

Abstract
G protein-coupled receptors (GPCRs) are of great importance to the pharmaceutical industry as they are involved in many human diseases and include well-validated targets for therapeutic intervention. Discovery of lead compounds, including small synthetic molecules, that specifically inhibit the receptor's function, is an important initial step in drug development and relies on sensitive, specific, and robust cell-based assays. Here, we describe a kinetic cellular assay with a fluorescent readout primarily designed to identify receptor-specific antagonists that inhibit the intracellular Ca2+ release evoked upon the activation of the CXC chemokine receptor 4 (CXCR4) by its endogenous ligand, the CXC chemokine ligand 12 (CXCL12). A key advantage of this method is that it also enables screening of compounds endowed with intrinsic agonistic properties (i.e., compounds eliciting an increase in intracellular Ca2+ concentration in the absence of CXCL12) or compounds modulating the receptor's function via interaction with allosteric binding sites (i.e., positive and negative allosteric modulators (PAMs and NAMs, respectively)). On the down side, autofluorescent compounds might interfere with the assay's readout, thereby hampering reliable data interpretation. Most likely this assay can be implemented, with minimal adaptations, as a generic drug discovery assay for many other GPCRs of which the activation leads to a release of intracellular Ca2+.
Keywords: Medicine, Issue 132, G protein-coupled receptor, chemokine, CXC chemokine receptor 4, fluorescence, cell-based assay, Ca2+ mobilization, agonist, antagonist, drug discovery, allosteric modulator, small molecules
Introduction
GPCRs are an important superfamily of cell surface proteins that activate signal transduction cascades upon extracellular ligand binding. They can be activated by a large variety of stimuli including peptides, protein hormones, biogenic amines, and lipids, which results in the initiation of diverse intracellular signaling pathways and eventually biological responses1,2. Furthermore, GPCRs are involved in many, if not all, developmental and physiological processes and many human diseases are associated with dysfunctional GPCR signaling or receptor overexpression. GPCRs are therefore amongst the most validated pharmacological targets in medicine1,3.
Typically, a GPCR drug discovery workflow starts with cellular screening assays enabling the identification of compounds such as small molecules, monoclonal antibodies, and peptides that can modulate the activity of a particular GPCR. In GPCR drug discovery many different types of assays exist to search for such compounds, most of which are compatible with mid- to high-throughput screening campaigns. The most used assays include receptor binding experiments, fluorescence or luminescence based assays detecting fluctuations in the level of so-called secondary messengers (e.g., Ca2+, cyclic adenosine monophosphate (AMP)), phenotypic screening assays, and β-arrestin recruitment assays4. The choice for a particular type of assay may depend on multiple factors, but is also determined by prior knowledge concerning the signaling properties of a given GPCR. Agonist binding to a GPCR induces a conformational change catalyzing the exchange of guanidine diphosphate (GDP) for guanidine triphosphate (GTP) on the α-subunit of heterotrimeric G proteins. Subsequently the Gα-GTP subunit dissociates from the Gβγ subunit and both subunits will initiate further signaling pathways. Hydrolysis of the GTP-molecule and subsequent re-association of the Gα-GDP and Gβγ subunits will restore the G protein into its inactive conformation5,6. Based on sequence similarity of the Gα subunit different types of G proteins are defined (Gs, Gi, Gq, G12/13)7. Signaling via the Gα subunit gives rise to several typical responses such as the increase (via Gs) or decrease (via Gi) of cyclic AMP production and intracellular Ca2+ mobilization (via Gq)5,7. Gβγ subunits are also able to induce intracellular effector pathways. For instance, upon activation of Gi-coupled GPCRs, Gβγ can directly stimulate phospholipase C (PLC-β) to produce inositol triphosphate (IP3) that triggers the release of Ca2+ from intracellular stores7. Following receptor activation, GPCRs are phosphorylated by GPCR kinases (GRKs) which promotes interaction with β-arrestins. This process terminates G protein signaling and leads to receptor desensitization and eventually internalization. β-arrestins are also able to form multi-molecular complexes that can trigger other signaling pathways independent of G protein signaling8.
Within the subfamily of chemokine receptors, the Gi-coupled CXCR4 is a GPCR that has raised much interest as a promising target for drug discovery. Given its established role as a major co-receptor for human immunodeficiency virus 1 (HIV-1) viral entry and infection, compounds targeting CXCR4 were initially developed as anti-HIV drug candidates9. More recently, a growing body of evidence has pointed to an important role for CXCR4 in tumorigenesis and cancer metastasis making it a well-validated therapeutic target in oncology as well10. CXCR4 is highly expressed in more than twenty types of human cancer and controls tumor cell survival, proliferation, and migration as well as tumor-related angiogenesis10. CXCR4 antagonists of different chemical classes have previously been described11,12, but only the small molecule AMD3100 is currently approved for use in the clinic as a stem cell mobilization agent used during treatment of lymphoma and myeloma patients13,14. Clinical trials are ongoing to evaluate the safety and efficacy of several other CXCR4 antagonists in different human diseases, but with a strong focus on oncology12. Given the many potential applications for CXCR4 antagonists, the search for novel compounds with improved pharmacokinetic properties, improved bioavailability, or potentially less side effects is warranted.
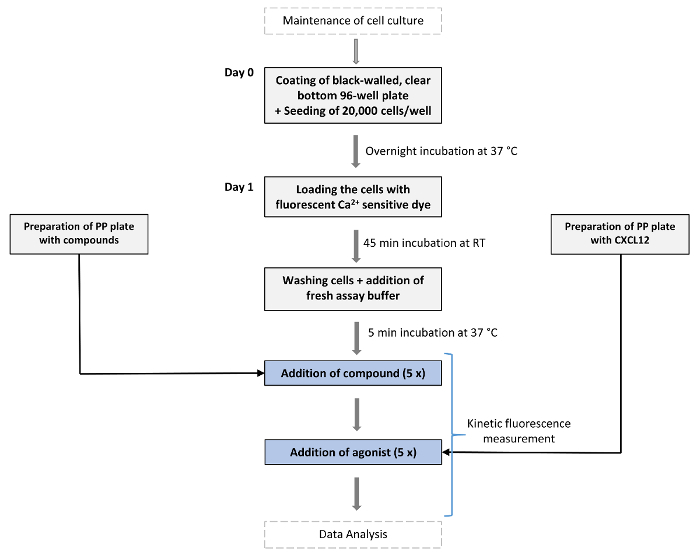
Herein, a kinetic fluorescence-based cellular assay primarily used to screen for compounds capable of inhibiting CXCR4 is described. The fluorescent measurement of this method is based on the transient increase of the intracellular Ca2+ concentration evoked upon CXCR4 activation by its endogenous agonist, the chemokine ligand CXCL12 (formerly known as stromal cell derived factor 1α (SDF1-α)), and the potential inhibition of this CXCL12-induced Ca2+ response by particular compounds. In this assay, U87 human glioblastoma cells stably expressing the human CXCR4 receptor are used. At the same time, these cells lack endogenous expression of CXCR7, a related chemokine receptor that also binds CXCL1215,16,17. CXCR7 has previously also been shown to be capable of forming heterodimers with CXCR4, thereby modulating the signaling properties of this latter receptor18. Fluctuations in the level of intracellular Ca2+ mediated by CXCR4 are monitored by loading the CXCR4+ cells with fluo-2 acetoxymethyl (AM) ester, a cell-permeable high affinity fluorescent Ca2+-binding dye. Fluo-2 AM is a single wavelength fluorescent molecule that can be excited at 490 nm while its emission fluorescence is measured at 520 nm. This emission fluorescence increases upon Ca2+ binding, with a large dynamic range between the Ca2+-bound and unbound state. The increase in fluorescent signal is transient, occurring within a time interval of a few minutes, and will decay afterwards. The peak height of the fluorescent emission further correlates with the level of receptor activation. The assay itself is performed using a fluorescence microplate reader equipped with an Intensified CCD (ICCD) camera that possesses an integrated pipetting system that allows standardization of the pipetting steps in the assay (see Table of Materials). In addition, the simultaneous measurement of the fluorescent signal in all wells of a microplate is another key advantage of the fluorescence reader that is used. During the first part of the assay the compounds under investigation (e.g., a panel of small molecules at a fixed concentration or in a dilution series) are added to the fluo-2 AM loaded CXCR4+ U87 cells followed by a ~ 10 min incubation period during which the potential agonistic effect of the compounds is continuously measured in real time. Then, the endogenous agonist (i.e., CXCL12) is added to the cells to evoke a CXCR4-mediated transient increase in the level of intracellular Ca2+. During this part of the assay the potential antagonistic activity of the tested compounds can be evaluated. A schematic overview of the assay's general workflow is presented in Figure 1.
Although this Ca2+ mobilization assay has primarily been used to identify and determine the inhibitory potency of competitive CXCR4 antagonists (i.e., compounds that prevent the endogenous agonist to bind and stimulate the receptor), it also can identify receptor agonists and, in addition, compounds that exert their function by binding at allosteric sites (i.e., sites that topographically differ from the orthosteric binding site occupied by the endogenous agonist). Examples of the latter category of compounds include allosteric agonists and PAMs and NAMs19,20. Whereas receptor-specific antagonists as well as NAMs would inhibit the CXCL12-induced Ca2+ response, PAMs would enhance this response (see also Discussion section). Although the assay described herein specifically targets CXCR4, it is anticipated that this method can be applied to other GPCRs with minimal optimization effort, at least if they signal via the release of intracellular Ca2+.
Protocol
NOTE: All steps described under sections 1 and 2 are carried out under sterile conditions in a laminar flow cabinet.
1. Maintenance of U87.CD4.hCXCR4 Cells
- Grow the cells in T75 culture flasks at 37 °C and 5% CO2 in a humidified incubator. NOTE: The in vitro cell line used in this protocol is a U87 glioblastoma cell line stably expressing cluster of differentiation 4 (CD4) and human CXCR4 and has been previously described17. Cell surface expression of CD4 and CXCR4 is continuously monitored by flow cytometry and expression levels remain constant over time (~ 100% CD4+ and ~ 100% CXCR4+ cells). A detailed description of the generation of the used cell line and of the flow cytometry procedure to investigate receptor expression levels is not within the scope of this protocol.
- Subculture cells at 80-85% confluency. Allow all reagents to reach room temperature (RT) before cell culturing.
- Remove the conditioned culture medium from the cells and wash the cell monolayer once with 5 mL phosphate buffered saline (PBS).
- Add 3 mL of 0.25% trypsin-EDTA and distribute it evenly over the cell monolayer. Then remove the excess of trypsin-EDTA and incubate up to 5 min at 37 °C until cells start to detach.
- Add 10 mL of fresh complete growth medium (Dulbecco's Modified Eagle's Medium (DMEM) + 10% Fetal Bovine Serum (FBS) + 0.01 M HEPES + appropriate selection agents). Resuspend the cells by gently pipetting up and down. Transfer the cell suspension to a sterile 50 mL tube.
- Count the number of viable cells using a method of choice. In the case of U87 glioblastoma cells, viability generally reaches ~ 100%. NOTE: Cell numbers can be determined in several ways. We routinely use an automated cell analysis system based on trypan blue staining (see Table of Materials) according to its standard handling procedures, but other manners should work equally well.
- Add 2 x 106 or 3 x 106 (viable) cells to a final volume of 25 mL fresh growth medium in a T75 culture flask and incubate at 37 °C and 5% CO2. NOTE: If 3 x 106 cells are used the cells will reach 80-90% confluency after 2 days. If 2 x 106 cells are used, they will reach the same confluency after 3 days. The growth rate of the cells should first be determined empirically if other cell lines are used.
2. Seeding of the Cells for the Ca2+ Mobilization Assay
- At the day of cell passaging (day 0), coat black-walled polystyrene 96-well plates with clear bottom (see Table of Materials) with a 0.1% gelatin solution to facilitate cell attachment. NOTE: Coating of the 96-well plates with gelatin might be omitted if pre-coated plates, which are commercially available (see Table of Materials), are used. It is recommended to evaluate the use of pre-coated plates instead of manual coating for each cell line under investigation before continuing with the protocol.
- Prepare gelatin solution by adding 1 g of gelatin to 100 mL PBS to obtain a 1% solution. Dilute by 10x with PBS before further use. To improve the solubility of gelatin, heat the solution to 37 °C.
- Add 100 µL of 0.1% gelatin solution per well of the 96-well plate using a multichannel pipette. Incubate for 2 h at RT.
- In the meantime, prepare the cells for seeding in the coated 96-well plate.
- Detach the cells from the culture flask, resuspend them in fresh growth medium, and count them using trypan blue staining method.
- Spin down the cells in a 50 mL tube for 5 min at 400 x g at RT.
- Resuspend the cells in fresh growth medium (DMEM + 10% FBS + 1% HEPES, no antibiotics) to obtain a cell density of 0.1 x 106 (viable) cells/mL.
Remove the gelatin solution from the black-walled plates with clear bottom by flipping over the plate and drying it on a tissue. Add 200 µL/well PBS to remove excess gelatin and flip over the plate again.
Dispense 200 µL of the cell suspension (corresponding to 0.2 x 105 cells) per well in the gelatin-coated 96-well plate (from now one this plate will be further referred to as the "measurement plate").
Incubate the plate overnight at 37 °C and 5% CO2.
3. Loading of the Cells with a Fluorescent Ca2+ -sensitive Dye
- At day 1 perform the actual Ca2+ mobilization assay, starting with loading of the seeded cells with the fluorescent Ca2+-binding dye fluo-2 AM.
- Prepare assay buffer by adding 40 mL HEPES (1 M) to 200 mL Hank's Balanced Salt Solution (HBSS, 10x, no phenol red, no sodium bicarbonate). Add ultrapure water to obtain a final volume of 2 L and add 4 g Bovine Serum Albumin (BSA). Dissolve the BSA via magnetic stirring. Adjust the pH to 7.4 (with NaOH) and filter the solution. NOTE: During all following steps the same buffer, referred to as "assay buffer", is used.
- Prepare a stock solution (4 mM in dimethyl sulfoxide (DMSO)) of the fluorescent Ca2+-sensitive dye fluo-2 AM. Avoid excessive exposure to light. Dissolve 1 mg fluo-2 AM (molecular weight: 1,061 g/mol) in 235.6 µL DMSO.
- Prepare a working solution of fluo-2 AM. For one 96-well assay plate mix 12.5 µL of fluo-2 AM stock solution (4 mM) and 12.5 µL nonionic surfactant polyol (e.g., pluronic f-127) solution (20% weight/volume in DMSO, see Table of Materials). Add 22 µL of this mixture to 11 mL assay buffer in a 15 mL tube. The final concentration of fluo-2 AM is 4 µM. NOTE: The nonionic surfactant is used as a dispersion agent to improve the aqueous solubility of fluo-2 AM and, in consequence, to enhance cell loading. To solubilize surfactant in DMSO, heat the sample at 37 °C and mix regularly to obtain a homogenous solution.
- Remove the growth medium from the previously seeded cells in the 96-well measurement plate by flipping over the plate and drying it on a paper towel.
- Add 100 µL of loading dye solution per well using a multichannel pipette and incubate for 45 min at RT in the dark.
4. Preparation of 96-well Polypropylene Plates Containing the Chemokine Ligand CXCL12 or the Compounds under Investigation
- Obtain round bottom polypropylene (PP) 96-well plates (see Table of Materials).
- Allow the stock solution of CXCL12 (1 mg/mL) and the stock solution of the compounds under investigation to equilibrate at RT. NOTE: The stock solution of CXCL12 is prepared in ultrapure water supplemented with 0.01% Tween20 and stored at -20 °C as single-use aliquots.
- Prepare a 5x concentrated solution of CXCL12 in assay buffer (250 ng/mL which equals 31.25 nM) and a 5x concentrated dilution of each compound (in assay buffer).
- Dispense the stock solutions into the appropriate 96-well plate according to a predefined plate layout. Dispense 75 µL/well in the plate containing CXCL12 (the "chemokine plate"), dispense 50 µL/well in the plate containing the compounds (the "compound plate"). NOTE: Both compounds and CXCL12 will finally become diluted 5x upon dispensing in the measurement plate. It is also important to include negative control wells containing only assay buffer in both the compound plate as well as the chemokine plate. Also positive control wells need to be included (i.e., wells with CXCL12 in the chemokine plate, but with assay buffer in the corresponding wells of the compound plate).
5. Protocol Settings on the Fluorescence Microplate Reader
NOTE: The fluorescence microplate reader used in this protocol is referred to in the Table of Materials.
Switch on the cooler unit of the fluorescence plate reader first, and then switch on the fluorescence reader itself. Let initiate for a few minutes and open the system's software.
- Create the assay's protocol using the drag-and-drop menu that includes the following steps:
- In the "Settings" box, select 'Read_Mode' with excitation wavelength: 470-495 nM; emission wavelength: 515-575 nM. NOTE: The selected wavelengths are appropriate for use with the Ca2+-sensitive dye fluo2.
- Include the "Mix with TF" (transfer fluid) box for automatic mixing of the compounds in the compound plate, that will be put at the source 2 position of the device (see step 6.4). Select 15 µL of solution in each well to be automatically aspirated and mixed three times. Adjust the height of the pipette tips at 20 µL below the liquid surface. Set the speed of aspiration and dispensing at 50 µL/s. NOTE: The height of the pipette tips refers to the volume that is left underneath the pipette tips. For instance, if the wells of a compound plate contain 50 µL, a height of 30 µL corresponds to 20 µL below the liquid surface. The volume of compound solution taken to mix, the speed of mixing, the position of the pipette tips during mixing, and the number of mixing cycles can all be adjusted using the software.
- In the "Transfer Fluid" box, define that 20 µL of each well from the compound plate will be transferred into the measurement plate. Position the pipette tips at 20 µL below the liquid surface i.e., at height 30 µL during aspiration of the compounds and at height 60 µL during dispensing in the measurement plate. Set the speed of aspiration at 50 µL/s and the speed of dispensing at 25 µL/s. NOTE: The compound plate contains 50 µL of solution per well (see step 4.1.3), thus a height of 30 µL corresponds to a position 20 µL below the liquid surface. The measurement plate will contain 80 µL of assay buffer per well (see step 6.3), thus a height of 60 µL corresponds to a similar position of the tips.
- Select the "Read with TF" button. During the first interval, select 60 reads (fluorescent measurements) with a read interval time of 1 s. Define that 10 reads are recorded before dispensing of the compounds into the measurement plate, and 50 reads afterwards. Define the second interval with a read interval time of 30 s and 18 reads. NOTE: During this step fluorescence will be measured kinetically (at the defined time intervals) for ~ 10 min in total.
- Include the "Wash Tips" button in the protocol three times. Within each washing step, select the fluid type: fluid A (ultrapure water) or fluid B (70% ethanol), the number of wash cycles (1), the pump speed (Fast), and the number of strokes (5) during each wash step. NOTE: During the first and last wash step fluid A is used, during the second wash step fluid B is used.
- Include the "Pause Pipettor" function. Set the pipettor to pause for 300 s. NOTE: By including this step in the protocol, the pipettor head, which is integrated in the fluorescence plate reader device, will pause for 5 min before continuing the protocol.
- Include the "Mix with TF" box to allow automatic mixing of the CXCL12 solution in the chemokine plate, that will be positioned at the source 3 position of the device (see step 6.4). Select 15 µL of solution in each well to be automatically aspirated and mixed three times. During aspiration and mixing, position the pipette tips 20 µL below the liquid surface. The speed of aspiration and dispensing is set at 50 µL/s. NOTE: Similar as for step 5.2.2, these parameters can be adjusted.
- In the subsequent "Transfer Fluid" box, define that 25 µL from each well of the chemokine plate will be transferred into the measurement plate. Position tips 20 µL below the liquid surface i.e., at height 55 µL during aspiration from the chemokine plate that contains 75 µL/well (see step 4.1.3), and at height 80 µL during dispensing in the measurement plate.Set the speed of aspiration at 50 µL/s and the speed of dispensing at 25 µL/s.
- Include the "Read with TF" button. During the first interval, select 145 reads with a read interval time of 1 s. Define that 5 reads are recorded before dispensing CXCL12 into the measurement plate, and 140 reads afterwards. Define the second interval with a read interval time of 6 s and select 20 reads. NOTE: Taken together, throughout the entire protocol 243 reads (fluorescent measurements) are recorded: 78 at step 5.2.4 and 165 during step 5.2.9.
- Similar to step 5.2.5, include the "Wash tips" button three times in the protocol. NOTE: Including this step at the end of the protocol allows that the tips can be re-used to perform another assay without needing to change the tips.
Set the temperature of the device at 37 °C by clicking the "Set stage temperature" button and selecting 37 °C.
6. Running the Fluorescence Assay
After the measurement plate has incubated with loading buffer for 45 min (see step 3.1.5), remove the buffer by flipping over the plate and dry it on a tissue.
Wash the seeded cells by adding 150 µL/well of assay buffer and incubate for 2 min.
Remove the buffer again by flipping over the plate. Add 80 µL/well of assay buffer with a multichannel pipette.
Put all the plates into the device at their appropriate position: the compound plate at the source 2 position, the chemokine plate at the source 3 position, and the measurement plate at the read position. Put a box of black tips at the source 1 tips position. Shut the device's door and incubate for 5 min before continuing the protocol.
Select the "Protocol signal test" button to determine the background relative light units (RLUs) and fluorescence variance over the plate. A new window will pop up. Select "Test signal". NOTE: During this step, background RLUs are determined by the ICCD camera, which is integrated in the optics compartment of the fluorescence reader device. These RLUs result from the excitation of the Ca2+-sensitive dye (fluo-2) by light emitting diodes (LEDs) (LED output wavelength 470-495 nm). Values of 8,000-10,000 RLUs are well suited for this application. RLUs can, if necessary, be adapted by changing the excitation intensity (Select Exc. Intensity), or the camera gate (Select Gate Open). The variance over the plate should ideally be less than 7.5%.
Select "Update" if background values of 8,000-10,000 RLUs are obtained. Save the main protocol by clicking the Save button. NOTE: By doing so, the settings from the protocol signal test will be carried over to the main protocol.
Run the assay by pushing the "RUN" button.
7. Data Analysis and Quality
- After the assay has finished, open the "Analysis" box in the system's software.
- Go to "configure corrections" and choose "response over base line". Define base line 1 to start at measurement 1 and end at measurement 5; the mean fluorescence will be calculated in each well between measurement 1 and 5. NOTE: All further measurements from a particular well are divided by this well-specific base line 1.
- Define base line 2 to start at measurement 78 until measurement 83 (CXCL12 is dispensed after measurement 83; again the mean fluorescence is calculated in each well between measurement 78 and 83 and this correction factor is applied to all measurements following measurement 83).
- Activate the tick box "show as percentage" and "subtract background".
- Go to "Configure Kinetic Reduction". To evaluate the inhibitory effect of compounds on the CXCL12-induced Ca2+ response, choose Max-Min starting from measurement 84 (i.e., the first measurement after which CXCL12 is dispensed) to 243 (i.e., the final measurement). NOTE: By choosing Max-Min the difference between the minimum and maximum response over baseline between measurement 84 and measurement 243 is reported. If the Max-Min is chosen starting between measurement 11 and measurement 78, the difference between the minimum and maximum response over baseline relative to baseline 1 is reported. This value can be used to analyze the potential agonistic activity of the compounds, see Discussion).
- The data will be visualized in the system's software. If needed, export the raw data for additional analysis and visualization in other common analysis software packages.
Representative Results
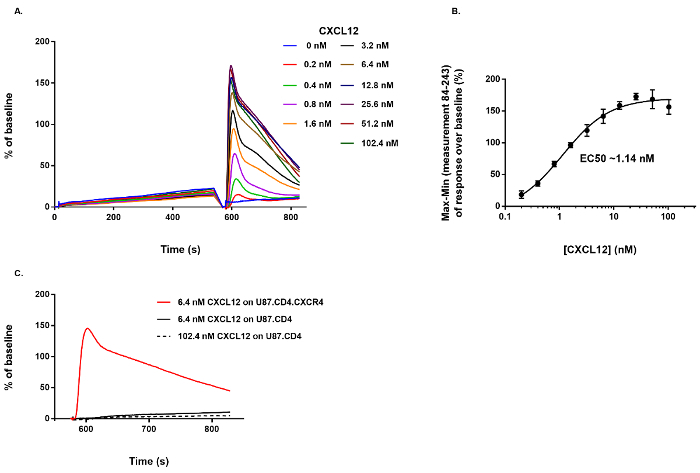
The effect of CXCL12 stimulation on the intracellular Ca2+ mobilization in U87.CD4.CXCR4+ and U87.CD4 cells was evaluated with the Ca2+ mobilization assay. Instead of 20 µL of test compound that would normally be added during the first pipetting step of the protocol (Figure 1), assay buffer was added to the fluo-2 AM loaded U87.CD4.CXCR4+ cells in the measurement plate. During the second dispensing step, different concentrations of CXCL12 (0.2-102.4 nM, final concentration) were dispensed in the measurement plate. A dose-dependent increase in fluorescence, correlating with a dose-dependent increase of the release of Ca2+, is demonstrated (Figure 2A). Here, the negative control sample represents those wells in which at both additions only assay buffer was added to the measurement plate (i.e., 0 nM CXCL12), resulting in the absence of a response (Figure 2A). Increasing amounts of CXCL12 induce increased levels of fluorescence that decay over time (Figure 2A). From these data, a dose response curve was generated based on the "Max-Min response over baseline" between measurement 84 (i.e., the first measurement after CXCL12 addition) and measurement 243 (i.e., the final measurement in the protocol). Using nonlinear regression, the EC50 value (i.e., the concentration needed to evoke the half maximal response) was determined and corresponds to 1.14 nM (Figure 2B). No fluorescent Ca2+-related response was evoked by CXCL12, even at high concentration (102.4 nM), when U87.CD4 cells lacking functional CXCR4 expression were used. This demonstrates the receptor-specificity of the measurement evoked by CXCL12 (Figure 2C).
A key application for this cell-based assay is the identification of small molecules specifically targeting CXCR4 and capable of inhibiting the CXCL12-induced Ca2+ mobilization. If active compounds ("hits") are identified, they can be further characterized by testing serial dilutions of the compound and analyzing the inhibitory potency in more detail. As an example, the effect of the bicyclam AMD3100, a well-established small molecule CXCR4-specific receptor antagonist with potent anti-HIV-1 activity21,22, is illustrated in Figure 3. A concentration series of AMD3100 (n = 4, 3 µM down to 1.37 nM final concentration in a 1/3 dilution series) was dispensed on U87.CD4.CXCR4+ cells during the first step of the protocol. The compound was then allowed to incubate with the cells for ~ 10 min during which the fluorescence signal was recorded continuously. Then 6.4 nM of CXCL12 (50 ng/mL, final concentration) was added to all wells of the cell plate simultaneously to induce the CXCR4-mediated Ca2+ mobilization. Dose-dependent inhibition of this response by the different dilutions of AMD3100 is shown in Figure 3A. In this case, only the part of the graph following addition of CXCL12 is shown. As a negative control (blue line) only buffer was applied in the assay (no pre-incubation with AMD3100, no CXCL12 added to the wells), resulting in the expected lack of response. The positive control (red line) corresponds to the samples in which 6.4 nM CXCL12 was added to wells without prior incubation of AMD3100 (Figure 3A). Based on these defined negative and positive controls the Z' value in this assay typically is between 0.5 and 1. In order to determine the inhibitory potency of AMD3100 a dose-response curve was generated by non-linear regression resulting into a calculated IC50 value of 770.8 nM (Figure 3B). To further illustrate the behavior of a non-active compound, maraviroc was included in this experiment. Maraviroc is a small molecule specifically inhibiting the CC chemokine receptor 5 (CCR5) thereby blocking (CCR5)-tropic HIV-1 infection23 . Even at high concentration (10 µM final concentration) no inhibitory effect of this compound was observed on the CXCR4-mediated Ca2+ response (Figure 3A).
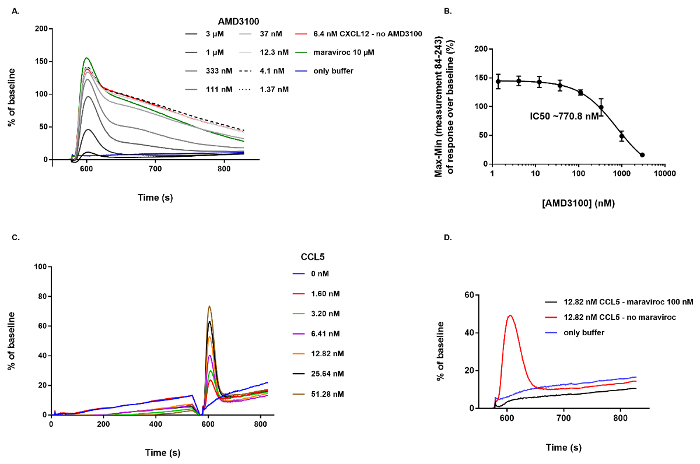
Finally, to demonstrate that this Ca2+ mobilization assay can also be applied to study other GPCRs, a serial dilution of the CC chemokine ligand 5 (CCL5), the endogenous ligand for CCR5, was added to cells expressing CCR5 (U87.CD4.CCR5+) using exactly the same experimental conditions and hardware settings. A dose-dependent fluorescent Ca2+ response is demonstrated after addition of a serial dilution of CCL5 (Figure 3C). In addition, and in contrast to its effect on CXCR4, pre-incubation with 100 nM (final concentration) of maraviroc strongly inhibited this response (Figure 3D).
Figure 1: Schematic overview of the assay's workflow. On day 0 cells expressing the GPCR of interest (in this case CXCR4) are seeded in black-walled 96-well plates with clear bottom and are grown overnight at 37 °C and 5% CO2. At day 1 the fluorescence-based Ca2+ assay is performed. Cells are first loaded with a fluorescent Ca2+-sensitive dye (fluo-2 AM) and incubated for 45 min at RT in the dark. A "chemokine plate" and "compound plate" are prepared (PP = Polypropylene). After incubation with loading dye, seeded cells are washed once with assay buffer (150 µL/well) after which 80 µL/well of assay buffer is added. All plates are then incubated for 5 min in the device at 37 °C before starting the assay. Then, compounds of interest (e.g., small molecules) are dispensed at the desired concentration into the wells of the measurement plate and allowed to incubate for ~ 10 min while fluorescence is continuously measured. Next, a fixed concentration of the endogenous agonist of the GPCR (here CXCL12) is added to evoke Ca2+ release and fluorescence is further recorded over time. Please click here to view a larger version of this figure.
Figure 2: Agonist activity of CXCL12 on CXCR4+ cells and specificity of the detected response. (A) The dose-dependent effect of CXCL12 on the Ca2+ mobilization in U87.CD4.CXCR4+ cells. (B) Based on the Max-Min response over baseline between measurement 84 and 243 a dose-response curve was generated and the EC50 value calculated (n = 4, mean ± SD). (C) No response was induced by CXCL12 when it was dispensed on U87.CD4 cells lacking CXCR4. Please click here to view a larger version of this figure.
Figure 3: Illustration of the effect of a CXCR4 antagonist (AMD3100) and a non-active compound (maraviroc) on CXCR4+ cells and activation of CCR5 by its endogenous agonist, CCL5. (A) Dose-dependent inhibitory effect of AMD3100 on the Ca2+ mobilization evoked by adding 6.4 nM CXCL12 to U87.CD4.CXCR4+ cells. Maraviroc (at 10 µM final concentration) showed no inhibitory effect on the CXCL12-induced Ca2+ response. (B) Based on the Max-Min response over baseline between measurement 84 and 243, an inhibitory dose-response curve was generated and the IC50 value was calculated to be 770.8 nM (n = 4, mean ± SD). (C) Dose-dependent activation of CCR5 by its endogenous agonist, CCL5. (D) Inhibition of the CCR5-mediated Ca2+ response by pre-incubation of the cells with 100 nM of maraviroc. Please click here to view a larger version of this figure.
Discussion
The Ca2+-mobilization assay described herein has previously been shown to be a valuable tool to identify and characterize receptor antagonists targeting CXCR417. It is, however, anticipated that this method can be more generally applied to a large group of other GPCRs that trigger a cytosolic Ca2+ release upon their activation, as illustrated for the related chemokine receptor CCR5. Whereas in the case of CCR5 exactly the same experimental conditions could be applied, several steps in the protocol (e.g., the cell number at plating, loading of the cells with a fluorescent dye) will need to be re-optimized before transferring the assay to other GPCRs in order to obtain a favorable signal-to-noise ratio. An important issue here is also the selection of a suitable in vitro cellular host allowing high-level expression of the GPCR of interest and functional coupling to the Ca2+ pathway. In case of CXCR4, human glioblastoma U87 cells were chosen because of: (1) the lack of endogenous expression of CXCR7 (a related chemokine receptor that occupies the same agonist as CXCR4), (2) their ability to couple CXCR4 activation to the Ca2+-signaling pathway, (3) the favorable dynamic range of the fluorescent response obtained after loading of the cells with the Ca2+-sensitive dye, and (4) their excellent adherence to the assay plates minimizing cell disturbance throughout the procedure. All of these parameters might need to be experimentally determined for each novel GPCR of interest and each cellular expression system.
When setting up a similar assay for another GPCR, several alternative steps or reagents in the protocol can be considered. For instance, other Ca2+-sensitive fluorophores (e.g., fluo-4, fluo-3) might be used as an alternative for the fluorophore (fluo-2) used in this protocol. In case of loosely adhering cells, washing of the cells might be omitted by introducing no-wash dyes to minimize cell dislodging24. In addition, excluding the washing step from the protocol would further increase the assay's throughput. Although this assay might also be performed with cells showing endogenous expression of CXCR4, for instance including several types of human cancer cell lines10,25, it should be noted that with stably transfected cell lines high GPCR expression levels that remain stable over time can be obtained. This will result in more pronounced assay measurements, a larger window for reliable data interpretation, and consistent assay performance over longer periods. When using cells endogenously expressing CXCR4 it will be much more difficult to achieve this result.
A major drawback of the assay is that it relies on a fluorescence measurement. Hence, autofluorescent compounds can interfere with the assay's measurement, which excludes them from analysis due to unreliable data interpretation. Also, this Ca2+ mobilization assay is ideally performed using a cellular screening system equipped with an integrated pipetting system that allows standardized mixing and dispension of compounds and receptor agonist into the measurement plate in all wells simultaneously. The assay could, however, be adapted for use with other, less expensive fluorescence microplate readers with kinetic measurement capabilities. The addition of compounds and agonist would then need to be performed manually using multichannel pipettes, which will increase hands-on-time and lowers the assay's throughput. Furthermore, obtaining a similar number of fluorescence measurements within a given time interval of a kinetic assay would be difficult to achieve as many microplate readers measure signals well-by-well whereas high-end screening systems measure all wells simultaneously.
Receptor antagonists preventing binding to the receptor and subsequent activation by the endogenous agonist (CXCL12) currently form the main category of compounds specifically targeting CXCR411,12. AMD3100, the small molecule that was used to illustrate the performance of the Ca2+ mobilization assay, is one of the most prominent examples of CXCR4 antagonists26. Besides receptor antagonists, molecules that regulate GPCR signaling differently raise general interest as well. These molecules include inverse agonists, as well as GPCR PAMs and NAMs19,20,27. An interesting feature of the assay described in this manuscript is that it not only can identify receptor antagonists, but also agonists and allosteric modulators. However, some minor adaptations to the assay and limitations need to be taken into account.
PAMs and NAMs bind to receptor sites topographically distinct from the orthosteric binding site occupied by the receptor's endogenous ligand. Whereas PAMs enhance the potency and/or efficacy of the receptor's endogenous ligand(s), NAMs inhibit the potency and/or efficacy of the endogenous ligand(s)19,20. PAMs have no intrinsic agonist activity. They are only active in the presence of the endogenous agonist, unlike so-called allosteric agonists. Because allosteric GPCR modulators would evoke less side effects than classical receptor antagonists when applied in clinical settings28,29, they are valuable pharmacological tools. In our assay, allosteric agonists would evoke a transient increase of the fluorescence signal immediately upon addition to the CXCR4+ cells. Receptor specificity of this signal should then be determined by testing the same compound with the same assay, but on cells lacking CXCR4. When specifically screening for PAMs, a lower concentration of endogenous agonist should be used in the assay to induce a fluorescent Ca2+ response. Although this lower amount of agonist will generate a smaller fluorescent signal, it leaves a larger window to detect additional receptor stimulation. Typically, an agonist concentration corresponding to the EC10-EC30 value of receptor stimulation is chosen30,31. Addition of a PAM during the first part of the assay would not result in an increased fluorescent measurement. However, the fluorescent Ca2+ signal after subsequent stimulation of the receptor with its endogenous agonist would increase. Similarly, a NAM does not alter the fluorescent measurement before agonist addition, but does inhibit the receptor's response after addition of the agonist. Hence, the activity profile of a receptor antagonist and a NAM will look similar. They both will result in the inhibition of the fluorescent response after agonist addition. Therefore, further experimental studies are required to discriminate between an antagonist and a NAM. Here, receptor binding studies whereby unlabeled compounds compete with a fixed amount of labeled CXCL12 for binding at CXCR417,32 can be a valuable strategy to discriminate between an antagonist, which would prevent the labeled agonist from binding to the receptor, and a NAM that would not as it would not share the same receptor binding site.
Ligand independent (or basal or constitutive) activity of GPCRs has been observed for numerous GPCRs33 although the level of basal activity is generally rather low when GPCRs are expressed recombinantly27. Basal GPCR activity can be enhanced by naturally occurring mutations33 and a point mutation leading to increased basal CXCR4 activity has been described earlier34. By coupling this constitutively active CXCR4 mutant to the pheromone response pathway in yeast and by [35S]GTPγS binding experiments, it was shown that T140, a peptide-based CXCR4 antagonist with potent anti-HIV activity17,35 significantly decreased this basal activity and thus was shown to behave as an inverse agonist34. Of note, in a Fura-2 fluorescence-based Ca2+ mobilization assay the same authors observed a small transient decrease in Ca2+-related fluorescence following addition of T140 to cells expressing mutant CXCR4, suggesting a decrease of basal receptor activity34. However, when analyzing an additional panel of cyclic pentapeptide-based CXCR4 inverse agonists designed from T140, detection of reduced basal activity with a Ca2+ mobilization assay was less straightforward36. When the effect of T140 on cells expressing wild type CXCR4 was analyzed using the Ca2+ mobilization assay as described in this manuscript, no change in basal fluorescence signal was observed17. Furthermore, fluorescence fluctuations are fast and transient and increases in basal Ca2+ are, for instance, not observed in cells expressing constitutively active Gαq-coupled receptors4. Taken together, the effect of inverse agonists would be very difficult, if not impossible to identify and evaluate reliably with our assay.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank Eric Fonteyn and Geert Schoofs for excellent technical assistance. This work has been supported by the KU Leuven (grant no. PF/10/018), the Fonds voor Wetenschappelijk Onderzoek (FWO, grant no. G.485.08), and the Fondation Dormeur Vaduz.
References
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- Zhang R, Xie X. Tools for GPCR drug discovery. Acta Pharmacol Sin. 2012;33:372–384. doi: 10.1038/aps.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G, Kostenis E. Heterotrimeric G-proteins: a short history. Br J Pharmacol. 2006;147(Suppl 1):S46–S55. doi: 10.1038/sj.bjp.0706405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja N. Signaling through G protein coupled receptors. Plant Signal Behav. 2009;4:942–947. doi: 10.4161/psb.4.10.9530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- Smith JS, Rajagopal S. The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. J Biol Chem. 2016;291:8969–8977. doi: 10.1074/jbc.R115.713313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. Discovery of non-peptide small molecular CXCR4 antagonists as anti-HIV agents: Recent advances and future opportunities. Eur J Med Chem. 2016;114:65–78. doi: 10.1016/j.ejmech.2016.02.051. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics. 2013;3:47–75. doi: 10.7150/thno.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou LK, et al. Harnessing CXCR4 antagonists in stem cell mobilization, HIV infection, ischemic diseases, and oncology. Med Res Rev. 2017. [DOI] [PubMed]
- Keating GM. Plerixafor: a review of its use in stem-cell mobilization in patients with lymphoma or multiple myeloma. Drugs. 2011;71:1623–1647. doi: 10.2165/11206040-000000000-00000. [DOI] [PubMed] [Google Scholar]
- DiPersio JF, et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin's lymphoma. J Clin Oncol. 2009;27:4767–4773. doi: 10.1200/JCO.2008.20.7209. [DOI] [PubMed] [Google Scholar]
- Balabanian K, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–35766. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- Burns JM, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hout A, D'Huys T, Oeyen M, Schols D, Van Loy T. Comparison of cell-based assays for the identification and evaluation of competitive CXCR4 inhibitors. PLoS One. 2017;12:e0176057. doi: 10.1371/journal.pone.0176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113:6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- Burford NT, Watson J, Bertekap R, Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem Pharmacol. 2011;81:691–702. doi: 10.1016/j.bcp.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Hendrix CW, et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004;37:1253–1262. doi: 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]
- Schols D, Este JA, Henson G, De Clercq E. Bicyclams, a class of potent anti-HIV agents, are targeted at the HIV coreceptor fusin/CXCR-4. Antiviral Res. 1997;35:147–156. doi: 10.1016/s0166-3542(97)00025-9. [DOI] [PubMed] [Google Scholar]
- Perry CM. Maraviroc: a review of its use in the management of CCR5-tropic HIV-1 infection. Drugs. 2010;70:1189–1213. doi: 10.2165/11203940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Mehlin C, Crittenden C, Andreyka J. No-wash dyes for calcium flux measurement. Biotechniques. 2003;34:164–166. doi: 10.2144/03341dd03. [DOI] [PubMed] [Google Scholar]
- Zhao H, et al. CXCR4 over-expression and survival in cancer: a system review and meta-analysis. Oncotarget. 2015;6:5022–5040. doi: 10.18632/oncotarget.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil) Biochem Pharmacol. 2009;77:1655–1664. doi: 10.1016/j.bcp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Milligan G. Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol. 2003;64:1271–1276. doi: 10.1124/mol.64.6.1271. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, et al. Discovery of positive allosteric modulators and silent allosteric modulators of the mu-opioid receptor. Proc Natl Acad Sci U S A. 2013;110:10830–10835. doi: 10.1073/pnas.1300393110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartfai T, Wang MW. Positive allosteric modulators to peptide GPCRs: a promising class of drugs. Acta Pharmacol Sin. 2013;34:880–885. doi: 10.1038/aps.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatse S, et al. Fluorescent CXCL12AF647 as a novel probe for nonradioactive CXCL12/CXCR4 cellular interaction studies. Cytometry A. 2004;61:178–188. doi: 10.1002/cyto.a.20070. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- Zhang WB, et al. A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40-4C are weak partial agonists. J Biol Chem. 2002;277:24515–24521. doi: 10.1074/jbc.M200889200. [DOI] [PubMed] [Google Scholar]
- Tamamura H, et al. A low-molecular-weight inhibitor against the chemokine receptor CXCR4: a strong anti-HIV peptide T140. Biochem Biophys Res Commun. 1998;253:877–882. doi: 10.1006/bbrc.1998.9871. [DOI] [PubMed] [Google Scholar]
- Wang Z-x, et al. In: Chemokine Biology - Basic Research and Clinical Application: Volume II: Pathophysiology of Chemokines. Neote K, Letts GL, Moser B, editors. Birkhäuser Basel; 2007. pp. 61–77. [Google Scholar]