

Abstract
Background
Two recombinant enzymes (agalsidase alfa 0.2 mg/kg/every other week and agalsidase beta 1.0 mg/kg/every other week) have been registered for the treatment of Fabry disease (FD), at equal high costs. An independent international initiative compared clinical and biochemical outcomes of the two enzymes.
Methods
In this multicentre retrospective cohort study, clinical event rate, left ventricular mass index (LVMI), estimated glomerular filtration rate (eGFR), antibody formation and globotriaosylsphingosine (lysoGb3) levels were compared between patients with FD treated with agalsidase alfa and beta at their registered dose after correction for phenotype and sex.
Results
387 patients (192 women) were included, 248 patients received agalsidase alfa. Mean age at start of enzyme replacement therapy was 46 (±15) years. Propensity score matched analysis revealed a similar event rate for both enzymes (HR 0.96, P=0.87). The decrease in plasma lysoGb3 was more robust following treatment with agalsidase beta, specifically in men with classical FD (β: −18 nmol/L, P<0.001), persisting in the presence of antibodies. The risk to develop antibodies was higher for patients treated with agalsidase beta (OR 2.8, P=0.04). LVMI decreased in a higher proportion following the first year of agalsidase beta treatment (OR 2.27, P=0.03), while eGFR slopes were similar.
Conclusions
Treatment with agalsidase beta at higher dose compared with agalsidase alfa does not result in a difference in clinical events, which occurred especially in those with more advanced disease. A greater biochemical response, also in the presence of antibodies, and better reduction in left ventricular mass was observed with agalsidase beta.
Keywords: fabry disease, enzyme replacement therapy, ert, agalsidase alfa, agalsidase beta
Introduction
Fabry disease (FD) (OMIM 301500) is an X linked lysosomal storage disorder characterised by the accumulation of globotriaosylceramide (Gb3) in several cell types due to deficiency of the enzyme alpha galactosidase A (aGAL) (enzyme commission number: 3.2.1.22).1 The disease is associated with potentially life-threatening complications such as renal failure, cardiac rhythm disturbances, heart failure and stroke. Circulating levels of plasma globotriaosylsphingosine (lysoGb3) can be used to differentiate between the severe classical phenotype and the more attenuated non-classical phenotype and as a biochemical marker to monitor treatment effects.2–4 Men with classical FD often have childhood onset of symptoms, and usually have one or more characteristic FD signs or symptoms such as cornea verticillata, neuropathic pain and clustered angiokeratoma,5 while non-classical FD generally has a later onset with more limited disease, often primarily affecting the heart. Despite the X linked inheritance pattern, women often have signs and symptoms of FD, but they are in general less severely affected.6 Classically affected women generally show a disease course that is similar to that of men with non-classical FD.7
Enzyme replacement therapy (ERT) with recombinant alpha galactosidase was the first available specific treatment for FD. In Europe and Canada, two ERTs have received marketing authorisation: agalsidase alfa (Replagal, Shire) and agalsidase beta (Fabrazyme, Sanofi Genzyme), while in the USA only agalsidase beta is licensed. Although the preparations are biochemically and structurally very similar,8–10 there is a fivefold difference in recommended dose (agalsidase alfa 0.2 mg/kg/every other week (EOW)11; agalsidase beta 1.0 mg/kg/EOW.12 Studies have shown that treatment with both enzymes can delay some of the clinical complications of the disease.13–15 However, only two clinical trials have directly compared the two agents: a small randomised controlled trial comparing both agents at a 0.2 mg/kg/EOW dose, which showed no clinically relevant differences,16 except for a dose-dependent decline of plasma lysoGb3 in a follow-up study17 and the Canadian Fabry Disease Initiative (CFDI), which showed no difference in event rate after a mean of 50 months of follow-up in 92 patients randomised to receive either agalsidase alfa or beta at licensed doses.18
Indirect comparisons of non-randomised observational studies using agalsidase alfa or beta are hampered by differences in inclusion criteria, end points definition and an absence of stratification for phenotype, which is an important predictor of the disease course.7 Also, development of neutralising antibodies directly against the enzymes has been associated with a smaller decrease of lysoGb3,19 while their effect on the occurrence of clinical manifestations has not been fully elucidated.19 20
The current study aims to compare clinical and biochemical outcomes of agalasidase alfa versus beta as part of a large international, collaborative project including three European centres of excellence combined with data from the CFDI.
Methods
Patients
Retrospective data from three European FD centres of excellence (Academic Medical Center (AMC), The Netherlands; Royal Free London NHS Foundation Trust, UK and the University Hospital Wuerzburg, Germany) were merged into one database. For the current analysis, these data were combined with the prospectively collected 8 years follow-up data from patients who newly started ERT (cohort 1b) in the CFDI.21 Data included diagnostic data, clinical, biochemical and imaging outcomes, comorbidities and medication use.
Patients were included who had a definite FD diagnosis according to previously developed criteria,22 were treatment naïve and treated with either agalsidase alfa at a dose of 0.2 mg/kg/EOW or agalsidase beta at a dose of 1.0 mg/kg/EOW for at least 9 months. Allocation of treatment was non-random, except for part of the patients from the CFDI. Initially, there was a 1:1 ratio within the CFDI but during the shortage of agalsidase beta most patients started on agalsidase alfa. In one of the European treatment centres, agalsidase alfa was preferentially prescribed, while in the other agalsidase beta was predominantly prescribed. In the third European centre, allocation of treatment was based on the preferences of the patient in close consultation with the treating physician, without the presence of strict treatment guidelines.
Baseline was defined as start of ERT. Follow-up ended at switch to the other enzyme preparation or to a different dose, discontinuation of ERT or the last recorded clinic visit.
Medication with ACE inhibitors (ACEi)/angiotensin receptor blockers (ARBs), antiplatelet therapy and antihypertensive therapy was applied according to the best practices at that time.
Phenotype
Patients were categorised as classical or non-classical on the basis of enzyme activity and the presence or absence of characteristic FD symptoms (neuropathic pain, clustered angiokeratoma and/or cornea verticillata.)5 A detailed description of the classification method has been published previously and can be found in online supplementary appendix S1.7 The CFDI database did not always capture the same criteria for phenotype, thus pedigree analysis was added for some uncertain cases.
jmedgenet-2017-104863supp001.pdf (297.1KB, pdf)
Clinical outcomes
We assessed the clinical event rate from start of therapy until first event or end of follow-up. Clinical and laboratory measurements were longitudinally analysed.
Clinical events
Clinical events were defined as follows:
Renal events: chronic kidney disease (CKD) category G5 (estimated glomerular filtration rate (eGFR) <15 mL/min/1.73 m2), renal transplantation or dialysis.
Cardiac events: implantation of an implantable cardiac defibrillator (ICD) or pacemaker (PM).
Cerebral events: stroke or transient ischaemic attack diagnosed by a neurologist
Death from any cause.
Renal function
Renal function was evaluated using the eGFR and the amount of protein excretion in urine. The eGFR was calculated using the CKD-EPI in adults and the Schwartz formula in children up to 18 years of age.23 24 The eGFR of patients who had received a renal transplant or were undergoing dialysis was set at 10 mL/min/1.73 m2. Albuminuria and proteinuria excretion was categorised according to Kidney Disease Improving Global Outcomes guidelines.24
Cardiac involvement
Cardiac involvement was assessed by echocardiography. Left ventricular mass index (LVMI) was calculated using the Devereux formula and was corrected for height (m2.7).25 The upper reference limit for men and women is 48 and 44 g/m2.7, respectively.25 Analysis of cardiac MRI data was not feasible due to the small number of patients with baseline and follow-up MRIs.
LysoGb3 and antibodies
Plasma lysoGb3 levels, as reflection of the accumulation of sphingolipids, were measured at the AMC with tandem mass spectrometry using glycine or isotope-labelled lysoGb3 as internal standard.26 27 Results from both internal standards correlated very well.7 Antibodies were measured as previously described.28 A titre of ≥6 was considered as antibody positive. Patients were considered antibody positive if all antibody measurements after treatment initiation were positive. Since antibody development in women is rare,19 antibodies were measured in men only. No lysoGb3 and antibody data were obtained from the CFDI.
Statistical analysis
R (V.3.1.5) was used for statistical analysis. Data are presented as mean and SD or median and range where appropriate. A Cox proportional hazard model was used to assess the clinical event rate defined as first event (renal, cardiac or cerebral), or death. ERT type (ie, agalsidase alfa or beta), baseline eGFR, sex, phenotype and the interaction between sex and phenotype were included as covariates (full model specifications: online supplementary appendix S2). Inclusion of baseline LVMI and a history of an event before initiation of ERT (stroke, dialysis, transplantation and/or ICD/PM implantation) did not improve the model. Patients were censored when an event occurred or at end of follow-up. The proportional hazard assumption was visually tested by using Schoenfeld residuals. In addition, we applied propensity score matching in a 1:1 ratio (package: MatchIt) in order to assess if an uneven distribution of covariates could bias the results, by using callipers of width equal to 0.1 SD of the estimated propensity score. Propensity scores were based on sex, phenotype, baseline LVMI measured on echocardiography, baseline eGFR, events before ERT and age at initiation of ERT. Subsequently, we performed a Cox proportional hazard model on the matched data.
jmedgenet-2017-104863supp002.pdf (283.3KB, pdf)
The proportion of patients with a decrease in LVMI 1 year after initiation of ERT was analysed by logistic regression. Mixed effect models (package: nlme) were used to analyse the eGFR, LVMI and lysoGb3 over time. Only adult patients were included in the analysis of the eGFR and LVMI. Both LVMI and lysoGb3 values showed a decrease in the first year followed by constant levels in the following years. In order to account for this non-linear relation between time and LVMI/lysoGb3, we used the change from baseline in LVMI and lysoGb3 in the respective models. A random slope and random intercept were included when appropriate. Time on ERT, ERT type, age at start of ERT, sex, phenotype, ACEi/ARBs, baseline eGFR, baseline LVMI and/or baseline lysoGb3 were included as covariates when appropriate (full model specifications: online supplementary appendix S2). Models were selected in a stepwise manner, and the Akaike Information Criterion (AIC) was used to evaluate the goodness of fit. Furthermore, eGFR analyses were stratified for low or high eGFR at baseline (eGFR <60 and ≥60 mL/min/1.73 m2, respectively). In the ∆LVMI analyses, results were stratified by the presence or absence of LVH at baseline. Differences in the prevalence of antibodies were assessed with the Fisher’s exact test. The relation between antibody formation and the change in lysoGb3 over time was evaluated in by a linear mixed effect model. Mixed effect model assumptions were visually tested by diagnostic plots. Variance inflation factor was used to explore potential multicollinearity. P values <0.05 were considered statistically significant. Where appropriate, 95% CI are given. Results are reported in accordance with the ‘The Strengthening the Reporting of Observational Studies in Epidemiology’ statement.29
Ethics statement
According to Dutch law, and after review of the AMC ethics committee, no approval of the study protocol was needed because of the observational nature of the study. All data were obtained from medical records. Patient records were anonymised and de-identified prior to analysis. All patients have provided consent for the use of their medical data and samples in accordance with local ethics requirements.
Results
Patients
In total, 283 European and 104 Canadian patients (54% females) were included in the analysis (table 1). Mean age at start of ERT was 46 (±15) years. Treatment consisted of agalsidase alfa in 248 and agalsidase beta in 139 patients with a median follow-up time of 4.9 (0.8–14.4) years. In general, patients treated with agalsidase beta were more likely to have classical disease, to have received a renal transplant or dialysis before start of therapy and to have higher lysoGb3 and lower eGFR at baseline (all P<0.05). Patient characteristics stratified for sex, phenotype and ERT type can be found in online supplementary table S1. Discontinuation of treatment (n=15) due to patient preferences or treatment failure, change in dose (n=48) or switch of ERT (n=37) occurred in 100 patients (26%), which resulted in censoring. The shortage of agalsidase beta was the main reason to reduce dose (n=47) or switch to agalsidase alfa (n=17).
Table 1.
Patient characteristics at start of ERT
| Agalsidase alfa (0.2 mg/kg) | Agalsidase beta (1.0 mg/kg) | P value | |
| Patients | 248 | 139 | |
| Men, classical* | 69 (28%) | 71 (51%) | <0.001 |
| Men, non-classical | 47 (19%) | 7 (5%) | 0.22 |
| Women, classical | 95 (38%) | 43 (30%) | 0.14 |
| Women, non-classical | 37 (15%) | 18 (13%) | 0.86 |
| ERT start <18 years of age | 15 (6%) | 3 (2%) | 0.13 |
| Follow-up time (years) | 5.2 (0.8–14.4) | 3.8 (0.8–12.1) | <0.001 |
| Events before initiation of ERT | |||
| Dialysis/renal transplant | 8 (3%) | 12 (9%) | 0.007 |
| PM/ICD | 21 (8%) | 9 (7%) | 0.87 |
| Stroke | 22 (9%) | 17 (12%) | 0.09 |
| Any of the above† | 46 (19%) | 31 (22%) | 0.08 |
| LysoGb3 (nmol/L) | 10 (0.7–146) | 80 (2.0–178) | <0.001 |
| eGFR (mL/min/1.73 m2) | 89 (10–159) | 86 (10–140) | 0.009 |
| CKD category A3 | 44/195 (23%) | 42/113 (37%) | 0.008 |
| LVMI (g/m2.7) | 49 (15–117) | 52 (20–148) | 0.14 |
| Use of ACEi/ARBs | 89/248 (36%) | 52/139 (37%) | 0.83 |
| Hypertension | 109/236 (39%) | 62/137 (45%) | 0.23 |
| BMI (kg/m2) | 26 (±4.9) | 25 (±5.6) | 0.30 |
| HDL cholesterol (mmol/L) | 1.5 (±0.4) | 1.5 (±0.4) | 0.92 |
| LDL cholesterol (mmol/L) | 2.7 (±0.9) | 2.7 (±0.8) | 0.76 |
| Total cholesterol (mmol/L) | 4.8 (±1.1) | 4.7 (±1.0) | 0.51 |
| Triglycerides (mmol/L) | 1.2 (0.2–5.9) | 1.2 (0.3–3.6) | 0.18 |
Continuous variables are presented as mean (±SD) or median (range). Hypertension is defined as a diagnosis of increased blood pressure by the treating physician. CKD category A3 is defined as AER > 300 g/day or equivalent. Missing values (percentage): lysoGb3 (54%), eGFR (5%), LVMI (18%), BMI (5%), HDL cholesterol (28%), LDL cholesterol (18%), total cholesterol (17%), triglycerides (17%). For baseline characteristics per sex and phenotype see online supplementary table S1, for detailed genotype-phenotype information see online supplementary table S2.
*Classical, strictly defined including the presence of at least one characteristic symptom (angiokeratoma, acroparesthesia or cornea verticillata).
†Reflects the number of patients with one or more events (either dialysis or renal transplant, PM/ICD and/or stroke).
ERT, enzyme replacement therapy; PM, pacemaker; ICD, implantable cardiac device; lysoGb3, globotriaosylsphingosine; eGFR, estimated glomerular filtration rate; CKDA, chronic kidney disease albuminuria categories; LVMI, left ventricular mass index measured by echocardiography; ACEi, ACE inhibitors; ARB, angiotensin receptor blocker; BMI, body mass index; HDL, high-density lipoprotein cholesterol; LDL, low-density lipoprotein cholesterol.
jmedgenet-2017-104863supp003.pdf (308.6KB, pdf)
jmedgenet-2017-104863supp004.pdf (279.1KB, pdf)
Clinical events
One or more events occurred in 103 patients (27%). In the agalsidase alfa group 65/248 (26%) patients developed a clinical event compared with 38/139 (27%) patients receiving agalsidase beta. Cardiac events (n=54) were most common as first event after initiation of ERT, cerebral events (n=25) and renal events (n=10) were less frequent. Ten patients died without experiencing any other event during treatment. Causes of death in these patients were congestive heart failure (n=2), sudden cardiac death (n=2), secondary complications of end-stage renal disease (n=2), stroke (n=1), hepatic encephalopathy (n=1), meningitis (n=1) and ovarian cancer (n=1).
The event rate of patients treated with agalsidase alfa or beta was similar when stratified for sex and phenotype and adjusted for age at initiation of ERT and baseline eGFR (HRalfa vs beta: 0.96, 95% CI 0.59 to 1.57, P=0.87). A sensitivity analysis with addition of a decrease in eGFR of ≥33% and an increase in LVMI of ≥20% to the definition of clinical events revealed similar results (HRalfa vs beta: 0.84, 95% CI 0.55 to 1.29, P=0.44). Likewise, neither the inclusion of LVMI (n=314) as covariate to the original analysis (HRalfa vs beta: 0.94, 95% CI 0.55 to 1.59, P=0.81), nor the exclusion of patients with a renal event before treatment initiation (n=20) (HRalfa vs beta: 0.84, 95% CI 0.50 to 1.40, P=0.50) changed the results. With propensity scores, 188 patients were matched in a 1:1 ratio. The subsequent Cox regression analysis showed similar results (HRalfa vs beta: 0.98, 95% CI 0.55 to 1.77, P=0.95) as the unmatched analyses (figure 1).
Figure 1.

Kaplan-Meier curve after propensity score matching. Kaplan-Meier curve for any first event (renal, cardiac or cerebral event or death) after propensity score matching.
Renal function
Longitudinal data on eGFR was available for 337 adult patients (figure 2). Adjusted for sex and phenotype, there was no difference in the slope of eGFR between agalsidase alfa and beta in patients with a baseline eGFR ≥60 (βslope alfa-beta: −0.12 mL/min/1.73 m2/year, 95% CI −0.76 to 0.51, P=0.70). Also, in patients with an eGFR <60 there was no difference in the rate of decline (βslope alfa-beta: −0.85 mL/min/1.73 m2/year, 95% CI −2.31 to 0.62, P=0.26). Adding the use of ACEi/ARBs and/or the presence of proteinuria at baseline as covariates to the model did not result in a better fit or different results. Our previous study30 showed that in this cohort (with exception of Canadian patients) of patients, the eGFR slope was −2.5 mL/min/1.73 m2/year (95% CI −2.9 to −2.1, P<0.001) for classical men with a baseline eGFR >60 mL/min/1.73 m2 and −4.5 mL/ min/1.73 m2/year (95% CI −5.6 to −3.3, P<0.001) for classical men with a baseline eGFR <60 mL/min/1.73 m2. The eGFR slope of women and non-classical patients with an eGFR >60 mL/min/1.73 m2 were between −1.4 and −1.6 (all P<0.001). For women and non-classical patients with an eGFR <60 mL/min/1.73 m2 only non-classical men (−3.3 mL/ min/1.73 m2/year, 95% CI −5.1 to −1.5, P<0.001) and non-classical women (−1.5 mL/ min/1.73 m2/year, 95% CI −2.8 to −0.2, P=0.04) showed a change in eGFR.
Figure 2.

Estimated glomerular filtration rate (eGFR) vs time on enzyme replacement therapy (ERT). Linear mixed model of eGFR adjusted for sex and phenotype, stratified for baseline eGFR <60 and ≥60 mL/min/1.73 m2. The larger lines represent the predicted values at group level, the smaller lines represent the predicted values at individual patient level.
Left ventricular mass
Two hundred and seventy-eight adult patients were included in the longitudinal analysis of LVMI. In patients without LVH at baseline (n=110), there was no change in LVMI after 1 year of treatment. In patients with LVH (n=168), there was a decrease after 1 year of treatment. The magnitude of the decrease depended on the LVMI at baseline (P<0.001) and was independent of sex and phenotype (figure 3). Patients with an LVMI above the reference value but <75 g/m2.7, that were treated with agalsidase beta showed a larger but non-significant decrease of LVMI over the first year compared with alfa (βalfa-beta: −3.31 g/m2.7, 95% CI −6.84 to 0.23, P=0.07), but no difference for the entire group was found (βalfa-beta: −2.26 g/m2.7, 95% CI −5.39 to 0.87, P=0.15). The decrease over the first year was followed by stabilisation of LVMI in the following years (βtime on ERT: 0.22, P=0.33). Hence, the observed difference between agalsidase alfa and beta over the first year persisted during the following years.
Figure 3.
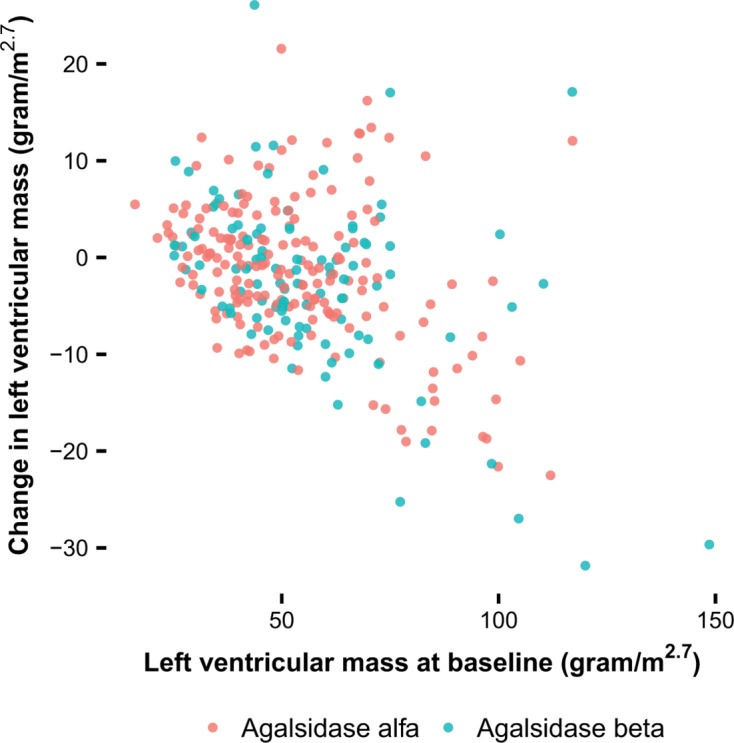
Change in left ventricular mass index (LVMI) in relation to the LVMI at baseline. Estimates of the change in LVMI from baseline after 1 year per patient, results from the linear mixed model of the change in LVMI.
The analysis on the number of patients that showed a decrease in LVMI after 1 year of treatment revealed that treatment with agalsidase beta resulted in a higher proportion of patients with a decrease in LVMI compared with agalsidase alfa (79% vs 62%) (OR 2.27, 95% CI 1.11 to 4.86, P=0.03), adjusted for the LVMI at baseline.
LysoGb3
Longitudinal data on lysoGb3 was available for 153 patients (figure 4). After initiation of ERT, lysoGb3 concentrations rapidly decreased, followed by stabilisation in all subgroups (men with classical FD: β: 0.83 nmol/L/year, P=0.08; men with non-classical FD and women: β: 0.03 nmol/L/year, P=0.94). After adjustment for baseline lysoGb3 concentration, sex and phenotype, the decrease in lysoGb3 (∆lysoGb3) in men with classical FD was more pronounced in those treated with agalsidase beta (βalfa-beta: −18.06 nmol/L, 95% CI −25.81 to −10.03, P<0.001). For example, in a classically affected man with a baseline lysoGb3 value of 100 nmol/L, the lysoGb3 concentration will be on average 45 nmol/L following 1 year of treatment with agalsidase alfa and 27 nmol/L after 1 year of treatment with agalsidase beta. In the other patients (women and non-classical men), this difference was also significant but smaller (βalfa-beta: −1.07 nmol/L, 95% CI −2.04 to −0.11, P=0.03).
Figure 4.

LysoGb3 vs time on enzyme replacement therapy (ERT) in men with classical Fabry disease. Linear mixed model of lysoGb3 adjusted for lysoGb3 at baseline. The figure presents data of men with the classical phenotype. The larger lines represent the predicted values at group level, the smaller lines represent the raw individual patient data.
Antibodies
One or more antibody assays were performed for 124 men. From 32 non-classical men (agalsidase alfa: n=27, agalsidase beta: n=5), only one developed transient antibodies. Antibody measurements were available for 92 classical men. Patients with a mixed antibody response over time (negative as well as positive antibody measurements) were excluded (n=11). Of the remaining 81 patients, persisting antibodies were established in 33 men, of whom 11 were treated with agalsidase alfa (11/39, 28%) and 22 with agalsidase beta (22/42, 52%) resulting in an OR of 2.8 (95% CI 1.02 to 7.88, P=0.041). Results did not change substantially if the first antibody measurements of patients with a mixed antibody response were included (OR 3.14, P=0.011). Antibody titres remained stable over time (figure 5).
Figure 5.

Antibody titres in men with classical Fabry disease. Antibody titres per patient. The solid dark line represent the cut-off, at an antibody titre of 6.
A comparison of the clinical event rate, LVMI and eGFR between patients treated with agalsidase alfa and agalsidase beta with or without antibodies was hampered by the uneven distribution of disease severity among groups. The limited number of patients (n=81) and events in these groups (n=29) makes extensive correction for disease severity variables impossible.
Analysis of the influence of antibodies on the decrease in lysoGb3 in men with classical FD treated with agalsidase alfa (n=21) or beta (n=35) revealed the following: in patients treated with agalsidase alfa the presence of antibodies was associated with a less prominent decrease in lysoGb3 following ERT, resulting in 34.77 nmol/L (95% CI 23.65 to 45.88, P<0.001, adjusted for baseline lysoGb3 concentrations) higher lysoGb3 concentrations in the antibody-positive group compared with the antibody-negative group. In patients receiving agalsidase beta, the decrease in lysoGb3 after ERT initiation was minimally affected by the presence or absence of antibodies (βAB+ vs AB−: 6.72 nmol/L, 95% CI −1.43 to 14.87, P=0.10) (figure 6).
Figure 6.

Effect of antibody formation on lysoGb3 in men with classical Fabry disease. Linear mixed model of the change in lysoGb3 adjusted for lysoGb3 at baseline, stratified for enzyme replacement therapy (ERT) type and antibody status. The larger lines represent the predicted values at group level (at the mean lysoGb3 concentration of 105 nmol/L in these patients), the smaller lines represent the predicted values at individual patient level.
Discussion
In this study, we systematically compared clinical outcomes and biochemical response in a large cohort of almost 400 patients with Fabry disease treated with either agalsidase alfa or agalsidase beta at authorised dose. There is no difference in clinical event rate between both enzymes, but treatment with agalsidase beta results in a larger decrease in lysoGb3 concentrations compared with agalsidase alfa. In addition, treatment with agalsidase beta has a better effect on left ventricular mass, in patients with a LVMI <75 g/m2.7. Fewer patients had an immunological response to agalsidase alfa as compared with agalsidase beta. There were considerable baseline differences between both treatment groups, which could be explained by differences in proportions of patients with classical FD as well as prescription behaviour between centres. In order to account for these differences, all analyses were adjusted for phenotype and disease severity.
In line with our findings, a small observational study showed that treatment with agalsidase beta resulted in a more pronounced decrease in lysoGb3 compared with agalsidase alfa.17 Furthermore, a slight increase in lysoGb3 was observed in men with classical FD who received a lower dose or switched to agalsidase alfa during the shortage of agalsidase beta.31 Also, a correlation between cumulative dose and podocyte Gb3 clearance has been reported.32 A dose effect on biochemical markers is further supported by the observations from this study on the influence of neutralising antibodies. We confirmed the higher prevalence of neutralising antibodies in patients treated with agalsidase beta.11 12 19 This may be related to the dose or the manufacturing method of the product (ie, human vs CHO cell line and the possible differences in glycosylation).8–10 The vast majority of neutralising antibodies in FD are IgG antibodies.28 33 IgE-mediated reactions are very infrequently observed, and have only been reported for patients treated with agalsidase beta.33 Severe anaphylaxis is rare and infusion-related reactions are usually well manageable with antipyretic drugs and/or low-dose corticosteroids prior to the infusion. The presence of antibodies in patients treated with agalsidase alfa is associated with a less prominent decrease in lysoGb3, while the decrease in lysoGb3 is almost unaffected by antibody formation during treatment with agalsidase beta, which has also been found in an earlier study.17 This is most likely caused by the fivefold higher dose, which overcomes the negative effects of antibody formation. In other lysosomal storage disorders, such as Gaucher disease, biochemical markers including glucosylsphingosine, correlate well with clinical disease parameters and can be used to monitor the effect of therapeutic intervention.4 34 In FD, previous studies have shown that plasma lysoGb3 is derived from the storage material and is related to phenotype and disease severity.3 7 35 In addition, lysoGb3 levels are associated with Mainz Severity Score index (MSSI) scores and LVMI in men and the presence of white matter lesions (WML) in women.36 Although most previous studies were unable to show differences in clinical outcomes between antibody-negative and antibody-positive patients,19 37 38 a more recent study showed larger LVMI and worse renal function in patients with antibodies.20 In that study, however, no differentiation was made by phenotype, and results were only adjusted for having a nonsense mutation. This has probably led to a confounding effect of phenotype, since the most severely affected patients (ie, classical patients) are most likely to develop antibodies. Correction for phenotype in the current study limits the risk of this bias and made it impossible to analyse differences in clinical outcome between those with and without antibodies.
No direct relationship between the decrease in lysoGb3 can be established with the clinical outcome. A reason for this is that the lysoGb3 merely reflects reduction in storage material. In some patients, the observed dose effect on plasma lysoGb3 may not always be followed by clinically relevant outcomes. Recently, we have described the absolute slopes for eGFR, LVMI of patients on treatment stratified for phenotype and sex which might be used as benchmark for future studies.30 This study also emphasises the fact that in some patients there is irreversible damage to organs such as the kidneys, which will not be reversed. It was shown that in patients with advanced kidney disease, the risk (HR 2.7) to develop new complications is high, despite treatment.38 Left ventricular hypertrophy has been shown to be reversible to some extent although the presence of fibrosis in the heart hampers an effect of any therapy.14 30 39 Hence, the direct relationship between biochemical and clinical response cannot be expected. We believe however, that an optimal biochemical response is a prerequisite for a clinical response. Indeed, this study shows that a better effect of agalsidase beta was established during the first year of treatment. However, no effect on eGFR or clinical event rate could be established. Indeed, the presence of irreversible organ damage might be the reason why disease progression has been observed in patients receiving ERT, leading to only a modest clinical effect of ERT. Although a clear effect of dose is observed on biochemical outcomes, the fact that several patients were in an advanced and irreversible disease state limits the potential to observe a similar magnitude of effect on clinical outcomes. It is also possible that the current study is still underpowered to detect a significant difference in event rate. The CFDI investigators previously calculated that almost 300 patients per group (ie, 600 patients in total) would be required to detect a 10% difference,18 which is more than the nearly 400 patients we were able to include, even after combining data from three European referral centres and the CFDI. Likewise, none of the studies that evaluated the effects of switching or dose reduction during the shortage of agalsidase beta was able to show a change in clinical event rate, since the cohorts were often small and had relatively short follow-up.31 40–42 Nonetheless, the results of one of the largest studies suggested a steeper decline in eGFR and higher disease severity scores in patients who had been switched or received a lower dose.42
Besides the possibility that the present study is underpowered to detect a difference in event rate, there are some other limitations. First of all, allocation of treatment was not random, except for part of the patients from the CFDI, although randomisation in this cohort was hampered by the shortage of agalsidase beta. Second, data were not collected through a uniform protocol and although data were mostly prospectively collected at the individual centres, the information was merged at a later time point. Finally, lysoGb3, antibody measurements and MRI scans were only available for a subset of patients. In conclusion, although we were unable to show a difference in event rate between patients treated with agalsidase alfa and beta, our results suggest a more pronounced reduction of storage materials, with agalsidase beta.
Acknowledgments
The authors thank the Fabry Support and Information Group of the Netherlands (FSIGN) for their cooperation and critical review. The authors would like to acknowledge Professor Dr F Weidemann (Katharinen-Hospital Unna, Unna and University Hospital Wuerzburg, Wuerzburg), Dr C Drechsler and Dr P Nordbeck (University Hospital Wuerzburg, Wuerzburg), Professor Dr AH Zwinderman (Academic Medical Center, Amsterdam) as well as all CFDI investigators for their support. In addition, the authors would like to thank Tracey Clarke, Kim de Gier, Shirley Klein, van Loon, Kaye LeMoine, Mark Mckie, Els Ormel, Matthew Reed and Irina Turkin for their help with the data and sample collection.
Footnotes
Contributors: MA: study design, data acquisition, data analyses, data interpretation, first draft of manuscript. MB and CEMH: study design, data interpretation, revision of manuscript. CW, SS, AM, DO, OTW, DGB, AK, MI, MLW, PME, FMV, ABPvK, DAH: data acquisition, data interpretation, revision of manuscript.
Funding: This study was supported by a grant (project number: 836011009) from the Ministry of Health of the Netherlands (ZonMw) to establish appropriate use of ERT. The funding source had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report and in the decision to submit an article for publication. The collection of data at Wurzburg University has been supported by the Bundesministerium für Bildung und Forschung of the Federal Republic of Germany (BMBF 01EO1504 MO2).
Competing interests: CW has received honoraria for lecturing from Sanofi Genzyme (Cambridge, Massachusetts, USA) and a grant to the institution from Sanofi Genzyme and Shire (Dublin, Ireland). SS, DGB, AK, MI and MLW have served on advisory boards, received fees for speaking or travel support and participated in other clinical trials and registries sponsored by Sanofi Genzyme and Shire. MLW has received travel funds, research funds or consultancy fees from Actelion, Alexion, Amicus, Excelsior, GlaxoSmithKline, Protalix and SumitomoPharma. AM has received honoraria for consultancies and educational activities as well as research support from Shire, Sanofi Genzyme, Protalix/Pfizer (New York City, New York, USA) and Amicus. DO has received speakers honoraria from Sanofi Genzyme and travel assistance from Sanofi Genzyme and Shire. PME has received speaker fees from Shire and consultancy and speaker fees from Sanofi Genzyme, Pfizer and Gilead Sciences (Foster City, California, USA). DAH has received honoraria for speaking and participating in advisory boards and support for research from Shire, Sanofi Genzyme, Amicus (Cranbury, New Jersey, USA) and Protalix (Carmiel, Israel). Also, DAH has a consultancy arrangement through UCL Consultants (London, UK) to support, in part, laboratory research. MB and CEMH have received travel support, honoraria for consultancies and educational grants from Sanofi Genzyme, Shire, Protalix, Actelion (Allschwil, Switzerland) and Amicus. All financial arrangements are made with the AMC Medical Research BV in accordance with the Research Code of the Academic Medical Center.
Patient consent: Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Desnick RJ, Ioannou YA, Eng CM, et al. . Α-galactosidase a deficiency: fabry disease : Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KMG, OMMBID - The Online Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill, 2013. [Google Scholar]
- 2. Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A 2008;105:2812–7. 10.1073/pnas.0712309105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smid BE, van der Tol L, Biegstraaten M, Linthorst GE, Hollak CE, Poorthuis BJ. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J Med Genet 2015;52:262–8. 10.1136/jmedgenet-2014-102872 [DOI] [PubMed] [Google Scholar]
- 4. Dekker N, van Dussen L, Hollak CE, Overkleeft H, Scheij S, Ghauharali K, van Breemen MJ, Ferraz MJ, Groener JE, Maas M, Wijburg FA, Speijer D, Tylki-Szymanska A, Mistry PK, Boot RG, Aerts JM. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood 2011;118:e118–27. 10.1182/blood-2011-05-352971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van der Tol L, Cassiman D, Houge G, Janssen MC, Lachmann RH, Linthorst GE, Ramaswami U, Sommer C, Tøndel C, West ML, Weidemann F, Wijburg FA, Svarstad E, Hollak CE, Biegstraaten M. Uncertain diagnosis of fabry disease in patients with neuropathic pain, angiokeratoma or cornea verticillata: consensus on the approach to diagnosis and follow-up. JIMD Rep 2014;17:83–90. 10.1007/8904_2014_342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001;38:769–75. 10.1136/jmg.38.11.769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, Elliott PM, Linthorst GE, Wijburg FA, Biegstraaten M, Hollak CE. Characterization of classical and nonclassical fabry disease: a multicenter study. J Am Soc Nephrol 2017;28:1631–41. 10.1681/ASN.2016090964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee K, Jin X, Zhang K, Copertino L, Andrews L, Baker-Malcolm J, Geagan L, Qiu H, Seiger K, Barngrover D, McPherson JM, Edmunds T. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology 2003;13:305–13. 10.1093/glycob/cwg034 [DOI] [PubMed] [Google Scholar]
- 9. Blom D, Speijer D, Linthorst GE, Donker-Koopman WG, Strijland A, Aerts JM. Recombinant enzyme therapy for Fabry disease: absence of editing of human alpha-galactosidase A mRNA. Am J Hum Genet 2003;72:23–31. 10.1086/345309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sakuraba H, Murata-Ohsawa M, Kawashima I, Tajima Y, Kotani M, Ohshima T, Chiba Y, Takashiba M, Jigami Y, Fukushige T, Kanzaki T, Itoh K. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured human Fabry fibroblasts and Fabry mice. J Hum Genet 2006;51:180–8. 10.1007/s10038-005-0342-9 [DOI] [PubMed] [Google Scholar]
- 11. Schiffmann R, Kopp JB, Austin HA, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001;285:2743–9. [DOI] [PubMed] [Google Scholar]
- 12. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ. International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human alpha-galactosidase a replacement therapy in Fabry’s disease. N Engl J Med 2001;345:9–16. 10.1056/NEJM200107053450102 [DOI] [PubMed] [Google Scholar]
- 13. Rombach SM, Smid BE, Bouwman MG, Linthorst GE, Dijkgraaf MG, Hollak CE. Long term enzyme replacement therapy for Fabry disease: effectiveness on kidney, heart and brain. Orphanet J Rare Dis 2013;8:47 10.1186/1750-1172-8-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weidemann F, Niemann M, Störk S, Breunig F, Beer M, Sommer C, Herrmann S, Ertl G, Wanner C. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med 2013;274:331–41. 10.1111/joim.12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, Finkel R, Packman S, Bichet DG, Warnock DG, Desnick RJ. Fabry Disease Clinical Trial Study Group. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007;146:77–86. [DOI] [PubMed] [Google Scholar]
- 16. Vedder AC, Linthorst GE, Houge G, Groener JE, Ormel EE, Bouma BJ, Aerts JM, Hirth A, Hollak CE. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One 2007;2:e598 10.1371/journal.pone.0000598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Breemen MJ, Rombach SM, Dekker N, Poorthuis BJ, Linthorst GE, Zwinderman AH, Breunig F, Wanner C, Aerts JM, Hollak CE. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta 2011;1812:70–6. 10.1016/j.bbadis.2010.09.007 [DOI] [PubMed] [Google Scholar]
- 18. Sirrs SM, Bichet DG, Casey R, Clarke JT, Lemoine K, Doucette S, West ML. CFDI investigators. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol Genet Metab 2014;111:499–506. 10.1016/j.ymgme.2014.01.014 [DOI] [PubMed] [Google Scholar]
- 19. Rombach SM, Aerts JM, Poorthuis BJ, Groener JE, Donker-Koopman W, Hendriks E, Mirzaian M, Kuiper S, Wijburg FA, Hollak CE, Linthorst GE. Long-term effect of antibodies against infused alpha-galactosidase a in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS One 2012;7:e47805 10.1371/journal.pone.0047805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lenders M, Stypmann J, Duning T, Schmitz B, Brand SM, Brand E. Serum-mediated inhibition of enzyme replacement therapy in Fabry disease. J Am Soc Nephrol 2016;27:256–64. 10.1681/ASN.2014121226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sirrs S, Clarke JT, Bichet DG, Casey R, Lemoine K, Flowerdew G, Sinasac DS, West ML. Baseline characteristics of patients enrolled in the Canadian Fabry disease initiative. Mol Genet Metab 2010;99:367–73. 10.1016/j.ymgme.2009.11.001 [DOI] [PubMed] [Google Scholar]
- 22. Smid BE, van der Tol L, Cecchi F, Elliott PM, Hughes DA, Linthorst GE, Timmermans J, Weidemann F, West ML, Biegstraaten M, Lekanne Deprez RH, Florquin S, Postema PG, Tomberli B, van der Wal AC, van den Bergh Weerman MA, Hollak CE. Uncertain diagnosis of Fabry disease: consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int J Cardiol 2014;177:400–8. 10.1016/j.ijcard.2014.09.001 [DOI] [PubMed] [Google Scholar]
- 23. Schwartz GJ, Muñoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, Furth SL. New equations to estimate GFR in children with CKD. J Am Soc Nephrol 2009;20:629–37. 10.1681/ASN.2008030287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl 2013;3:1–150. [Google Scholar]
- 25. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ. Chamber Quantification Writing Group American Society of Echocardiography’s Guidelines and Standards Committee European Association of Echocardiography. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr 2005;18:1440–63. 10.1016/j.echo.2005.10.005 [DOI] [PubMed] [Google Scholar]
- 26. Gold H, Mirzaian M, Dekker N, Joao Ferraz M, Lugtenburg J, Codée JD, van der Marel GA, Overkleeft HS, Linthorst GE, Groener JE, Aerts JM, Poorthuis BJ. Quantification of globotriaosylsphingosine in plasma and urine of fabry patients by stable isotope ultraperformance liquid chromatography-tandem mass spectrometry. Clin Chem 2013;59:547–56. 10.1373/clinchem.2012.192138 [DOI] [PubMed] [Google Scholar]
- 27. Krüger R, Tholey A, Jakoby T, Vogelsberger R, Mönnikes R, Rossmann H, Beck M, Lackner KJ. Quantification of the Fabry marker lysoGb3 in human plasma by tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2012;883-884:128–35. 10.1016/j.jchromb.2011.11.020 [DOI] [PubMed] [Google Scholar]
- 28. Linthorst GE, Hollak CE, Donker-Koopman WE, Strijland A, Aerts JM. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int 2004;66:1589–95. 10.1111/j.1523-1755.2004.00924.x [DOI] [PubMed] [Google Scholar]
- 29. von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP. STROBE Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med 2007;147:573–7. 10.7326/0003-4819-147-8-200710160-00010 [DOI] [PubMed] [Google Scholar]
- 30. Arends M, Biegstraaten M, Hughes DA, Mehta A, Elliott PM, Oder D, Watkinson OT, Vaz FM, van Kuilenburg ABP, Wanner C, Hollak CEM. Retrospective study of long-term outcomes of enzyme replacement therapy in Fabry disease: Analysis of prognostic factors. PLoS One 2017;12:e0182379 10.1371/journal.pone.0182379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smid BE, Rombach SM, Aerts JM, Kuiper S, Mirzaian M, Overkleeft HS, Poorthuis BJ, Hollak CE, Groener JE, Linthorst GE. Consequences of a global enzyme shortage of agalsidase beta in adult Dutch Fabry patients. Orphanet J Rare Dis 2011;6:69 10.1186/1750-1172-6-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tøndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, Svarstad E. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol 2013;24:137–48. 10.1681/ASN.2012030316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deegan PB. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J Inherit Metab Dis 2012;35:227–43. 10.1007/s10545-011-9400-y [DOI] [PubMed] [Google Scholar]
- 34. de Fost M, Hollak CE, Groener JE, Aerts JM, Maas M, Poll LW, Wiersma MG, Häussinger D, Brett S, Brill N, vom Dahl S. Superior effects of high-dose enzyme replacement therapy in type 1 Gaucher disease on bone marrow involvement and chitotriosidase levels: a 2-center retrospective analysis. Blood 2006;108:830–5. 10.1182/blood-2005-12-5072 [DOI] [PubMed] [Google Scholar]
- 35. Ferraz MJ, Marques AR, Appelman MD, Verhoek M, Strijland A, Mirzaian M, Scheij S, Ouairy CM, Lahav D, Wisse P, Overkleeft HS, Boot RG, Aerts JM. Lysosomal glycosphingolipid catabolism by acid ceramidase: formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett 2016;590:716–25. 10.1002/1873-3468.12104 [DOI] [PubMed] [Google Scholar]
- 36. Rombach SM, van den Bogaard B, de Groot E, Groener JE, Poorthuis BJ, Linthorst GE, van den Born BJ, Hollak CE, Aerts JM. Vascular aspects of Fabry disease in relation to clinical manifestations and elevations in plasma globotriaosylsphingosine. Hypertension 2012;60:998–1005. 10.1161/HYPERTENSIONAHA.112.195685 [DOI] [PubMed] [Google Scholar]
- 37. Vedder AC, Breunig F, Donker-Koopman WE, Mills K, Young E, Winchester B, Ten Berge IJ, Groener JE, Aerts JM, Wanner C, Hollak CE. Treatment of Fabry disease with different dosing regimens of agalsidase: effects on antibody formation and GL-3. Mol Genet Metab 2008;94:319–25. 10.1016/j.ymgme.2008.03.003 [DOI] [PubMed] [Google Scholar]
- 38. Bénichou B, Goyal S, Sung C, Norfleet AM, O’Brien F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab 2009;96:4–12. 10.1016/j.ymgme.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 39. Weidemann F, Sanchez-Niño MD, Politei J, Oliveira JP, Wanner C, Warnock DG, Ortiz A. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis 2013;8:116 10.1186/1750-1172-8-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsuboi K, Yamamoto H. Clinical course of patients with Fabry disease who were switched from agalsidase-β to agalsidase-α. Genet Med 2014;16:766–72. 10.1038/gim.2014.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pisani A, Spinelli L, Visciano B, Capuano I, Sabbatini M, Riccio E, Messalli G, Imbriaco M. Effects of switching from agalsidase Beta to agalsidase alfa in 10 patients with anderson-fabry disease. JIMD Rep 2013;9:41–8. 10.1007/8904_2012_177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lenders M, Canaan-Kühl S, Krämer J, Duning T, Reiermann S, Sommer C, Stypmann J, Blaschke D, Üçeyler N, Hense HW, Brand SM, Wanner C, Weidemann F, Brand E. Patients with Fabry Disease after Enzyme Replacement Therapy Dose Reduction and Switch-2-Year Follow-Up. J Am Soc Nephrol 2016;27:952–62. 10.1681/ASN.2015030337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zon MW. Treatment of patients with Fabry disease with agalsidase alfa and agalsidase beta: phenotypic diversity necessitates the development of individualized treatment guidelines. 2013. https://www.zonmw.nl/nl/onderzoek-resultaten/doelmatigheidsonderzoek/programmas/project-detail/goed-gebruik-geneesmiddelen/treatment-of-patients-with-fabry-disease-with-agalsidase-alfa-and-agalsidase-beta-phenotypic-divers/ (accessed 15 Jan 2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmedgenet-2017-104863supp001.pdf (297.1KB, pdf)
jmedgenet-2017-104863supp002.pdf (283.3KB, pdf)
jmedgenet-2017-104863supp003.pdf (308.6KB, pdf)
jmedgenet-2017-104863supp004.pdf (279.1KB, pdf)