

Abstract
The quantification of X-ray fluorescence (XRF) microscopy maps by fitting the raw spectra to a known standard is crucial for evaluating chemical composition and elemental distribution within a material. Synchrotron-based XRF has become an integral characterization technique for a variety of research topics, particularly due to its non-destructive nature and its high sensitivity. Today, synchrotrons can acquire fluorescence data at spatial resolutions well below a micron, allowing for the evaluation of compositional variations at the nanoscale. Through proper quantification, it is then possible to obtain an in-depth, high-resolution understanding of elemental segregation, stoichiometric relationships, and clustering behavior.
This article explains how to use the MAPS fitting software developed by Argonne National Laboratory for the quantification of full 2-D XRF maps. We use as an example results from a Cu(In,Ga)Se2 solar cell, taken at the Advanced Photon Source beamline 2-ID-D at Argonne National Laboratory. We show the standard procedure for fitting raw data, demonstrate how to evaluate the quality of a fit and present the typical outputs generated by the program. In addition, we discuss in this manuscript certain software limitations and offer suggestions for how to further correct the data to be numerically accurate and representative of spatially resolved, elemental concentrations.
Keywords: Chemistry, Issue 132, X-ray fluorescence, quantification, synchrotron, fitting, solar cell, defects, impurities, software, MAPS
Introduction
Synchrotron-based XRF has been used across multiple disciplines for many decades. For example, it has been used in biology on studies such as that done by Geraki et al., in which they quantified trace amounts of metal concentrations within cancerous and non-cancerous breast tissue 1. More generally, quantitative XRF has been applied to a wide array of biology studies concerned with metal concentrations in cells and tissues, as described by Paunesku et al.2. Similarly, marine protists were studied for trace elements 3,4 and even micro- and macronutrient distributions were observed within plant cells 5. Work by Kemner et al.6, which identified distinct differences in morphology and elemental composition in single bacteria cells, was also made possible through quantitative XRF analysis. Additionally, and specifically relevant to the example disclosed herein, materials scientists studying solar cell devices have made use of high-resolution XRF for studies on the existence of sub-micron metal impurities in silicon semiconductors 7,8, correlative work on how elemental distributions affect electrical performance in solar devices 9,10, and identifying depth-dependent gradients of CIGS thin film solar cells via grazing incidence X-ray fluorescence (GIXRF) 11.
Many of these studies make use not only of the high-resolution capabilities of synchrotron X-ray fluorescence to study spatial distribution, but also the quantification of the information for drawing numerical conclusions. In many studies it is critical to know the elemental concentrations associated with the aforementioned spatial distributions. For instance, in the work by Geraki et al., the study required quantifying the difference in concentrations of iron, copper, zinc, and potassium in cancerous and non-cancerous breast tissues, to better understand what concentrations become harmful to human tissues 1. Similarly, work by Luo et al. made use of quantified XRF to identify small amounts of chlorine incorporated in perovskite solar cells when synthesized both with and without chlorine-containing precursors 12. Therefore, for certain studies in which the concentrations of elements are needed, proper quantification is a necessary and critical step.
The process of quantifying elemental concentrations from X-ray fluorescence (XRF) measurements translates fluorescence intensity counts into mass concentrations (e.g. µg/cm2). The raw spectra present the number of photons collected by the energy dispersive fluorescence detector as a function of energy. The spectra are first fit and then compared to a standard measurement to calculate the quantified data. In particular, the first step of fitting fluorescence spectra is critical even for the qualitative analysis of the elements. This is because prior to fitting, counts are binned based on their energy, which becomes a problem when two elements with similar fluorescence transitions are contained in the sample. In this situation, counts may be incorrectly binned and thus associated with the wrong element.
It is often also necessary to quantify XRF spectra in order to accurately draw conclusions on relative quantities of elements in a sample. Without proper quantification, counts of heavy elements and lighter elements will be compared directly, ignoring differences in capture cross section, absorption and fluorescence probability, attenuation of the fluorescence photons, and the distance of the element's absorption edge from the incident energy, which all affect the number of photons striking the detector. Therefore, the process of fitting the spectra for each map and comparing peak intensities to the standard, both of which are done in the following procedure, is critical for the accurate quantification of each of the elemental concentrations.
We demonstrate how to convert the raw counts of fluorescence photons to units of micrograms per square centimeter (µg/cm2) by first fitting an integral spectrum, or a summed spectrum of all the individual spectra produced at each measurement spot or pixel in a 2D map. This spectrum demonstrates the relative intensities of the different elements contained in the sample. The distance the absorption edge of a certain element is from the incident beam energy influences the intensities of their fluorescence peaks. In general, the closer the two energies are, the greater the intensity produced for those elements, although this is not always the case. Figure 4 in Ref 13 shows the dependence of the absorption length of X-ray photons, which directly relates to the resulting intensity, for the majority elements in a methylammonium lead iodide perovskite solar cell. This demonstrates the fluorescence response of elements with respect to energy, and shows that it is not a continual decrease in response with increasing distance from the incident energy, but rather that it is also dependent upon the element itself.
The result of this relationship is that raw elemental concentrations may appear higher for element channels with excitation energies closer to the incident energy, even if the true quantities of those elements are lower in relation to other elements with excitation energies farther from the incident. Therefore, the energy dependence of intensity, along with other factors such as fluorescence yield variations, different absorption edges, detector sensitivity, and measurement background, etc., is why fitting the data is very important prior to drawing conclusions on the observed elemental quantities. We then apply a fitting algorithm to the integral spectrum, where the user defines the elements and parameters to fit via a text document.
The algorithm, created by Vogt et al. 14, makes use of regions of interest (ROI) filtering, in which it integrates over certain elements' peak regions, and principle component analysis (PCA). First, PCA is done to identify only the elements and peaks that are very strongly apparent. This allows for the separation of noise from the true signal. Next, the principle components identified are numerically quantified, which is important for deconvoluting different element peaks with the same excitation energy, for example overlapping Au Mα and P Kα. Finally, ROI filtering may be applied to the numeric data by integrating over specified regions.
To relate counts to elemental concentrations, a well-quantified reference (often referred to as "standard") is measured under the same measurement conditions, geometry and energy, as the sample under study. This standard is often from Dresden AXO or from the National Institute of Standards and Technology (NIST). They cover a variety of different elements and come with tabulated elemental distributions. The normalization of the measured counts of the sample of interest to the counts of the standard under the same measurement conditions provides the basis for the elemental quantification for the sample of interest.
More specifically, MAPS identifies the elements and their concentrations of the standard either by the fact that the standard information is known by the program (as is the case for the AXO and NIST standards) or through data entered into a separate file (in the case of a different standard being used). From this information, the program relates the measured intensities of the standard elements under the measurement settings to the anticipated concentration embedded in MAPS. It then creates a scaling factor to adjust for any offset and extrapolates this scaling factor to all the remaining elements not included in the standard. The scaling factor then includes the offset from the measurement settings and the information provided within MAPS for the linear conversion of raw counts to areal density in µg/cm2.
Here, we demonstrate how to make use of the program, MAPS, developed by Dr. S. Vogt, to quantify data acquired from fluorescence-capable beamlines at Argonne National Laboratory (ANL) 14. The data used for the demonstration was acquired at sector 2-ID-D of ANL using the measurement setup shown in Figure 1 of 10. The fitting procedure may also be applied to data taken from other beamlines, however, note that certain characteristics of the ANL beamlines are embedded in the program and will need to be updated.
Protocol
NOTE: Prior to beginning the fitting, it is important to know a few things about the measurements taken: the number of detector elements used - different beamlines use different detectors which are sometimes segmented into smaller sections from which the counts are read and compiled; the incident energy used; and the standard measured. This information will be applied throughout different aspects of the procedure.
1. Setting up the program
- Download the IDL and the MAPS program NOTE: Links for downloading MAPS and IDL can be found at http://www.stefan.vogt.net/downloads.html and http://www.harrisgeospatial.com/ respectively. Together, the program files may be expected to take up to approximately 20 MB. The actual space requirements for the fitting process, however, will actually be much larger and depend on the number of data files to be fit and their file sizes. The minimum device requirements for fitting are roughly 8 GB RAM, dual- or quad-core processor with speeds above 2.0 GHz. Again, the requirements increase dramatically with the file sizes being fit.
- To download IDL Virtual Machine, go to the link provided and create an account. After receiving email confirmation, log in to the website and select "Downloads" under the tab "My account". Within downloads, there should be an option for the most recent IDL software for Windows, Mac, or Linux. For the video, the version 8.5 of IDL Virtual Machine was used. NOTE: For the video, version 1.7.3.12 of MAPS was used. Version 1.7.3.12 and above can be used for the fitting of data acquired in both stepscan and flyscan modes.
- Click the link for the most recent version of MAPS and download the zip folder. The ZIP file should contain the MAPS IDL binary file as well as the files "compound.dat", "henke.xdr", and "xrf_library.csv".
- Move the latter three files to the folder "lib" in the IDL folder created in the documents upon installation. These files contain X-ray fluorescence-specific information needed for MAPS. For instance, the XRF library file stores all of the excitation energies for all of the elements.
- Place the icon for the MAPS program anywhere, however, it is often easiest to place it on the desktop. Doing so makes it easy to locate and run the graphical user interface (GUI).
- Gather necessary files for preliminary fitting NOTE: The files that are required before performing a fitting are named "maps_settings.txt", "maps_fit_parameters_override.txt", data files ending in ".mda" and a standard file ending in ".mca". An example set of these files may be found in the supporting documents. The standard file provided is the AXO standard that was measured at sector 2-ID-D of Argonne National Laboratory using an incident X-ray beam energy of 10.4 keV. The same settings were used for the measurements of the data shown in this demonstration.
- Transfer the fit parameters file, the data files, and the standard files in the directory used during data acquisition to the computer on which the fitting with MAPS will be performed. Additionally, the maps settings file provided in the supplementary information may be used for any situation.
- Create a folder on the desktop. For example, create "C:\Users\user\Desktop\Fittingfiles". It is very important that no files or folders included in the pathway for the fitting contain a space, as MAPS will not be able to access them.
- Create a subfolder of "Fittingfiles" named "mda" and place all of the ".mda" data files acquired from the X-ray fluorescence measurements inside. Be sure to include the file for the representative map that will be used to generate the fit. For instance, in the video, the file "2idd_0220.mda" is used because it is most similar in dimensions and resolution to the majority of the remaining maps to be fit.
- Place the remaining files from Step 1.2 in the parent folder. These files will be edited in the steps described below.
- Place the standard file(s) (ending in ".mca") in the folder "Fittingfiles". NOTE: A well-suited standard should contain at minimum two elements in common with the samples being studied. In cases where no suitable commercial standard exists, a representative sample containing elements of interest in known quantities may be fabricated and used as a standard. In this situation, the created sample should be measured in the same way as any other standard.
- When using the AXO Dresden standard 15, rename the file to be "axo_std.mca". For a 1 element detector, only one file is required and should end with ".mca". However, if a 4-quadrant detector was used, there should be four standard files ending with ".mca0" through ".mca3".
- If using the National Institute of Standards and Technology (NIST) standard either 1833 or 1832 for thin film samples, name the file anything as the NIST standard is later selected manually rather than referenced within the program. NOTE: At this time, only one standard may be applied for the fitting at a time. However, if there is concern that a certain standard does not cover enough elements that are also contained in the sample of interest, it is possible to rerun the fittings using different standards each time. The data from each standard may then be compared for accuracy.
- If more than one standard is used, either relocate the fit ".h5" files from each run to a distinct folder labeled for each standard or rename the files, for instance ending in "_ <standard used>"
- If neither the AXO or the NIST standard is sufficient for the samples being studied, measure any other sample as a standard as long as it contains a known quantity of elements. In order for MAPS to know to use this newly created standard, add the file "maps_standardinfo.txt" to the parent folder, "Fittingfiles". This file has been included in the supplementary information and may be adapted directly for use.
- To use the created standard, open the standard info file and enter the file name for the standard, which may be named anything ending in the proper ".mca" formats, to be used in the line "FILENAME:" (if using multiple quadrants, enter only the name ending in ".mca0"). Next, list the elements that are in the standard separated by commas in the line "ELEMENTS_IN_STANDARD:".
- By multiplying the density of each element in the standard by the standard's thickness, calculate the areal density for each element in µg/cm2. List these values in order with respect to the order of the elements listed in step 1.2.4.4. in the line "WEIGHT:", also separated by commas.
- Match the number of files named "maps_fit_parameters_override.txt" for the number of detector elements used. If only one detector was used, the file maintains the format "maps_fit_parameters_override.txt".
- If a four-quadrant detector was used, copy and paste the parameters file four times into the parent folder and then rename the extensions to range from ".txt0" to ".txt3".
- Check in the file "maps_settings.txt" that it lists the correct number of detector elements in the line "DETECTOR_ELEMENTS:" (it should be either 1 or 4).
2. Performing the fitting
- Run MAPS
- Double click on the "maps.sav" icon located on the desktop and then click anywhere on the IDL pop-up window to continue.
- On the first window after opening, called "start up: MAPS v <version being used>", select "press to change parent dir" and then select the "Fittingfiles" folder created previously.
- Select "O.K. and go to configuration".
- Generate a spectrum from a representative map to use for fitting
- From the configuration window, which can be accessed through "config + generate maps" → "general configuration", change the beamline setting to correspond to the beamline used for the measurements.
- If measurements were completed at a different beamline, verify which option in the program is for a beamline most similar in parameters to that of the used setup. Beamline capabilities for the Advanced Photon Source (APS) at ANL can be found at https://www1.aps.anl.gov/Beamlines/Directory. Comparing certain parameters such as resolution and flux can determine which ANL setting in the configuration window is most similar to the beamline used for the measurements.
- Choose "select mda files (and directory)" and the folder should open up to the "Fittingfiles" folder containing all necessary files. Next select a ".mda" file that corresponds to a representative map (it should be a map of similar composition and measurement parameters to the majority of the scans that will be fit using the parameters established from that representative map) and click "open". NOTE: If it does not open to the correct folder, it can be changed by selecting "parent dir" and the appropriate folder.
- Type in the incident energy used in keV.
- Select "start processing". When the process is done, various new folders have been created in the directory, and within the "img.dat" folder, there should be 1 or 5 files (depending on the number of detectors used) named ".h5". NOTE: In the case of a multi-element detector, one h5 file is generated for each segment of the detector (for example, files ".h50" through ".h53" for a 4-element detector) and one file that is the average of the individual elements.
- To view certain elements as processed, but not fit or quantified, enter these elements in the line "ELEMENTS_TO_FIT:" of the "maps_fit_parameters_override.txt" file. Include elements with L- or M-line peaks as "In_L" or "Au_M" for example. Otherwise, the processed file will only contain map information for the elements that were included in the fit parameters file prior to processing.
- To view the spectrum, go to "file" → "Open XRF image - avg or single element only" and locate in the "img.dat" folder the generated ".h5" file that will be used for the fitting of the remaining maps, then select "open". NOTE: There are a variety of ways to view the processed file, which contains the integral spectrum of the raw data, or the sum of the individual spectra acquired at each pixel within a map, as well as the full maps of the unfit and unquantified data. In most cases, it is best to view the data normalized to the upstream ion chamber, which can be done by going to the second drop down menu of the MAPS directory and selecting "US-IC".
- View the generated data by selecting "viewing" → "multi-element view (M)", which will show the processed maps for each of the elements. NOTE: At this point, the data appear to have been quantified, as they are represented in units of µg/cm2, however, they have not yet been fit and therefore do not represent the properly quantified data. When the actual fitting is completed, it will override the ".h5" file, so it is extremely important to keep track of which files have been fit and which have simply been converted to ".h5" format, as was described in step 2.2.4.
- For the sake of the fitting, view the integral spectrum, so go to "viewing" → "plot integral spectrum". Figure 1 shows an example of what an integral spectrum (in white) will look like.
- Save the integrated spectrum data to a ".txt" file by selecting "generate output" → "export raw integrated spectra series (long)". Close the window.
- To save the image, use a screen capture program, as MAPS itself does not have the option to save the image.
- To replot the data, plot the exported integrated spectrum data against the energy range using any data visualization software. NOTE: The file with the integrated spectrum will be named "intspec <beamline>_ <samplenumber>.h5.txt" and will be found in the parent directory within the folder named "output".
- Prepare the override file for fitting
- Go to "Tools" → "spectrum tool" then select the ".txt" file named "intspec <beamline>_ <samplenumber>.h5.txt"
- Open the file named "maps_fit_parameters_override.txt", which should now be located in the main directory as described in step 1.2.3., and enter the known settings as discussed below. This file is used for all situations, irrespective of the number of detector elements.
- In the line "DETECTOR_ELEMENTS:", make sure the number is the appropriate number for the number of detector elements used for the beamline.
- In the line "ELEMENTS_TO_FIT:", enter any missing anticipated elements, labeling L and M lines with suffixes "_L" and "_M". K-lines are assumed by default.
- In the line "COHERENT_SCT_ENERGY:", enter the incident energy used. In the following two lines, include upper and lower bounds for the energy range, as MAPS will fit for the exact incident energy. Usually, a range of ± 0.2 keV to ± 0.5 keV of the incident energy is sufficient.
- In the line "MAX_ENERGY_TO_FIT:", enter a value no less than 1 keV above the incident energy and similarly, in the line "MIN_ENERGY_TO_FIT:", enter a value no less than 1 keV.
- Towards the bottom of the file, there are the options "SI_ESCAPE_ENABLE:" AND "GE_ESCAPE_ENABLE:". Depending on the material of the detector element used (Si or Ge), enter a 1 for the element used and a 0 for the element not used. This will incorporate an escape factor for the detector element into the fit.
- Beneath the lines for 2.3.2.5, enter detector element names. It is very important that this information is correct, otherwise the quantification will be incorrect because it did not reference the proper channels during the process.
- To find the appropriate file names, open the ".mda" file by selecting "file" → "open mda file" then going to "viewing" → "multi element view (M)" and selecting on the bottom right hand side "select elements/detectors". This will open a window containing all the channels stored in the ".mda" file. Copy and paste the process variable (PV) names for the SRCurrent (e.g. SRCURRENT:S:SRcurrentAI.VAL), upstream and downstream ion chambers (e.g. US_IC:2idd:scaler1_cts1.C and DS_IC:2idd:scaler1_cts1.B) and four files for the elapsed live time, the elapsed real time, the input count rate, and the output count rate (e.g. ELT1:2iddXMAP:ElapsedLive, ERT1:2iddXMAP:ElapsedReal, ICR1:2iddXMAP:dxp1.InputCountRate, OCR1:2iddXMAP:dxp1.OutputCountRate). Finally, include the naming for the airpath (e.g. AIRPATH:0.0). NOTE: If it is unclear what names to use, ask the beamline scientist for assistance, as this step is critical for the procedure's accuracy.
- Save the file.
- Identifying fitting parameters
- Try the fit by going to "analysis" → "fit spectrum".
- First, check that the energy range in the top of the window is inclusive of all the elements of interest. Generally, it is easiest to use the same range as set in step 2.3.2.3. NOTE: Next, for the first fitting, it is fine for the "no of iters", or number of iterations of the fitting, to be zero. This will be increased later on in the fitting process to improve the quality of the fit.
- Finally, at the bottom of the window where it says "do BATCH fit of ALL spectra", select the third button from the top labeled "w/ free E, FWHM, scatter, fixed tails" in order to run the first fit. In the main directory folder, a new file named "average_resulting_maps_fit_parameters.txt" will be created.
- On the left of the spectrum tool, select all of the drop down menus named "spec" to read "none" except for one, which should read "fitted". This will show only the fit overlaid on top of the spectrum, with the colored line representing the fit and the white line representing the measured spectrum respectively. From this image, it may become clear that certain major peaks do not overlap very well with the spectrum or even that the fit does not contain a peak at all where the spectrum clearly demonstrates one. If this is the case, changes will need to be made in the fit parameters file.
- Start by using the feature "add" → "element" and the (+) and (-) signs at the bottom of the spec tool to search for missing elements. This can be done by lining up the energy lines with peaks that the fit does not appear to include.
- If it is not entirely clear what elements might be missing, run the fit with a greater number of iterations to improve the fit and help elucidate which improvements still need to be made. This can be done by changing in the "spec fit" window the option "no of iters". Usually, doing at least 50 iterations will improve the fit significantly enough such that other needed improvements can be more easily identified.
- Add identified missing elements to the "maps_fit_parameters.txt" file (note: not the "average_resulting" file mentioned before) and save the file. NOTE: If there still appears to be some peaks not being included in the fitting, it is possible there are elements with pile ups. An element pile up occurs when two XRF photons (often from the same element with the same energy) strike the detector at the same time, being read as one photon with an energy that is the sum of the two original photons.
- To search for and include pile-up elements, use the same procedure as in 2.4.3, but instead by selecting "add" → "element+pileup". Once pile-ups are identified, add the element combinations to the line "ELEMENTS_WITH_PILEUP:" with the elements included in one pile-up separated by an underscore (e.g. Si_Si for a silicon-silicon pile-up or Si_Cl for a pile-up of Si and Cl photons).
- Evaluate the branching ratios of the L-line elements. Most K-line elements are included in the fit parameters file already and these are largely accurate and cited in literature 16,17. L-lines, however, often need improvement as they have been found to vary with excitation energy 14. Currently, there is no such procedure for M-lines.
- Look at the L-line elements in need of improved branching ratios both in the sections "BRANCHING_FAMILY_ADJUSTMENT_L:"and BRANCHING_RATIO_ADJUSTMENT_L:". NOTE: "BRANCHING_FAMILY_ADJUSTMENT_L:" describes the accessible energy levels L1, L2, L3, which depend on the incident energy. For instance, if L1 > incident energy > L2, then 0, 1, 1 should be used for the respective adjustment values.
- To adjust the branching family, start by creating individual lines formatted with the element and then three 1.'s, as shown below. BRANCHING_FAMILY_ADJUSTMENT_L: In_L, 1., 1., 1. BRANCHING_FAMILY_ADJUSTMENT_L: Pb_L, 1., 1., 1.
- Adjust these numbers to shift the ratios of the peak intensities belonging to each group in relation to each other in an attempt to best match the intensity of the fit to the peak intensity of the integrated spectrum. NOTE: "BRANCHING_RATIO_ADJUSTMENT_L:" alternatively describes each individual L-shell transition, each with a unique energy. Changing the branching ratio changes the value in reference to the literature-sourced value imbedded in the MAPS program by multiplying the two values.
- To adapt these values, form a comparison of the height of the reference line (blue, yellow, or pink usually, depending on the branching family) observed through the "add" → "element" feature and the actual peak height and then calculate the branching ratio from rough estimation. NOTE: For example, if the Lβ1 line is represented twice as high as the actual peak appears, then a branching ratio for that line should be 0.5 or 50% of the literature value.
- Save the fitparameters file and repeat the steps starting with step 2.4.2 until the fit appears to be reasonable. When the fit appears to be within reason, run the fitting once more but with at least 10k iterations. NOTE: This is done because during the quantification process, it is not possible to change the number of iterations used. Therefore, increasing the number of iterations at this stage of the process ensures that the information in the average resulting fit parameters file, which is updated with each fit attempt and later employed for the quantification procedure, is as accurate as possible.
3. Run the fitting
- Rename files
- As the "average_resulting_maps_fit_parameters_override.txt" file is updated with each fitting, use this file for the actual fitting that will be applied to all of the ".mda" files that need quantification. First, rename the "maps_fit_parameters_override.txt" file that was being used for the fitting as "maps_fit_parameters_override_input.txt". Then rename the "average_resulting_maps_fit_parameters_override.txt" file as "maps_fit_parameters_override.txt".
- If using a 4-element detector, copy and paste the newly named "maps_fit_parameters_override.txt" file to create four additional files, and then change the extension of each such that they are named "maps_fit_parameters_override.txt0" through "maps_fit_parameters_override.txt3".
- Create configuration file
- Go to "config + generate maps" → "general configuration and generate maps" and change the settings as in the beginning of the procedure to reflect the measurement setup.
- Select the used beamline or representative beamline and check "use fitting".
- Click "select mda files (and directory)" and select all of the ".mda" files to be fit from the "mda" folder. These files may have been added to the folder previously in step 1.2.3.), but if not, they may be copied and pasted into the folder at this time.
- On the right-hand side of the window, using the (+) and (-) signs to move right and left, scroll through the elements and check off those that are included in the fit parameters file that will be used for the fitting.
- Add elements missing from this window by checking off a box of an unused element, and then renaming the element in the field "ROI name", including "_L" or "_M" just as it is written in the fit parameters file. In addition to renaming the element, enter the energy for the center position of the line with the highest energy (i.e. if creating a tab for In_L, enter the value for InLα1 energy) from literature. NOTE: An easy way to find this value is by using the Hephaestus program which can be downloaded online as part of the "Demeter with Strawberry Perl" package, which can be found currently at https://bruceravel.github.io/demeter/ 18. Another option is to download the X-ray data booklet from the page http://xdb.lbl.gov/. In the Hephaestus program, it is possible to select an element from the periodic table, and scroll down to find the proper energy (note the unit difference between Hephaestus and MAPS).
- Also check off the boxes for "s_i", "s_e", "s_a", "TFY", and "Bkgnd".
- Once all the changes have been made, select "write settings to config file" in the top right corner. The file can be named anything, but must end in the extension ".cfg" and must be placed in the main folder, "Fittingfiles". This file will save all the settings entered in step 3.2.) for future use. NOTE: Selecting "read settings from config file" allows the user to open the file containing the stored fit settings again in the future. Additionally, the saved settings may be changed and updated by opening a ".cfg" file, entering the changes, and then selecting "write settings to config file" once more.
- Finally, if using the NIST standard (1832 or 1833), click "NBS" for the correct standard and select the standard file that should be used for the fitting.
- Applying the fitting
- Select start processing. A series of boxes will pop-up, one for each standard element that is fit (depending on the number of detector elements used). This will not happen, however, if a standard other than the NIST or AXO is used. NOTE: Depending on the number and size of the ".mda" files being fit, the program may take anywhere from 30 minutes to hours to complete.
4. Checking the fit
Open the files for the fitted spectra by going to "file" → "Open XRF image - avg or single element only" and select under the "img.dat" folder one of the ".h5" files that have been created.
- Following the same procedure as described in step 2.2.5.1, open the multi-element viewing window and normalize the data to the upstream ion chamber.
- To select which elements/channels are displayed, use the drop down menus for "# images X:" and "# images Y:" to select the dimensions of maps to be displayed. Next, select "select elements/detectors" and a pop-up menu will be displayed with check boxes for all of the channels contained in the fit ".h5" file.
Check that the values for the elements known to be in the sample make sense. An estimation for the concentration can be calculated if the sample thickness is known. To calculate, use the following equation: Concentration = elemental density × sample thickness NOTE: For example, an estimation of the concentration of copper in a 2 µm thick CIGS solar cell is about 18 µg/cm2. Comparing this to the values for copper in Figure 2, it can be seen that the estimation is on the lower end, but that the quantification is of the right order of magnitude.
To be able to click through multiple scans that have been fit, go to "file" → "Update List of Current Files from Directory". This will place all of the files in the "img.dat" folder in the third drop down menu from the left of the MAPS window.
To export the data to a spreadsheet file or an Igor file, select "make combined ASCII files of maps" or "make combined IGOR files of maps" to export the data for each currently selected channel being shown in the window. Therefore, in order to export the data for all the channels, first select enough rows and columns for the number of elements and detectors that want to be viewed and exported and then click "select elements/detectors" to choose them. If it is desirable to have each of the channels stored as separate files, use the option "make separate…" for the respective file type.
To export the data for a batch of files, go to "file" →"Update list of Current Files from Directory". This will show all of the files in the "img.dat" folder. Then, having selected the elements to be exported, the option "export combined (separate) ASCII files of maps" will generate spreadsheet files for all of the ".h5" files in the folder. NOTE: The exported data will be saved within the "output" folder within the parent directory.
Representative Results
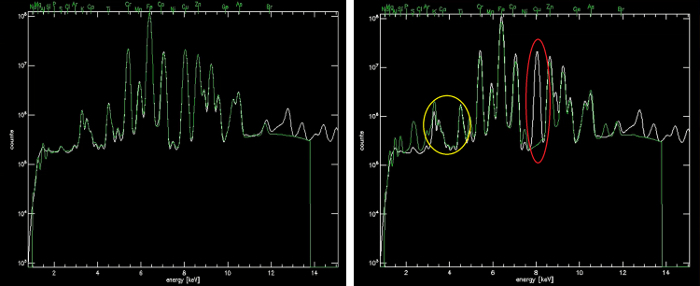
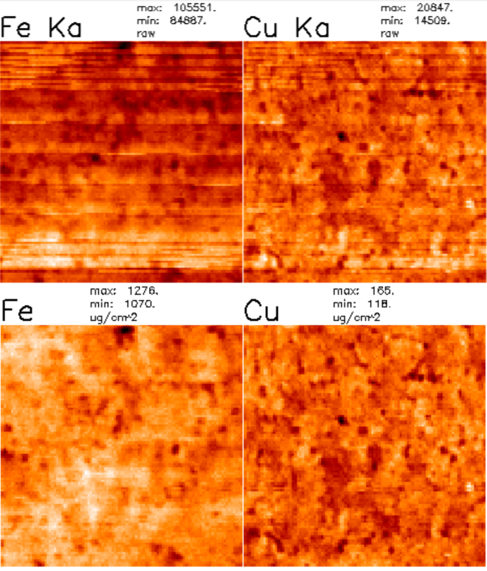
An example of proper fitting results can be seen in the following Figures. Firstly, in Figure 1 a direct comparison is shown between a poor fit and a good fit for the integral spectrum. The bad fit is reparable by both ensuring no elements are missing, for instance copper, which has a clear peak in Figure 1(left) but is not being included in the fit, and adjusting the branching ratios of the L and K lines to improve the accuracy. Figure 2 instead shows a comparison between the element channels before and after fitting. The first noticeable difference is the units for the values changes from "raw" to "ug/cm^2", suggesting that the data have been quantified. Additionally, number ranges should align with those expected from the suggested calculation in section 4.2. These values should generally not go to zero. If they do, this is almost always a sign that there is an error in the fitting.
Both the iron and copper channels can be seen both before and after the fitting. Beyond observing the value changes, it is also clear that the images are better resolved and the stripes that appear in the raw data are gone upon fitting. This resolution increase comes as a result of the peak deconvolution done by the fitting for the separation of elements with overlapping peaks. It is just one of the benefits of fitting and quantifying the data, providing the ability to more accurately qualitatively and quantitatively analyze the fluorescence data. In the particular example of a CIGS solar cell, one of the properties that researchers are interested in is the distribution of the three cations, copper, indium, and gallium, throughout the device. Statistical research has been done to study the change in their concentrations within grains and grain boundaries 16. Such a study requires the improved resolution within the map so that boundaries can be more easily identified using a watershed technique. Additionally, having the capability to study the correlation and anti-correlation of the elements provides an outlook on sample homogeneity and how to improve it.
While the quantified data can now be used to relate elemental concentrations, the fitting procedure is not perfect. There will always be a degree of error introduced from the various procedure steps, including, but not limited to, the quality of the fit, the selection, matrix homogeneity, measurement, and extrapolation of the standard, and the influence of other factors such as secondary fluorescence and sample thickness variation that are not taken into consideration by MAPS. These errors can be minimized by selecting a homogenous standard with multiple common elements with the sample and improving branching ratios as much as possible, however, note that some of these, such as controlling fit quality, are systematic errors that are hard to eradicate completely. Although it is not possible to directly quantify the error incurred, purchased standards will provide an error estimation for the concentrations of the elements, which are often quite high and should be considered when trying to analyze and propagate errors.
Correcting the data further for certain issues such as thickness variation, beam attenuation, and secondary fluorescence can help reduce the error even further. Methodologies available to do such corrections are described in the discussion section.
Figure 1. Demonstration of before (left) and after (right) the proper adjustments have been made to the fitting file to produce an accurate fitting (shown in green) of the integral spectrum (shown in white) and later, to convert raw counts to µg/cm2 accurately. Two common types of error are circled in (a): the red circle identifies a missing element, in this case Cu, and the yellow circle identifies a problem with the branching ratios for the In_L line. Please click here to view a larger version of this figure.
Figure 2. Demonstration of before (top) and after (bottom) fitting and quantification of the fluorescence channels of interest. Most notable is the change in units from "raw" to "ug/cm^2". The quantified values for iron and copper in a 2 µm thick CIGS solar cell on a 500 µm thick stainless steel substrate are on the order of 1000 and 100 µg/cm2.
Discussion
The figures show the importance of fitting data using this procedure. Figures 1 (right) and 2 (bottom) show a representative result that should arise from a proper fitting. If there is an insufficient fit, the integral spectrum image will look noticeably off and the resulting quantified data will have errors in it, although these will be hard to detect in most cases. For certain sample types for which the standard is not representative of the elements in the sample, particularly in that the samples do not contain any of elements in the standard, the quantification relies solely on extrapolating information for all the elements of interest. In a situation like this, the fitting will appear to be accurate when analyzed using the integral spectrum, however, upon quantifying, the values will appear dramatically incorrect. In this situation, it becomes necessary to use a standard of known quantities that is more similar to the sample. The process of choosing and comparing standards is well demonstrated in a study by Twining et al., which made use of the NIST standard, as well as some synthesized standards, for quantifying biological samples 3. By doing so, the authors were able to verify the appropriateness of each standard and compare the areal densities produced by using each standard for the fitting. Most importantly, the paper shows the reduction of error that results from choosing an appropriate standard and oppositely, the strong impact that using the wrong standard can have on quantification.
In addition to the fitting and quantification, there are other corrections that can be done to ensure the accurate interpretation of quantified data produced by XRF. One example is described by West et al. 20, which uses attenuation calculations provided by DeBoer 21 to further correct the quantified data for thickness variations and beam attenuation within a multi-layer sample. The article uses CIGS thin film solar cells in a case study for demonstrating the importance of using such attenuation corrections prior to forming conclusions on the observed concentration variations. The correction addresses the issue that in a multilayer sample with assumed thickness variation within the layers, regions prior to correction may suggest higher relative quantities of a certain element due to increased thickness rather than increased elemental concentration. The correction also accounts for beam attenuation of the incoming and outgoing beam in multilayer samples, as well as self-attenuation of elements within the specific layer of interest. This is one example of additional analysis necessary for the accurate quantification of X-ray fluorescence microscopy data. However, the corrections applied in 20 are based on assumptions that do not hold for all types of samples, and further corrections may need to be considered depending on the sample material and structural characteristics.
It is important to note that the raw and fitted data also include counts from secondary fluorescence occurring from fluorescence photons of one element providing enough energy to produce fluorescence photons of other elements with lower binding energies 21,22. The isolation of primary fluorescence photons from others is not possible by the fitting program, resulting in over- and under-quantification of certain elements. More specifically, the elements with higher fluorescence energies provide energy to elements of lower binding energy and are therefore not counted by the detector. Meanwhile, the atoms being excited by secondary fluorescence photons may be counted more than once because they first release photons due to the incident beam and then again release photons from the other sample elements. The data, therefore, requires additional treatment if these interactions are anticipated to have a significant impact on the quantification of elements of interest. Currently, the best approach for handling secondary fluorescence is through modeling and estimating the yield, such as what is described in 23. Information on the regime over which secondary fluorescence becomes significant and additional equations for estimation are provided in 22.
This work has demonstrated the first steps necessary for the quantification of X-ray fluorescence data. Although the process still requires many improvements and issues can arise that are specific to the type of sample studied (semiconductors, plant cells, human tissues, etc.), the technique is a reliable method for extracting meaningful quantitative information from the qualitative raw data acquired from XRF measurements.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We acknowledge funding from the U.S. Department of Energy under contract DE-EE0005948. Use of the Center for Nanoscale Materials and the Advanced Photon Source, both Office of Science user facilities, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. This material is based upon work supported in part by the National Science Foundation (NSF) and the Department of Energy (DOE) under NSF CA No. EEC-1041895. Video editing was done by VISLAB at Arizona State University. Any opinions, findings and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect those of NSF or DOE. T.N. is supported by an IGERT-SUN fellowship funded by the National Science Foundation (Award 1144616).
References
- Geraki K, Farquharson MJ, Bradley DA. X-ray fluorescence and energy dispersive x-ray diffraction for the quantification of elemental concentrations in breast tissue. Phys. Med. Biol. 2004;49:99–110. doi: 10.1088/0031-9155/49/1/007. [DOI] [PubMed] [Google Scholar]
- Paunesku T, Vogt S, Maser J, Lai B, Woloschak G. X-ray fluorescence microprobe imaging in biology and medicine. J. Cell. Biochem. 2006;99(6):1489–1502. doi: 10.1002/jcb.21047. [DOI] [PubMed] [Google Scholar]
- Twining BS, et al. Quantifying Trace Elements in Individual Aquatic Protist Cells with a Synchrotron X-ray Fluorescence Microprobe. Anal. Chem. 2003;75(15):3806–3816. doi: 10.1021/ac034227z. [DOI] [PubMed] [Google Scholar]
- de Jonge MD, et al. Quantitative 3D elemental microtomography of Cylotella meneghiniana at 400-nm resolution. Proc. Natl. Acad. Sci. 2010;107(36):15676–15680. doi: 10.1073/pnas.1001469107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duĉić T, et al. Enhancement in statistical and image analysis for in situ µSXRF studies of elemental distribution and co-localization, using Dioscorea balcanica. J. Synchrotron Rad. 2013;20:339–346. doi: 10.1107/S0909049512050170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemner KM, et al. Elemental and Redox Analysis of Single Bacterial Cells by X-ray Microbeam Analysis. Science. 2004;306(5696):686–687. doi: 10.1126/science.1103524. [DOI] [PubMed] [Google Scholar]
- Bertoni MI, et al. Nanoprobe X-ray fluorescence characterization of defects in large-area solar cells. Energy Environ. Sci. 2011;4(10):4252–4257. [Google Scholar]
- Fenning DP, et al. Iron distribution in silicon after solar cell processing: Synchrotron analysis and predictive modeling. Appl. Phys. Lett. 2011;98(162103) [Google Scholar]
- Buonassisi T, et al. Quantifying the effect of metal-rich precipitates on minority carrier diffusion length in multicrystalline silicon using synchrotron-based spectrally resolved x-ray beam induced current. Appl. Phys. Lett. 2005;87(044101) [Google Scholar]
- Stuckelberger M. Engineering solar cells based on correlative X-ray microscopy. J. Mat. Res. 2017;32(10):1825–1854. [Google Scholar]
- Streeck C, et al. Grazing-incidence x-ray fluorescence analysis for non-destructive determination of In and Ga depth profiles in Cu(In,Ga)Se2 absorber films. Appl. Phys. Lett. 2013;103(113904) [Google Scholar]
- Luo Y, et al. Spatially Heterogeneous Chlorine Incorporation in Organic-Inorganic Perovskite Solar Cells. Chem. Mater. 2016;28:6536–6543. [Google Scholar]
- Stuckelberger M. Charge Collection in Hybrid Perovskite Solar Cells: Relation to the Nanoscale Elemental Distribution. IEEE J. Photovolt. 2017;7(2):590–597. [Google Scholar]
- Vogt S, Maser J, Jacobsen C. Data analysis for X-ray fluorescence imaging. J. Phys. IV France. 2003;104:617–622. [Google Scholar]
- Applied X-ray Optics: AXO Dresden. 2017. Available from: http://axo-dresden.de/mainframe_products.htm.
- West BM. Grain engineering: How nanoscale inhomogeneities can control charge collection in solar cells. Nano Energy. 2017;32:488–493. [Google Scholar]
- Krause MO. Atomic Radiative and Radiationless Yields for K and L Shells. J. Phys. Chem. Ref. Data. 1979;8(2):307–327. [Google Scholar]
- Hubbell JH, et al. A Review, Bibliography, and Tabulation of K,L, and Higher Atomic Shell X-Ray Fluorescence Yields. J. Phys. Chem. Ref. Data. 1994;23(2):339–364. [Google Scholar]
- Ravel B, Newville M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- West BM, et al. X-ray fluorescence at nanoscale resolution for multicomponent layered structures: a solar cell case study. J Synchrotron Rad. 2017;24 doi: 10.1107/S1600577516015721. [DOI] [PubMed] [Google Scholar]
- De Boer DKG. Calculation of X-Ray Fluorescence Intensities from Bulk and Multilayer Samples. X-Ray Spectrom. 1990;19(3):145–154. [Google Scholar]
- Lachance GR, Claisse F. Quantitative X-ray Fluorescence Analysis: Theory and Application. John Wiley & Sons; 1995. [Google Scholar]
- Sokaras D, Karydas AG. Secondary Fluorescence Enhancement in Confocal X-ray Microscopy Analysis. Anal. Chem. 2009;81(12):4946–4954. doi: 10.1021/ac900688n. [DOI] [PubMed] [Google Scholar]