

Abstract
Angiogenesis is the growth of new vessels from pre-existing vasculature and is an important component of many biological processes, including embryogenesis and development, wound healing, tumor growth and metastasis, and ocular and cardiovascular diseases. Effective in vitro models that recapitulate the biology of angiogenesis are needed to appropriately study this process and identify mechanisms of regulation that can be ultimately targeted for novel therapeutic strategies. The bead angiogenesis assay has been previously demonstrated to recapitulate the multiple stages of endothelial sprouting in vitro. However, a limitation of this assay is a lack of endothelial – mural cell interactions, which are key to the molecular and phenotypic regulation of endothelial cell function in vivo. The protocol given here presents a methodology for the incorporation of mural cells into the bead angiogenesis assay and demonstrates a tight association of endothelial cells and pericytes during sprouting in vitro. The protocol also details a methodology for effective silencing of target genes using siRNA in endothelial cells for mechanistic studies. Altogether, this protocol provides an in vitro assay that more appropriately models the diverse cell types involved in sprouting angiogenesis, and provides a more physiologically-relevant platform for therapeutic assessment and novel discovery of mechanisms of angiogenesis regulation.
Keywords: Developmental Biology, Issue 132, Endothelial cells, HUVEC, Pericytes, Sprouting, Angiogenesis, Bead Assay, Mural cells
Introduction
Angiogenesis is vital to appropriate embryogenesis and wound healing, and it also plays key roles in numerous diseases including cancer progression1 and coronary artery disease.2,3 Having a better understanding of how angiogenesis occurs during normal development, and how it is reactivated in pathologic contexts, is critical for the development of novel, effective therapeutics. Faithful in vitro models that recapitulate the important stages and cell types involved in angiogenesis in vivo are needed to allow researchers to better characterize the molecular mechanisms driving angiogenesis and make novel discoveries in endothelial regulation.
Nakatsu and Hughes have optimized a sprouting bead assay that they have demonstrated undergoes the many known stages of sprouting angiogenesis.4,5 The purpose of the method presented here is to build upon the assay optimized by Nakatsu and Hughes by incorporating perictyes into the assay, so that the paracrine and juxtracrine roles of mural cells in endothelial cell sprouting can be incorporated in novel angiogenesis studies. Pericytes are mural cells that are defined by their role as cells that maintain close physical contact with endothelial cells due to their being embedded in the vascular basement membrane.6 Pericytes and endothelial cells engage in complex cross-talk via signaling pathways including Notch signaling, Ang-Tie2, PDGFRβ, TGFRβ, and many others.7,8 Mouse models deficient in these signaling pathways demonstrate poor pericyte coverage of developing vasculature in embryogenesis, leading to poor vascular remodeling and dysfunctional vasculature.7 In addition, the role of pericytes in pathologic angiogenesis is important but oftentimes under-appreciated. For example, a unique feature of tumor vasculature is that the vessels are more immature, leaky, and dysfunctional due to poor pericyte coverage.9 It has been proposed that the presence or absence of pericytes dramatically impacts the phenotype of tumor blood vessels and is an important mediator of responses to antiangiogenic and antitumor therapies.9 Thus, including the role of pericytes in in vitro assays is key to more completely capture the important mechanisms of endothelial regulation.
Although there are many in vitro and ex vivo assays currently employed to study angiogenesis, there are shortcomings to consider in each. Some, such as endothelial proliferation and endothelial migration assays, are overly simplified and focus on one endothelial function in an isolated setting on tissue culture plastic.10 Other assays occur in a more 3 dimensional (3D) setting, such as the Matrigel tube formation assay,10 but these assays are still oversimplified and focus more on the ability of endothelial cells to migrate and form de novo vascular structures, as opposed to sprouting from pre-existing vasculature. Furthermore, none of these assays incorporate mural cell types. There are ex vivo models such as the ring aorta assay that do incorporate pericytes present in the host organ, but genetic manipulation of these models is much more challenging due to the necessity of generating knockout or transgenic mouse models of the pathways of interest. The bead sprouting assay is ideal because it models endothelial sprouting, proliferation, migration, and even anastomosis and lumen formation in a 3D matrix.4 The assay faithfully allows for mechanistic assessment of the many different stages of sprouting, while still allowing for direct genetic modification of either the endothelial cells or pericytes in a more controlled setting. The fibrin clots containing the sprouting beads can be easily fixed, stained, and imaged at different stages of sprouting; these sprouts can also be placed in a live imaging chamber to perform real-time imaging of sprouting. The methodology presented here is ideal for studying basic mechanisms of angiogenesis through in depth phenotyping and thorough analysis of the pathways activated during angiogenesis.
Protocol
Day 1:
1. Transient Transfection of Endothelial Cells
If desired, perform a transient transfection of Human Umbilical Vein Endothelial Cells (HUVEC) using gene-regulatory oligonucleotides (e.g. micro-RNAs or small interfering RNA (siRNA)) and the appropriate lipofectamine reagent according to manufacturer's instructions. NOTE: This protocol has had great success performing reverse transfections with custom siRNA sequences and a commercial transfection reagent (See Materials Table). The authors have not tested other transfection protocols and reagents with this assay. The steps for the reverse transfection using the reagents listed are as follows:
Reconstitute lyophilized siRNAs or microRNAs at a concentration of 20 µM in nuclease free water.
Make transfection solutions at the following ratio: 1 mL of any serum free media with 9 µL of siRNA or microRNA.
Agitate the solution and allow it to incubate for 5 min at room temperature.
Add 18 µL of transfection reagent to the solution and agitate.
Allow the solution to incubate at room temperature for 20-40 min, agitating the solution every 10 to 15 min for the duration of the incubation.
While the transfection solution is incubating, detach HUVEC using a cell detachment solution (e.g., Accutase). For a T175 flask, wash the flask with 10 mL of Phosphate Buffered Saline (PBS), and then add 5 mL of cell detachment solution and incubate the flask at 37 °C for 10 min.
Add 5 mL of complete media to the T175 flask, remove the cell suspension from the flask and centrifuge at 1200 rpm for 3 min.
Remove the supernatant using a vacuum aspirator and resuspend the cell pellet at a density of 1 X 106 cells per mL in EGM2. NOTE: This protocol has had great success using Endothelial Cell Growth Medium (EGM2) containing the standard growth factor bullet kit as well as an additional 10% FBS.
Once the transfection solution has finished incubating, plate 1 mL of solution in a 10-cm plate, and add 1 mL of HUVEC suspension on top.
Add an additional 7 mL of complete media to bring the final volume in the 10-cm plate to 9 mL.
Make up at least two 10 cm plates worth of cells for each transfection condition to ensure that there are enough cells available for later steps in the assay. NOTE: These conditions result in a final working concentration of gene regulatory oligonucleotide of 20 nM. The authors have found these conditions to result in a 60-80% knockdown of gene expression for their targets of interest. Other researchers may need to optimize transfection conditions according to their experimental needs.
Exchange the media 4-6 h post addition of siRNA complexes with fresh complete media. NOTE: This protocol was developed using passage 1 - 2 HUVEC.
Day 2
2. Coating of Microcarrier Beads with Endothelial Cells
Reconstitute microcarrier beads (e.g., Cytodex 3) at a concentration of 60,000 beads/mL in PBS and autoclave. NOTE: microcarrier beads come at a concentration of approximately 3 x 106 beads per gram. Reconstitute the beads at a ratio of 20 mg of beads per mL of PBS to get a bead density of approximately 60,000 beads/mL.
Aliquot 1 mL of complete media into the desired number of round-bottomed Fluorescence Activated Cell Sorting (FACS) tubes.
Pipette 20 µL of microcarrier bead suspension into each tube (take care to ensure that the bead solution is well resuspended prior to pipetting). NOTE: There will likely be dramatic variability in the final working concentration of beads present in a given microcarrier bead suspension. Beads can easily adhere to the sides of plastic and glass containers which can greatly alter bead concentration in the solution. The most appropriate volume of bead solution added to the FACS tube for each batch of microcarrier bead suspension will need to be determined empirically by the investigator. The numbers presented here are meant to serve more as a starting point. Ideally, conditions should be optimized so there are 10-20 beads embedded within the fibrin gel per well in the 24 well plate at the end of the assay. See further discussion in the discussion section below regarding optimization of beads numbers for the assay.
- Detach transfected HUVEC 24 h post transfection using the cell detachment solution as follows:
- Wash each 10-cm plate of cells with 5 mL of PBS, and then add 2 mL of the cell detachment solution and incubate the cells at 37 °C for 10 min.
- Resuspend the cells at a concentration of 1 X 106 cells/mL in complete media.
Add 1 mL of cell suspension (i.e. 1 X 106 HUVEC) to each FACS tube containing beads.
Gently agitate the FACS tubes every 20 min for the duration of 4 h at 37 °C (if proceeding to step 3.4) or 2.5 to 3 h (if proceeding to step 3.2). NOTE: This duration and frequency of agitation has been empirically determined by previous researchers who have optimized the basic bead sprouting assay.4 Other frequencies of agitation and durations of the bead coating step have not been tested by the authors.
3. Sequential Coating of Pericytes on HUVEC-Coated Microcarrier Beads
- Begin detaching 10T1/2 cells (pericytes) using the cell detachment solution (trypsin) as follows:
- Wash a T175 flask with 10 mL of PBS, then add 5 mL of trypsin and incubate at 37 °C for 10 min.
- Resuspend 10T1/2 at a concentration of 2 X 105 cells/0.5 mL in complete media. NOTE: Cells were purchased from the American Type Culture Collection (ATCC). NOTE: Culture 10T1/2 cells in Minimum Essential Medium (MEM) supplemented with 10% FBS and 1% Penicillin Streptomycin.
After 2.5 to 3 h of agitating the bead + HUVEC solution, wait 5 min to allow the beads and free-floating HUVEC to settle to the bottom of the tube.
Remove 0.5 mL of complete media from each tube using a P1000 pipette and add 2 x 105 10T1/2 in complete media in a volume of 0.5 mL to each FACs tube. Agitate the cell and bead solution and then continue to incubate at 37 °C. Continue to agitate every 20 min until the beads have been agitated for a total of 4 h. NOTE: The ratio of 5 HUVEC: 1 pericyte on the beads is essential to maintain appropriate pericyte coverage of the vessels. NOTE: The following alternative steps can be followed instead at this point in the protocol:
Agitate beads and HUVEC only for 4 h.
- Once the 4 h of HUVEC coating of the microcarrier beads is complete, agitate the tubes one last time, wait 30-60 s, then immediately remove 1.5-1.75 mL of solution from the FACS tube. NOTE: This should allow enough time for most of the beads to settle while many free-floating HUVEC in the media will be removed.
- Add 2 X 105 10T1/2 in complete media and bring the volume of the FACS tube up to 2 mL.
- Gently agitate the FACS tubes every 20 min for an additional hour.
Once the agitating is complete, gently agitate the tubes one last time and immediately remove the entire solution (2 mL), and add it to a 6-cm plate. Add an additional 3 mL of complete media to each plate and place the plates in the 37 °C incubator overnight.
Day 3
4. Preparation of Fibrin Gel Solutions
Prepare working solutions of aprotinin at a concentration of 1 mg/mL in PBS and thrombin at 50 units/mL. NOTE: A large stock of each of these solutions can be made and stored at 4 °C for at least a year.
- Make a fresh solution of fibrinogen at a concentration of 2 mg/mL. Make enough solution to have 2.5 mL of solution per agitated FACS tube.
- Warm the solution in a 37 °C water bath for 10 min to allow all of the fibrinogen to go into solution.
- Add 37.5 µL of aprotinin solution per 1 mL of fibrinogen solution.
- Sterile-filter the solution and set aside at room temperature.
5. Preparing Beads for Gel Implantation
Examine the dishes containing the microcarrier-coated beads under the microscope using the 20X objective to ensure that all beads have been sufficiently coated by endothelial cells.
- Vigorously pipette the plated solution of cells to detach them from the 6-cm plate and place the solution into a tube. Wash the plate 2X additional times with complete media and transfer to the tube to collect all residual beads that may be adhered to the plate.
- Avoid introducing air bubbles and use a p1000 pipette to minimize shear stress on the endothelial cell-coated beads.
- If assessing siRNA knockdown efficiency in the endothelial cells is needed, in step 2, be sure to have a plate of HUVEC-only coated beads. Then during this step (5.2), detach the beads and collect the residual HUVEC attached to the plate for RNA isolation and qPCR analysis.
Wait 3 min to allow the beads in the solution to settle to the bottom of the tube.
Remove as much of the supernatant as possible without disturbing the bead pellet using a p1000 pipette tip (do not use a vacuum aspirator).
Add 1 mL of complete media to each tube and resuspend the beads.
Wait 30-60 s to allow the beads to settle to the bottom. Then remove as much of the supernatant as possible using a P1000 pipette without disturbing the bead pellet. Do not use a vacuum aspirator as the likelihood of accidentally removing the bead pellet is greatly increased.
Repeat Steps 5.5 and 5.6 two additional times. The goal is to remove as much of the free-floating HUVEC that may have been detached from the 6-cm plate as possible, while not removing too many HUVEC-coated beads, which should settle in the FACS tube more quickly than free-floating cells.
Add 1 mL of complete media to the beads.
6. Embedding Coated Beads in a Fibrin Gel
Remove as much media as possible without disturbing the bead pellet in each FACS tube using a P1000 pipette.
Resuspend the beads in 2.5 mL of fibrinogen solution.
Pipette 13 µL of thrombin solution into each desired well of a 24-well glass-bottomed plate. Plating in a glass-bottomed plate is essential for enabling optimal imaging of the sprouts. NOTE: Do not leave thrombin sitting in a well for more than 5-10 min before the next step.
- Add 0.5 mL of bead/fibrinogen solution to each well.
- Be sure to resuspend the bead solution well prior to pipetting the solution each time.
- Take caution to carefully pipette directly into the thrombin already present in the well. Slowly pipette up and down 2-3 times to thoroughly mix the solution, taking caution to not introduce any air bubbles.
- Be sure to change the pipette tip between wells.
- Take caution to not move or disturb the plate at any point during initial fibrin clotting, as any movement may disrupt the gel formation.
Leave the plate sitting in the hood for 30 min at room temperature.
Carefully transfer the plate to a 37 °C incubator for an additional 1.5-2 h to allow the gel to fully solidify
7. Plating Fibroblasts on Top of the Fibrin Gel
- During the last 30 min of the gel solidifying, detach Normal Human Lung Fibroblasts (NHLF) as follows:
- Wash a T175 flask of cells with 10 mL of PBS, then add 5 mL of cell detachment solution and incubate at 37 °C for 10 min.
- Resuspend NHLF at a concentration of 20,000 cells/mL in complete media. NOTE: NHLF cells can be cultured with Dulbecco Modified Eagle Medium (DMEM) supplemented with 10% FBS and 1% Penicillin Streptomycin.
Plate 1 mL of the NHLF cell suspension onto the gel of each well and place back in the incubator.
Change complete media every other day for the duration of the assay.
8. Observe sprouting over the course of 2-7 days following bead implantation in the fibrin gel. The length of time required for sprouting will depend on the passage and lot number of HUVEC.
9. Fixation of Sprouting Assay
Once sufficient sprouting has occurred to be able to determine a phenotypic difference between treatment groups, wash all wells one time with 2 mL of PBS.
Add 0.5 mL of cell detachment solution to each well and place the plate in a 37 °C incubator for 5 min.
Remove the plate from the incubator and tap the sides of the plate. NOTE: The purpose of this step is to remove as many of the NHLF cells growing on top of the fibrin gel as possible to facilitate antibody penetration into the gel as well as imaging of the gel. The total incubation time with cell detachment solution may need to be adjusted to optimize NHLF removal. Researchers are cautioned to not incubate the gel for excessive periods of time as the cell detachment solution may penetrate into the gel and begin disrupting the endothelial structures within.
Once a sufficient amount of NHLF cells have been removed, quench the cell detachment solution with 1 mL of complete media.
Aspirate the detached NHLF and media using a vacuum aspirator, and wash the wells once with 1 mL of PBS.
Add 300 µL of 2% paraformaldehyde solution in PBS to each well and incubate at room temperature for 10 min.
- Remove the 2% paraformaldehyde solution and wash each well 3 times with 1 mL of PBS for 3 min at room temperature. NOTE: Please dispose of 2% paraformaldehyde solution appropriately in accordance with Environmental Health and Safety (EHS) guidelines.
- Add 1 mL of PBS to each well and store at 4 °C until ready to stain or image the sprouts.
10. Staining of Sprouting Assay
Add 300 µL of 0.5% Triton-X 100 in PBS to each well and incubate for 30 to 45 min at room temperature.
Using a vacuum aspirator, remove Triton-X100 and wash 3 times in PBS for 5 min each.
Add 500 µL of blocking solution to each gel and incubate overnight at 4 °C. NOTE: The blocking solution is made up of the following components: 1% bovine serum albumin (BSA), 5% FBS, 0.1% tween-20, and 1% antiserum (use the same species from which your secondary antibodies are derived) in PBS.
Remove blocking solution from each well and add 300 µL of staining solution containing primary antibody to each well. NOTE: The staining solution is the same as the blocking solution, but without the antiserum present. The authors have had success incubating with primary antibodies at dilutions of 1:200 to 1:400.
Incubate primary antibodies in staining solution for 24 h at 4 °C.
Aspirate primary antibody solution and wash 3 times in TBS with 0.5% tween for 20 min each at room temperature with gentle rocking. After the third wash, replace with fresh wash buffer again, and store at 4 °C overnight.
Incubate with fluorescent secondary antibody diluted 1:1000 in staining solution for 2 h at room temperature.
Remove secondary antibody staining solution and wash 5 times for 20 min each with TBS containing 0.5% tween at room temperature with gentle rocking.
Dilute nuclear stain in PBS and incubate overnight at 4 °C. NOTE: This protocol has had great success with Hoechst diluted 1:10000 in PBS. NOTE: There is no need to remove nuclear staining solution prior to imaging.
Image within a few days of completing the staining to capture optimal fluorescent signal.
11. Sprouting Assay Quantification
After the sprouts have been imaged, import the image files into ImageJ and use the Count tool to count the number of sprouts, or the Line tool to measure sprout lengths. NOTE: Sprouts are defined as endothelial protrusions with a length greater than or equal to the diameter of the bead from which they are sprouting.11 Sprout length can be measured as the distance from the point at which the sprout begins protruding off of the bead to the tip of the sprout. Average sprout length per bead and average number of sprouts per bead are useful metrics to assess phenotypic effects of gene silencing in sprouting angiogenesis. The average number of beads containing at least one sprout per treatment group can also be used as a metric to assess the robustness of sprouting.
Representative Results
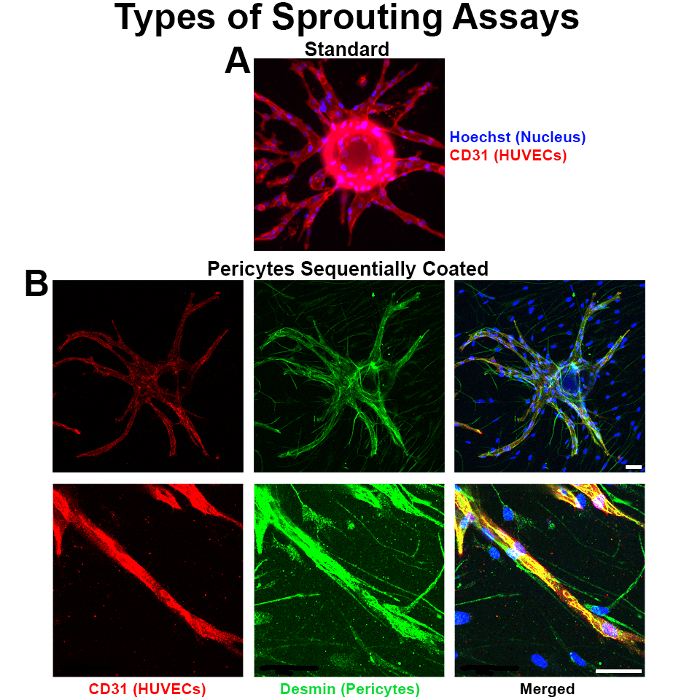
This protocol allows for a tight association of the two cell types in vitro, and the presence of the pericytes complements the occurrence of sprouting (Figure 1A, B). The protocol also enables effective silencing (e.g. via RNA interference) of a gene of interest in a cell type of interest (such as VEGFA specifically in endothelial cells or PDGFRβ in pericytes)7,12 during the assay, which can translate to inhibition of sprouting phenotype in the assay. Researchers employing this protocol are encouraged to perform a standard endothelial cell-only sprouting assay in parallel to the pericyte-coated sprouting assay to more thoroughly investigate the unique contributions of pericytes in the vascular functions and phenotypes being studied.
The conditions described within this protocol are important to adhere to in order to obtain sufficiently-coated beads (Figure 2A, B) that will be able to undergo robust sprouting in the fibrin gel. The authors have found that an excess of pericytes (such as pericytes coated at a ratio of 1 10T1/2 to 1 HUVEC) result in pericyte overgrowth of the entire well and subsequent inability to discern distinct vascular structures within the well.
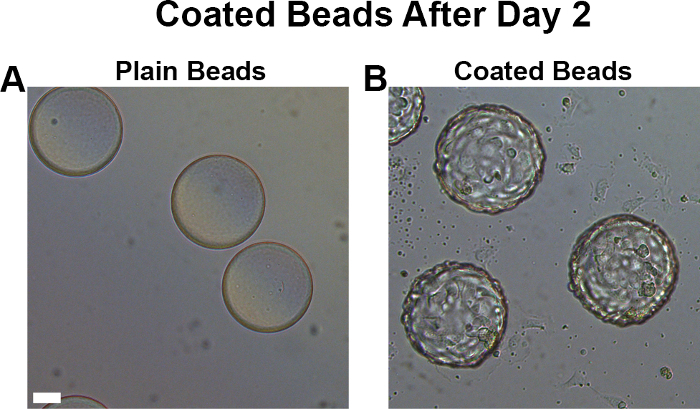
Figure 1. Pericytes tightly associate with endothelial vessels in the bead sprouting assay. (A) Standard sprouting assay with HUVEC only coated beads. (B) Confocal microscopy images of the pericyte sprouting assay demonstrate that pericytes wrap around endothelial vessels in the sprouting assay, but do not hinder their sprouting. Blue is Hoechst (nuclear stain); red is CD31 (endothelial stain), and green is Desmin (pericyte stain). All scale bars, 50 µm. Please click here to view a larger version of this figure.
Figure 2. Coated beads have a rough, golf-ball like appearance. Naked microcarrier beads (A) have a completely smooth surface, whereas appropriately-coated beads ready for fibrin gel implantation (B) have a rough, golf ball-like appearance. Scale bar, 50 µm. Please click here to view a larger version of this figure.
Discussion
This protocol presents a method for characterizing the complex stages and heterotypic cellular interactions of sprouting angiogenesis by enabling the researcher to employ genetic and imaging approaches to conduct thorough mechanistic investigations. When performing the assay, it is essential that efficient endothelial coating of the beads takes place during the bead agitation steps. Poor endothelial coating will be made evident, if the beads do not appear to have a golf ball-like rough surface the next morning prior to gel implantation and instead appear completely smooth. Take care to ensure that the beads are sufficiently resuspended during each agitation step by vigorously agitating the tubes to allow maximum exposure of the bead surface to the endothelial cells, but not so aggressively that it causes the endothelial cells already attached to become detached from the beads.
When adding pericytes into the assay, it is essential that the ratio of 5 endothelial cells to 1 pericyte is maintained. An excess of pericytes causes them to overgrow and overtake the endothelial cell populations during the assay, and a deficiency in pericytes causes poor coverage of the vessels. If the appropriate cell density is maintained but poor pericyte coverage of the vessels is still observed, agitation time with pericytes may need to be modified. It is not recommended that the cell numbers are altered. An alternative approach may be to coat the beads with endothelial cells only for 4 h, then remove the endothelial cells from the media by resuspending the bead and cell solution and removing the media once the beads have settled but before the cells have settled, then adding in pericytes and agitating every 20 min for an additional hour.
When embedding the coated beads in the fibrin clot, it is also essential to not disturb the integrity of the gel by disrupting the plate during clot formation. Disruption of the matrix formation may result in aberrant sprouting. It is also essential to ensure that the appropriate bead density is maintained in the gel. If the population of beads is too dense, then beads in close proximity to one another will affect the sprouting of neighboring beads and will begin to sprout towards one another and anastomose. Depending on the ultimate goal of the researcher and interest in characterizing vessel-vessel interactions, the bead density may need to be modified. However, a heterogeneous population of isolated beads and beads in close proximity to one another within the gel may give largely variable sprouting phenotypes. Bead density can be adjusted by altering the volume of bead solution added to the FACS tubes during the agitation step of the protocol, or by altering the volume of fibrin solution used to resuspend the beads prior to gel implantation.
It is also essential that healthy NHLFs at a density of 20,000 cells per mL are plated on top of each fibrin clot. Please note that the continuous supply of growth factors supplied by the actively dividing NHLFs are necessary for robust sprouting during the assay. NHLF-conditioned EGM2 is not a sufficient alternative.13 If poor sprouting is observed during the assay, it is recommended that a fresh bottle of complete media and a new, healthy passage of NHLF cells are used.
Although this assay begins to address the cellular and phenotypic complexity of angiogenesis at a greater resolution, it still represents an oversimplified system relative to the true, in vivo context. In addition, the sprouting angiogenesis occurring in this assay is more analogous to a healthy, physiological context as opposed to a pathologic context such as tumor angiogenesis. Future applications of this method may include the introduction of additional cell types (such as immune cell populations that modulate angiogenic responses) to further recapitulate the heterotypic cellular interactions involved in this complex process. The introduction of external stressors - such as a tumor embedded in close proximity to the sprouting vessels in the fibrin gel - to simulate pathologic angiogenesis in vitro may also be employed to allow more thorough, mechanistic characterization of the molecular pathways activated in pathological contexts. These advances will be important for improving the ability of researchers to study the complex stages of angiogenesis in a more controlled setting, ultimately enabling discovery and more in-depth characterization of novel therapeutic pathways for targeting pathologic angiogenesis.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Drs. Victoria Bautch and Joshua Boucher for helpful discussions and advice on optimizing standard bead sprouting assay conditions and a sprouting assay staining protocol. S.H.A. was supported in part by a grant from the National Institute of General Medical Sciences under award 5T32 GM007092.
References
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med. 2003;54:17–28. doi: 10.1146/annurev.med.54.101601.152418. [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8(5):491–500. doi: 10.1016/S1474-4422(09)70061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu MN, Hughes CC. An optimized three-dimensional in vitro model for the analysis of angiogenesis. Methods Enzymol. 2008;443:65–82. doi: 10.1016/S0076-6879(08)02004-1. [DOI] [PubMed] [Google Scholar]
- Nehls V, Drenckhahn D. A novel, microcarrier-based in vitro assay for rapid and reliable quantification of three-dimensional cell migration and angiogenesis. Microvasc Res. 1995;50(3):311–322. doi: 10.1006/mvre.1995.1061. [DOI] [PubMed] [Google Scholar]
- Sims DE. The pericyte--a review. Tissue Cell. 1986;18(2):153–174. doi: 10.1016/0040-8166(86)90026-1. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Tattersall IW, et al. In vitro modeling of endothelial interaction with macrophages and pericytes demonstrates Notch signaling function in the vascular microenvironment. Angiogenesis. 2016;19(2):201–215. doi: 10.1007/s10456-016-9501-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Tahergorabi Z, Khazaei M. A review on angiogenesis and its assays. Iran J Basic Med Sci. 2012;15(6):1110–1126. [PMC free article] [PubMed] [Google Scholar]
- Popson SA, et al. Interferon-induced transmembrane protein 1 regulates endothelial lumen formation during angiogenesis. Arterioscler Thromb Vasc Biol. 2014;34(5):1011–1019. doi: 10.1161/ATVBAHA.114.303352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438(7070):937–945. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- Newman AC, Nakatsu MN, Chou W, Gershon PD, Hughes CC. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell. 2011;22(20):3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]