
Burkholderia thailandensis is a less infective member of the Burkholderia genus of proteobacteria that is often used as a model for more virulent members of this genus (Haraga et al., 2008 ▸). Burkholderia are nonfermenting motile Gram-negative bacteria, and infectious members such as B. mallei, B. pseudomallei and B. cepacia are highly antibiotic-resistant and as such could be used as biological agents targeting humans and animals (Yabuuchi et al., 1992 ▸; Sawana et al., 2014 ▸). B. cepacia and 16 other closely related species known as the B. cepacia complex (BCC) cause severe and often fatal pulmonary infections in cystic fibrosis patients and other immunocompromised individuals (Mahenthiralingam et al., 2002 ▸; Agodi et al., 2002 ▸; Speert et al., 2002 ▸). Another species, B. pseudomallei, causes up to 20% of all community-acquired septicemias, notably a lethal form of melioidosis (Limmathurotsakul & Peacock, 2011 ▸). Other Burkholderia species cause infections in animals (B. mallei) and plants (B. glumae and B. gladioli), and the BCC species are infectious by inhalation (Whitlock et al., 2007 ▸; Cui et al., 2016 ▸). New antibacterials against Burkholderia need to be developed owing to the importance of these species. One promising approach is to target enzymes that have shown promise in other bacteria and are absent in mammals, for example the enzymes of the shikimate pathway, which is important for the biochemical synthesis of aromatic amino acids, folic acid, ubiquinone and other aromatic compounds (Mousdale & Coggins, 1991 ▸; Abell, 1999 ▸; Haslam, 1993 ▸; Coggins et al., 2003 ▸). The first committed step in the shikimate pathway is the Claisen rearrangement of chorismate to prephenate, which is catalysed by chorismate mutase, which is also known as hydroxyphenylpyruvate synthase. Chorismate mutases occur in two classes: the monofunctional AroH class and the more common, sometimes bifunctional AroQ class. Members of the AroH class have a trimeric α/β-barrel topology, while the AroQ class form dimeric helical bundles. Subclasses of the AroQ class include AroQγ and AroQα, with roles in virulence (Haslam, 1993 ▸; Roberts et al., 1998 ▸). Here, we present the recombinant production and structural analysis of an AroQγ chorismate mutase from B. thailandensis.
2. Materials and methods
2.1. Macromolecule production
Chorismate mutase from B. thailandensis was cloned, expressed and purified by the Seattle Structural Genomics Center for Infectious Disease (SSGCID; Myler et al., 2009 ▸; Stacy et al., 2011 ▸) following standard protocols described previously (Bryan et al., 2011 ▸; Choi et al., 2011 ▸; Serbzhinskiy et al., 2015 ▸). Briefly, genomic DNA from B. thailandensis E264 (Kim et al., 2005 ▸) was kindly provided by Dr Joseph Mougous, University of Washington. The signal-peptide-cleaved mature protein (UniProt Q2SY64) encoding amino acids 32–202 was PCR-amplified from genomic DNA using the primers shown in Table 1 ▸. The resultant amplicon was cloned into the ligation-independent cloning (LIC; Aslanidis & de Jong, 1990 ▸) pET-14b-based expression vector pBG1861, which was kindly provided by Wesley Van Voorhis, University of Washington, encoding a noncleavable 6×His fusion tag (MAHHHHHHM-ORF). Plasmid DNA was transformed into chemically competent Escherichia coli BL21(DE3)R3 Rosetta cells. The cells were tested for expression and a 2 l culture was grown using auto-induction medium (Studier, 2005 ▸) in a LEX Bioreactor (Epiphyte Three Inc.). The expression clone was assigned the SSGCID target identifier ButhA.00160.a.B2, and the clone and purified protein are available at https://apps.sbri.org/SSGCIDTargetStatus/Target/ButhA.00160.a.
Table 1. Macromolecule-production information.
| Source organism | B. thailandensis E264 (strain E264/ATCC 700388/DSM 13276/CIP 106301) |
| DNA source | Genomic DNA provided by Dr Joseph Mougous, University of Washington |
| Forward primer | CTCACCACCACCACCACCATATGGCCGACGGCGACGATACC |
| Reverse primer | ATCCTATCTTACTCACTTAGCCGACGGCGCTGGCGCC |
| Cloning vector | pBG1861 |
| Expression vector | pBG1861 |
| Expression host | E. coli BL21(DE3)R3 Rosetta |
| Complete amino-acid sequence of the construct produced† | MAHHHHHHMADGDDTALTNLVALASQRLALAEPVAHWKWINRKPISDPPREAALLTDVEKRATANGVDPAYARTFFDDQIAASKQLQNALFATWRATHGPEGPAPDLATSTRPQLDRLTQSLIAALARVAPLRDAPDCPSRLARSIANWKTLTRYDSAQKDALGTALSHVCAAGGASAVG |
The noncleavable 6×His fusion tag is underlined.
The protein was purified by a two-step protocol consisting of immobilized metal (Ni2+)-affinity chromatography (IMAC) followed by size-exclusion chromatography (SEC). All chromatography runs were performed on an ÄKTApurifier 10 (GE) using automated IMAC and SEC programs according to previously described procedures (Bryan et al., 2011 ▸). The final SEC was performed on a HiLoad 26/600 Superdex 75 column (GE Healthcare) using a mobile phase consisting of 300 mM NaCl, 20 mM HEPES pH 7.0, 5% glycerol, 1 mM TCEP. Peak fractions eluted as a single peak consistent with a monomeric enzyme. The peak fractions were pooled and analysed for the presence of the protein of interest using SDS–PAGE. The peak fractions were concentrated to 58.13 mg ml−1 using an Amicon purification system (Millipore). Aliquots of 200 µl were flash-frozen in liquid nitrogen and stored at −80°C until use for crystallization.
2.2. Crystallization
The protein was crystallized using established crystallization approaches at the SSGCID. Briefly, the protein was diluted to 20 mg ml−1 and single crystals were obtained by vapor diffusion in sitting drops directly from JCSG-plus condition C3 (Table 2 ▸).
Table 2. Crystallization.
| Method | Sitting-drop vapor diffusion |
| Plate type | 96-well Compact 300, Rigaku |
| Temperature (K) | 290 |
| Protein concentration (mg ml−1) | 20 (diluted from 58.13) |
| Buffer composition of protein solution | 20 mM HEPES pH 7.0, 300 mM NaCl, 5% glycerol, 1 mM TCEP |
| Composition of reservoir solution | JCSG-plus condition C3: 20%(w/v) PEG 3350, 200 mM ammonium nitrate |
| Volume and ratio of drop | 0.4 µl:0.4 µl |
| Volume of reservoir (µl) | 80 |
2.3. Data collection and processing
Data-collection and processing information is reported in Table 3 ▸. Data were integrated and scaled with XDS and XSCALE (Kabsch, 2010 ▸). X-ray diffraction images are available for this entry, which was previously refined as PDB entry 4oj7 and is now updated as PDB entry 6cnz, at https://proteindiffraction.org/project/4oj7/.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Diffraction source | Beamline 21-ID-G, APS |
| Wavelength (Å) | 0.97857 |
| Temperature (K) | 100 |
| Detector | MAR Mosaic 300 mm CCD |
| Crystal-to-detector distance (mm) | 275 |
| Rotation range per image (°) | 1.0 |
| Total rotation range (°) | 180 |
| Exposure time per image (s) | 1.5 |
| Space group | P21 |
| a, b, c (Å) | 51.71, 121.38, 88.67 |
| α, β, γ (°) | 90, 99.99, 90 |
| Mosaicity (°) | 0.2 |
| Resolution range (Å) | 50.00–2.15 (2.21–2.15) |
| Total No. of reflections | 221537 (16462) |
| No. of unique reflections | 57764 (4233) |
| Completeness (%) | 98.7 (98.6) |
| Multiplicity | 3.80 (3.89) |
| 〈I/σ(I)〉 | 18.47 (2.78) |
| R r.i.m. | 0.061 (0.54) |
| Overall B factor from Wilson plot (Å2) | 25.7 |
| PDB code | 6cnz |
2.4. Structure solution and refinement
The structure was solved by molecular replacement with MOLREP (Vagin & Teplyakov, 2010 ▸; Lebedev et al., 2008 ▸). The search model was chorismate mutase from Mycobacterium tuberculosis (PDB entry 2f6l; 31% sequence identity; Kim et al., 2006 ▸). Initial refinement was carried out with REFMAC 5.8.0049 (Murshudov et al., 2011 ▸) with TLS and manual refinement in Coot (Emsley & Cowtan, 2004 ▸; Emsley et al., 2010 ▸). The quality of the structure was checked by MolProbity (Chen et al., 2010 ▸). The coordinates were initially deposited as PDB entry 4oj7; after initial review of the manuscript, further model building and structure refinement were performed using the PHENIX package (Terwilliger et al., 2008 ▸) and the coordinates were updated as PDB entry 6cnz. The resulting structure-refinement data are provided in Table 4 ▸. All structural figures were generated using PyMOL (http://www.pymol.org).
Table 4. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Resolution range (Å) | 50.0–2.15 (2.204–2.150) |
| Completeness (%) | 98.7 (98.6) |
| No. of reflections, working set | 57741 (3941) |
| No. of reflections, test set | 1968 (139) |
| Final R cryst | 0.164 (0.218) |
| Final R free | 0.216 (0.276) |
| Cruickshank DPI | 0.2392 |
| No. of non-H atoms | |
| Protein | 7226 |
| Nitrate | 76 |
| Cryoprotectant (EDO) | 28 |
| Water | 470 |
| Total | 7800 |
| R.m.s. deviations | |
| Bonds (Å) | 0.007 |
| Angles (°) | 0.865 |
| Average B factors (Å2) | |
| Protein | 43.0 |
| Nitrate | 61.6 |
| Cryoprotectant (EDO) | 56.1 |
| Water | 42.4 |
| Ramachandran plot | |
| Most favored (%) | 98.21 |
| Allowed (%) | 1.16 |
3. Results and discussion
The crystal structure of chorismate mutase from B. thailandensis was solved in the monoclinic space group P21 with three homodimers per asymmetric unit (Fig. 1 ▸). The homodimer buries 2340 Å2 of surface. The dimer offers opportunities for the future study of allostery and chorismate biosynthesis in Burkholderia as previously investigated for Geobacillus sp. (Nazmi et al., 2016 ▸) and Mycobacterium tuberculosis (Munack et al., 2016 ▸). The structures most similar to chorismate mutase from B. thailandensis were identified by PDBeFold (http://www.ebi.ac.uk/msd-srv/ssm) analysis using the default threshold cutoffs of 70% for the percentage of the secondary structure of the target chain identified in the query protein and of the secondary structure of the query chain (Krissinel & Henrick, 2004 ▸). Of the 133 990 entries in the PDB, only three unique matches were identified at the default cutoff: chorismate mutases from M. tuberculosis (Okvist et al., 2006 ▸; Kim et al., 2006 ▸, Qamra et al., 2006 ▸), Yersinia pestis (Kim et al., 2008 ▸) and B. phymatum (PDB entry 5ts9; Asojo et al., 2018 ▸).
Figure 1.

The asymmetric unit of the 2.15 Å resolution crystal structure of chorismate mutase from B. thailandensis contains three homodimers.
The overall structure of each protomer of chorismate mutase from B. thailandensis follows the AroQγ topology as observed in the secreted chorismate mutases from M. tuberculosis (PDB entry 2fp2, 32% sequence identity; PDB entry 2f6l, 31% sequence identity; PDB entry 2ao2, 32% sequence identity; Okvist et al., 2006 ▸; Kim et al., 2006 ▸; Qamra et al., 2006 ▸), Y. pestis (PDB entry 2gbb, 20% sequence identity; Kim et al., 2008 ▸) and B. phymatum (PDB entry 5ts9, 33% sequence identity). The AroQγ topology is composed entirely of helices connected by short loops (Fig. 2 ▸). Despite having only 30% sequence identity, the members of the AroQγ subclass have a similar overall topology and conserved core helices (Fig. 2 ▸). The r.m.s.d.s on superposing Cα atoms of the B. thailandensis protomer with the homologues from B. phymatum, M. tuberculosis and Y. pestis are 1.17 Å (over 158 amino acids), 1.18 Å (over 154 amino acids) and 1.36 Å (over 154 amino acids), respectively. As shown in the primary-sequence alignment (Fig. 3 ▸) and the structural overlay (Fig. 4 ▸), the E. coli chorismate mutase (Lee et al., 1995 ▸) with AroQα topology has longer helices. These extended helices are in regions of low sequence homology between the different chorismate mutase subfamilies and are distant from the substrate-binding site.
Figure 2.
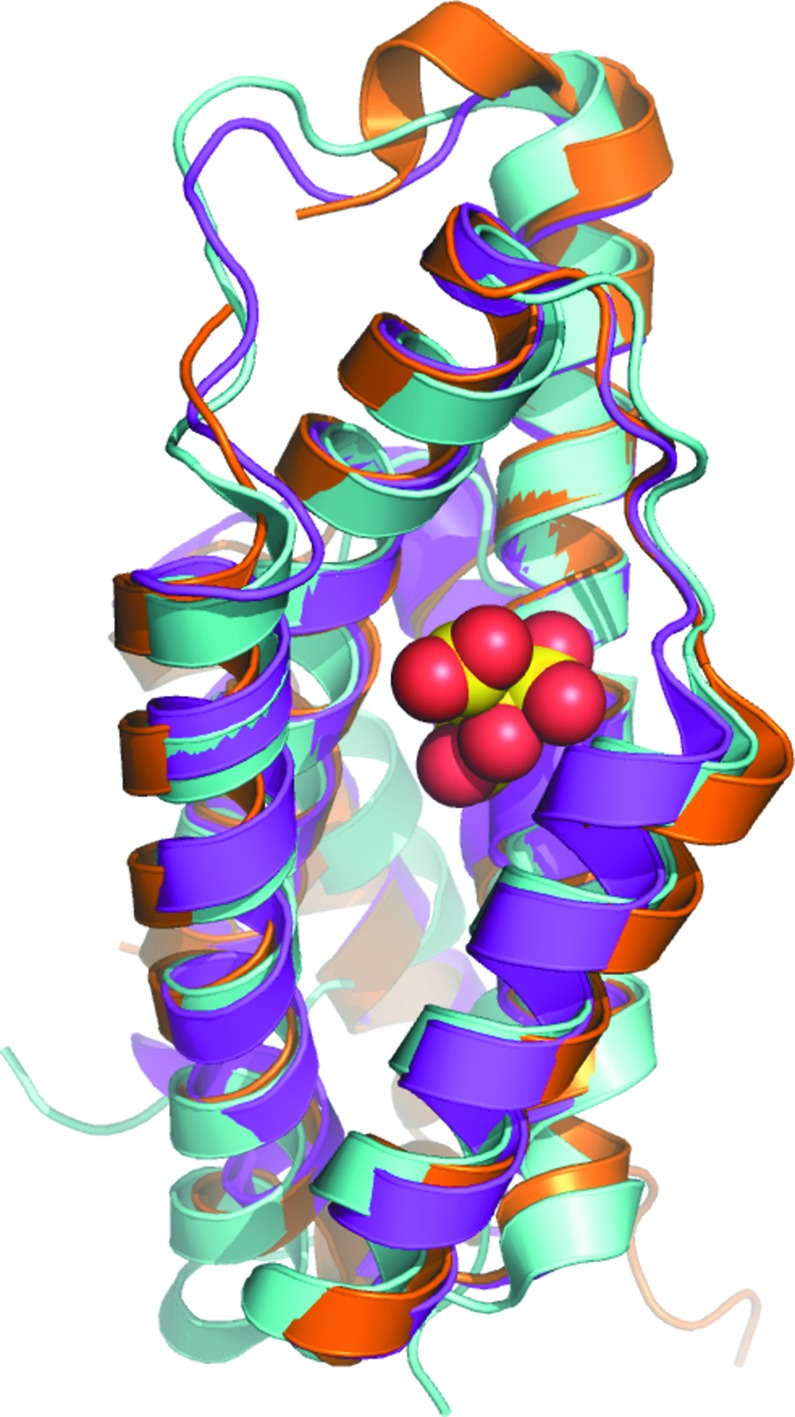
Superposed protomers of chorismate mutases with AroQγ topology from B. thailandensis (magenta; current study), M. tuberculosis (gold; PDB entry 2fp2; Okvist et al., 2006 ▸) and Y. pestis (cyan; PDB entry 2gbb; Kim et al., 2008 ▸). A citrate molecule (yellow and red spheres) sits in the substrate-binding cavity of the Y. pestis crystal structure.
Figure 3.

Structural and primary-sequence alignment of chorismate mutases from B. thailandensis (PDB entry 6cnz), M. tuberculosis (PDB entry 2fp2; Okvist et al., 2006 ▸), Y. pestis (cyan; PDB entry 2gbb; Kim et al., 2008 ▸) and E. coli (PDB entry 1ecm; Lee et al., 1995 ▸). The secondary-structure elements shown are α-helices (α), 310-helices (η), β-strands (β) and β-turns (TT). Identical residues are shown in white on a red background and conserved residues are shown in red. This figure was generated using ESPript (Gouet et al., 1999 ▸, 2003 ▸).
Figure 4.
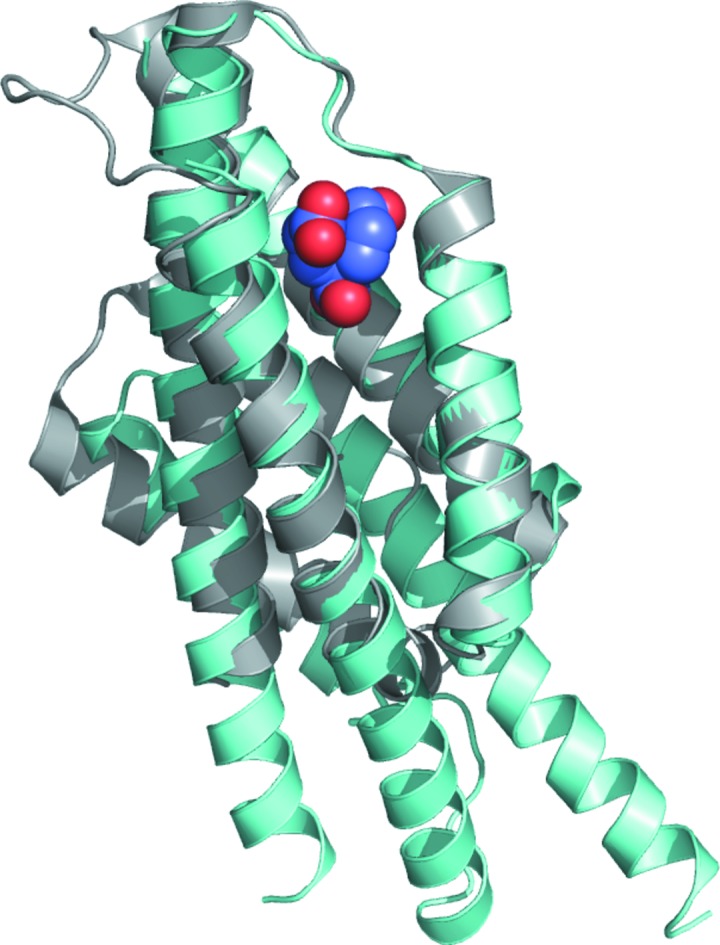
Superposition of chorismate mutase from E. coli (cyan; PDB entry 1ecm; Lee et al., 1995 ▸) with that from B. thailandensis (gray); the transition-state analogue (blue and red spheres) in the active site is shown.
While no reported structures of AroQγ chorismate mutases have substrates or substrate analogues in the active site, the crystal structure of the AroQα chorismate mutase from E. coli (PDB entry 1ecm; Lee et al., 1995 ▸) was determined with a transition-state analogue (TSA). The E. coli protein does not have any appreciable sequence similarity to the B. thailandensis protein, but the amino-acid residues that make up the active site and are involved in substrate binding are conserved (Figs. 4 ▸ and 5 ▸). A LigPlot (Laskowski & Swindells, 2011 ▸; Wallace et al., 1995 ▸) analysis of the interactions of the active site of E. coli chorismate mutase bound to a TSA and a model of B. thailandensis chorismate mutase generated via an overlay reveals a well conserved binding cavity. For example, based on this overlay with E. coli chorismate mutase bound to a TSA, we can predict that conserved residues Arg49, Lys60, Asp69, Gln73, Ser105 and Gln109 should bind to the TSA. Arg134 of the B. thailandensis chorismate mutase structure occupies a similar space to Arg11 of the E. coli enzyme, despite occurring in a different position in the primary sequence. The conservation of the active site of the enzyme suggests that broad-spectrum active-site inhibitors can be designed to target both AroQγ and AroQα chorismate mutases. We observed nitrate molecules bound in our crystal structure; for example, in all six protomers present in the asymmetric unit we observed a nitrate bound to Ser47, Trp171 and Gln181 and a second nitrate bound to Arg134. While the first site is not conserved and is therefore unlikely to be relevant for broad-spectrum drug development, the second site at Arg134 is significant since the equivalent residue in M. tuberculosis interacts with one of the carboxylates of the TSA. Thus, a nitrate isostere could be incorporated into the scaffold of a compound intended to mimic this interaction.
Figure 5.

LigPlot diagrams reveal conserved residues in the substrate-binding sites of chorismate mutase from E. coli bound to a transition-state analogue (PDB entry 1ecm; Lee et al., 1995 ▸) and of B. thailandensis chorismate mutase (PDB entry 6cnz) generated via an overlay with the E. coli structure.
4. Conclusions
Despite sharing less than 33% sequence identity with homologous proteins, the structure of chorismate mutase from B. thailandensis reveals that it has a conserved AroQγ topology and substrate-binding cavity and could be targeted by inhibitors developed for these homologs.
Supplementary Material
PDB reference: chorismate mutase from Burkholderia thailandensis, 6cnz
X-ray diffraction images. URL: http://dx.doi.org/10.18430/M34OJ7
Acknowledgments
We thank the SSGCID cloning and protein-production groups at the Center for Infectious Disease Research and at the University of Washington.
Funding Statement
This work was funded by National Institutes of Health, National Institute of Allergy and Infectious Diseases grants HHSN272200700057C, HHSN272201200025C , and HHSN272201700059C. U.S. Department of Energy, Office of Science grant DE-AC02-06CH11357. Michigan Economic Development Corporation grant 085P1000817.
References
- Abell, C. (1999). Comprehensive Natural Products Chemistry, edited by O. Meth-Cohn, D. Barton & K. Nakanishi, Vol. 1, pp. 573–607. Amsterdam: Elsevier.
- Agodi, A., Barchitta, M., Gianninò, V., Collura, A., Pensabene, T., Garlaschi, M. L., Pasquarella, C., Luzzaro, F., Sinatra, F., Mahenthiralingam, E. & Stefani, S. (2002). J. Hosp. Infect. 50, 188–195. [DOI] [PubMed]
- Aslanidis, C. & de Jong, P. J. (1990). Nucleic Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed]
- Asojo, O. A., Subramanian, S., Abendroth, J., Exley, I., Lorimer, D. D., Edwards, T. E. & Myler, P. J. (2018). Acta Cryst. F74, 187–192. [DOI] [PMC free article] [PubMed]
- Bryan, C. M., Bhandari, J., Napuli, A. J., Leibly, D. J., Choi, R., Kelley, A., Van Voorhis, W. C., Edwards, T. E. & Stewart, L. J. (2011). Acta Cryst. F67, 1010–1014. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Choi, R., Kelley, A., Leibly, D., Nakazawa Hewitt, S., Napuli, A. & Van Voorhis, W. (2011). Acta Cryst. F67, 998–1005. [DOI] [PMC free article] [PubMed]
- Coggins, J. R., Abell, C., Evans, L. B., Frederickson, M., Robinson, D. A., Roszak, A. W. & Lapthorn, A. P. (2003). Biochem. Soc. Trans. 31, 548–552. [DOI] [PubMed]
- Cui, Z., Zhu, B., Xie, G., Li, B. & Huang, S. (2016). Rice Sci. 23, 111–118.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Gouet, P., Robert, X. & Courcelle, E. (2003). Nucleic Acids Res. 31, 3320–3323. [DOI] [PMC free article] [PubMed]
- Haraga, A., West, T. E., Brittnacher, M. J., Skerrett, S. J. & Miller, S. I. (2008). Infect. Immun. 76, 5402–5411. [DOI] [PMC free article] [PubMed]
- Haslam, E. (1993). Shikimic Acid: Metabolism and Metabolites. Chichester: John Wiley & Sons.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kim, H. S., Schell, M. A., Yu, Y., Ulrich, R. L., Sarria, S. H., Nierman, W. C. & DeShazer, D. (2005). BMC Genomics, 6, 174. [DOI] [PMC free article] [PubMed]
- Kim, S.-K., Reddy, S. K., Nelson, B. C., Robinson, H., Reddy, P. T. & Ladner, J. E. (2008). FEBS J. 275, 4824–4835. [DOI] [PubMed]
- Kim, S.-K., Reddy, S. K., Nelson, B. C., Vasquez, G. B., Davis, A., Howard, A. J., Patterson, S., Gilliland, G. L., Ladner, J. E. & Reddy, P. T. (2006). J. Bacteriol. 188, 8638–8648. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Laskowski, R. A. & Swindells, M. B. (2011). J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed]
- Lebedev, A. A., Vagin, A. A. & Murshudov, G. N. (2008). Acta Cryst. D64, 33–39. [DOI] [PMC free article] [PubMed]
- Lee, A. Y., Karplus, P. A., Ganem, B. & Clardy, J. (1995). J. Am. Chem. Soc. 117, 3627–3628.
- Limmathurotsakul, D. & Peacock, S. J. (2011). Br. Med. Bull. 99, 125–139. [DOI] [PubMed]
- Mahenthiralingam, E., Baldwin, A. & Vandamme, P. (2002). J. Med. Microbiol. 51, 533–538. [DOI] [PubMed]
- Mousdale, D. M. & Coggins, J. R. (1991). Target Sites for Herbicide Action, edited by R. C. Kirkwood, pp. 29–56. New York: Plenum.
- Munack, S., Roderer, K., Ökvist, M., Kamarauskaite, J., Sasso, S., van Eerde, A., Kast, P. & Krengel, U. (2016). J. Mol. Biol. 428, 1237–1255. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Myler, P. J., Stacy, R., Stewart, L., Staker, B. L., Van Voorhis, W. C., Varani, G. & Buchko, G. W. (2009). Infect. Disord. Drug Targets, 9, 493–506. [DOI] [PMC free article] [PubMed]
- Nazmi, A. R., Lang, E. J. M., Bai, Y., Allison, T. M., Othman, M. H., Panjikar, S., Arcus, V. L. & Parker, E. J. (2016). J. Biol. Chem. 291, 21836–21847. [DOI] [PMC free article] [PubMed]
- Okvist, M., Dey, R., Sasso, S., Grahn, E., Kast, P. & Krengel, U. (2006). J. Mol. Biol. 357, 1483–1499. [DOI] [PubMed]
- Qamra, R., Prakash, P., Aruna, B., Hasnain, S. E. & Mande, S. C. (2006). Biochemistry, 45, 6997–7005. [DOI] [PubMed]
- Roberts, F., Roberts, C. W., Johnson, J. J., Kyle, D. E., Krell, T., Coggins, J. R., Coombs, G. H., Milhous, W. K., Tzipori, S., Ferguson, D. J., Chakrabarti, D. & McLeod, R. (1998). Nature (London), 393, 801–805. [DOI] [PubMed]
- Sawana, A., Adeolu, M. & Gupta, R. S. (2014). Front. Genet. 5, 429. [DOI] [PMC free article] [PubMed]
- Serbzhinskiy, D. A., Clifton, M. C., Sankaran, B., Staker, B. L., Edwards, T. E. & Myler, P. J. (2015). Acta Cryst. F71, 594–599. [DOI] [PMC free article] [PubMed]
- Speert, D. P., Henry, D., Vandamme, P., Corey, M. & Mahenthiralingam, E. (2002). Emerg. Infect. Dis. 8, 181–187. [DOI] [PMC free article] [PubMed]
- Stacy, R., Begley, D. W., Phan, I., Staker, B. L., Van Voorhis, W. C., Varani, G., Buchko, G. W., Stewart, L. J. & Myler, P. J. (2011). Acta Cryst. F67, 979–984. [DOI] [PMC free article] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L.-W., Read, R. J. & Adams, P. D. (2008). Acta Cryst. D64, 61–69. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Wallace, A. C., Laskowski, R. A. & Thornton, J. M. (1995). Protein Eng. 8, 127–134. [DOI] [PubMed]
- Whitlock, G. C., Estes, D. M. & Torres, A. G. (2007). FEMS Microbiol. Lett. 277, 115–122. [DOI] [PubMed]
- Yabuuchi, E., Kosako, Y., Oyaizu, H., Yano, I., Hotta, H., Hashimoto, Y., Ezaki, T. & Arakawa, M. (1992). Microbiol. Immunol. 36, 1251–1275. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: chorismate mutase from Burkholderia thailandensis, 6cnz
X-ray diffraction images. URL: http://dx.doi.org/10.18430/M34OJ7