

Abstract
Understanding of the hematopoietic stem and progenitor cell biology has important implications for regenerative medicine and the treatment of hematological pathologies. Despite the most relevant data that can be acquired using in vivo models or primary cultures, the low abundance of hematopoietic stem and progenitor cells considerably restricts the pool of suitable techniques for their investigation. Therefore, the use of cell lines allows sufficient production of biological material for the performance of screenings or assays that require large cell numbers. Here we present a detailed description, readout, and interpretation of proliferation and differentiation assays which are used for the investigation of processes involved in myelopoiesis and neutrophilic differentiation. These experiments employ the 32D/G-CSF-R cytokine dependent murine myeloid cell line, which possesses the ability to proliferate in the presence of IL-3 and differentiate in G-CSF. We provide optimized protocols for handling 32D/G-CSF-R cells and discuss major pitfalls and drawbacks that might compromise the described assays and expected results. Additionally, this article contains protocols for lentiviral and retroviral production, titration, and transduction of 32D/G-CSF-R cells. We demonstrate that genetic manipulation of these cells can be employed to successfully perform functional and molecular studies, which can complement results obtained with primary hematopoietic stem and progenitor cells or in vivo models.
Keywords: Developmental Biology, Issue 132, 32D/G-CSF-R cells, murine myeloid precursor cells, liquid culture, differentiation, neutrophils, proliferation, cytokines, IL-3, G-CSF
Introduction
The hematopoietic stem and progenitor population supplies the organism with a large range of mature cells, including cells from the myeloid lineage (neutrophils, eosinophils, basophils, and monocytes). The process that drives the production of myeloid cells from hematopoietic stem cells is known as myelopoiesis, and adequate production of mature myeloid cells in response to changing demands is a prerequisite for proper coping of the organism with stress conditions, such as infections and blood loss. Insufficient production of mature myeloid cells may lead to inability to eliminate pathogens, reduced blood coagulation, and other life-threatening conditions1,2. In addition, alterations in myeloid lineage development may also be associated with hematological malignancies, such as acute myeloid leukemia (AML)3. Alterations in myelopoiesis may occur due to various reasons, such as defects in cell surface receptors4, altered expressions of transcription factors5, impaired signaling pathways6, mutations resulting in formation/activation of oncogenes7, or inactivation of tumor suppressor genes8.
Various methods have been developed to study myeloid development, and assess the effect of specific genetic alterations in this process. Common approaches used to study myelopoiesis involve primary cells and transgenic mice. Though these models allow acquisition of biologically relevant data, they have certain limitations. The use of primary cells encounters a limited number of cells and a restricted period of culture, narrowing the possibilities to alter gene expression and subsequent biological or biochemical analysis. Transgenic mice are costly and require a reasonable degree of biological justification. In addition, working with in vivo models adds a degree of complexity into understanding the role of a gene of interest in a given process. Therefore, alternative approaches to circumvent these limitations are needed. Cell lines have indisputable advantages: (1) they possess unlimited proliferation capacity that allows generating enough material for biochemical and biological studies, (2) they are susceptible to genetic manipulations (knockdown, knockout, overexpression), (3) the cost is relatively low, and (4) they allow a degree of biological simplification required in certain experimental approaches.
The parental IL-3 (Interleukin-3) dependent 32D cell line was established in 1983 by Greenberger and colleagues by infection of bone marrow cells from C3H/HeJ mice with Friend murine leukemia virus9. Several 32D clones were described in literature: cl-239, cl-310, and cl-1011. The 32D cl-3 cells were shown to proliferate in IL-3 and undergo neutrophilic differentiation upon treatment with granulocyte-colony stimulation factor (G-CSF)10. On the contrary, 32D cl-10 cells, while being IL-3 dependent, originally were not differentiating in response to G-CSF treatment. In 1995 the group of Dr. Ivo Touw retrovirally transduced 32D cl-10 cells with wild type and mutant forms of G-CSF receptor (G-CSF-R), in order to identify functionally important regions of this receptor11. This study resulted in generation of the 32D/G-CSF-R cells, which are similarly dependent on IL-3, but within 6 to 10 days after replacement of IL-3 with G-CSF, cells stop to proliferate and irreversibly differentiate into mature neutrophils. These properties make 32D cl-3 and 32D/G-CSF-R cells simplified models of murine neutrophilic differentiation that can be modulated by two well-defined growth and differentiation factors - IL-3 and G-CSF. During the last decades multiple groups have used 32D/G-CSF-R cells to study the role of particular genes in proliferation and differentiation of myeloid cells in culture12,13,14,15,16, and to study G-CSF signaling17,18. Importantly, the results obtained using this cell line correlated with data obtained with primary cells and transgenic mice16,19,20,21. Consequently, we believe that 32D/G-CSF-R cells, being a widely used and well-established model, represent a valuable system to study myeloid differentiation which can be used in parallel with other approaches addressing this question.
Here, detailed protocols describing handling of the 32D/G-CSF-R cell line, which cover expansion, differentiation, and assessment of proliferation and differentiation of these cells is presented. Detailed information for genetic modification of 32D/G-CSF-R cells, either by retroviral or lentiviral transduction, as well as protocols for virus titration are provided. In addition, several representative results that demonstrate potential applications of 32D/G-CSF-R cells are provided.
Protocol
NOTE: Steps describing expansion, differentiation, and transduction of 32D/G-CSF-R cells are presented below.
1. Preparation
- Media preparation
- Prepare 250 mL of culture medium: RPMI (Roswell Park Memorial Institute) 1640 medium supplemented with 10% heat inactivated FBS (fetal bovine serum) and murine IL-3 (10 ng/mL).
- Alternatively, use home-made IL-3. To produce home-made IL-3, transduce HEK293 cells with IL-3 expressing vector and collect IL-3 containing supernatant22. NOTE: Antibiotics, such as penicillin G (100 IU/mL), streptomycin (100 µg/mL), and gentamicin (40 µg/mL), can be used for cell culturing at any step of the protocol unless otherwise stated.
- Prepare 50 mL of differentiation medium: RPMI 1640 medium supplemented with 10% FBS, and human G-CSF (100 ng/mL). NOTE: (Important) Not all serum batches support differentiation of 32D/G-CSF-R cells. Prior to experiment start test different batches of serum. It is recommended to test at least 4 different batches and select the optimal based on the ability of the 32D/G-CSF-R cells to differentiate. See 32D/G-CSF-R differentiation protocol below.
- Prepare 10 mL of freezing medium: RPMI 1640 medium supplemented with 40% FBS, and 10% DMSO (dimethyl sulfoxide).
- Production of retrovirus
- Plate a single cell suspension of Bosc23 cells (6 × 106) in a 16 cm Petri dish and cultivate in 18 mL of DMEM (Dulbecco's Modified Eagle's Medium) containing 10% FBS until the confluency of the culture reaches 80 % (24 h). NOTE: Cells should grow in a monolayer and not form clumps in culture. Cell counting can be performed using different methods (valid for the rest of the protocols presented here). In case of low number of samples, manual counting under the microscope using Bürker chamber is recommended. In this case, use Trypan blue for exclusion of dead cells. Mix cells 1:1 with 0.4% Trypan Blue in PBS. To perform counting of large number of samples, an automatic cell counter can be employed.
- Combine 40 µg of retroviral construct (such as MSCV), 20 µg of pCL-Eco (packaging vector)23, 80 µl of PEI (polyethylenimine), and 2 mL of reduced-Serum medium (e.g. Opti-MEM) medium (without antibiotics). Incubate this mixture for 20 min at room temperature (RT).
- Carefully replace Bosc23 cells medium with 16 mL DMEM supplemented with 2% FBS. Pre-warm the medium to 37 °C before use. NOTE: Do not use antibiotics during transfection, since it may reduce transfection efficiency.
- Add the mixture prepared in 1.2.2. dropwise and carefully to the Bosc23 culture, and incubate for 4 h at 37 °C. Limit incubation to less than 6 h due to PEI toxicity.
- After incubation, change Bosc23 medium to 18 mL pre-warmed DMEM containing 10% FBS, and cultivate cells for 48 h at 37 °C. Place dishes in a biosafety level 2 laboratory, keep here from this step on, and follow standard safety procedures.
- Collect Bosc23 medium (containing ecotropic retroviral particles) using a 25-mL serological pipette into a 50-mL conical tube, and store at 4 °C. NOTE: (Important) Transfection efficiency should reach 90% to produce a high titer virus.
- Add 18 mL pre-warmed DMEM 10% FBS to Bosc23 cells and cultivate for 24 h.
- Repeat step 1.2.6. NOTE: Retroviral supernatants from 1.2.6 and 1.2.8 can be pooled once they reach the same temperature (4 °C) to preserve viral integrity.
- To avoid Bosc23 contamination in viral supernatants, spin collected virus at 1500 × g for 10 min at 4 °C. Aliquot virus, snap freeze using dry-ice or liquid nitrogen, and store at -80 °C. However, note that freshly prepared virus is of higher quality in terms of infection efficiency than frozen stocks. NOTE: (Important) Avoid repetitive freezing/thawing of virus, since it leads to viral degradation.
- Production of lentivirus
- Plate a single cell suspension of HEK293T (Human embryonic kidney 293T) cells (6 × 106) in a 16-cm Petri dish and cultivate in 18 mL of DMEM containing 10% FBS until the confluency of the culture reaches 80 % (24 h). NOTE: Cells should grow in a monolayer and not form clumps in culture.
- Combine 15 µg of lentiviral construct (such as pGhU6), packaging plasmids pCMVdR8.74 (encoding for gag/pol, 12 µg) and pMD2.VSVG (encoding for VSV-G, 1.4 µg), 85.2 µL PEI, and 2 mL of reduced-serum medium (without antibiotics). Incubate this mixture for 20 min at RT.
- Carefully replace HEK293T medium with 16 mL of DMEM supplemented with 2% FBS. Pre-warm the medium to 37 °C before use. Do not use antibiotics during transfection, since it may reduce transfection efficiency.
- Carefully drop the mixture prepared in 1.3.2 to the HEK293T cell culture and incubate for 4 h at 37 °C. Limit incubation duration to within 6 h to prevent PEI toxicity.
- Change medium to 18 mL pre-warmed DMEM 10% FBS and cultivate cells for 48 h at 37 °C. Place dishes in a biosafety level 2 laboratory, keep here from this step on, and follow standard safety procedures.
- Collect HEK293T medium (containing amphotropic lentiviral particles) using a 25-mL serological pipette into a 50 mL conical tube and store it at 4 °C. NOTE: (Important) Transfection efficiency should reach 90% to produce a high titer virus.
- Carefully add 18 mL of pre-warmed DMEM 10% FBS to the HEK293T cells. Cultivate for 24 h at 37 °C.
- Repeat step 1.3.6. NOTE: Lentiviral supernatants from 1.3.6 and 1.3.8 can be pooled once they reach the same temperature (4 °C) to preserve viral integrity.
- To avoid contamination of viral supernatant with HEK293T cells, spin collected virus at 1500 × g for 10 min at 4 °C. Aliquot virus, snap freeze using dry-ice or liquid nitrogen, and store it at -80 °C. However, notice that freshly prepared virus is of higher quality in terms of infection efficiency than frozen stocks. NOTE: (Important) Avoid repetitive freezing/thawing of virus, since it leads to viral degradation.
- Titration of virus containing a GFP reporter
- Seed 1 × 105 NIH/3T3 cells in 300 µL of DMEM 10% FBS per well into a 24-well plate. 7 wells will be employed to titer one virus.
- Thaw virus at 37 °C and add 1, 5, 10, 50, 100, and 500 µL virus to NIH/3T3 cultures. Do not add any virus into negative control well. Cultivate cells in the presence of the virus for 48 h at 37 °C.
- Harvest NIH/3T3 cells using 30 µL of trypsin/EDTA (ethylenediaminetetraacetic acid) (0.25% Trypsin, 0.01% EDTA in PBS) and place cells in 250 µL PBS in a FACS (fluorescence activated cell sorting) tube.
- Determine the frequency of GFP+ cells by flow cytometry in each sample24. Exclude dead cells by addition of a viability dye, such as Hoechst 33258.
- Calculate the total numbers of GFP+ cells (based on the number of plated cells and percentage of GFP+ cells), and plot them against the volume of virus used for infection. NOTE: (Important) Efficiency of infection reaches a plateau when large viral doses are applied. Therefore, it is important to estimate the titer based on viral concentrations at which infection efficiency occurs in a linear range (example shown in Table 1). The number of GFP+ cells indicates the number of functional viral particles (TU - transforming units) in a given virus volume. Recalculate the number of TU per mL.
- Titration of virus containing a puromycin reporter
- Seed 5 × 104 NIH/3T3 cells in 3 mL of DMEM 10% FBS per well in a 6-well plate; employ 6 wells to titer one virus. Culture overnight at 37 °C.
- Next day dilute virus as indicated in Table 2. To dilute virus, use pre-warmed DMEM 10% FBS. Do not vortex.
- Remove culture medium from NIH/3T3 cells and add 1 mL of diluted virus.
- Incubate overnight at 37 °C.
- Gently replace the virus-containing medium with 2 mL of pre-warmed DMEM 10% FBS, and incubate overnight at 37 °C.
- Gently replace NIH/3T3 medium with 2 mL of pre-warmed DMEM 10% FBS containing puromycin (2 µg/mL).
- Culture at 37 °C and carefully refresh medium with puromycin every 3 days or when medium turns yellow.
- At 10-12 days after infection, aspirate medium from each well and wash gently with PBS.
- Stain with 1 mL of crystal violet solution (0.5% crystal violet in 20% ethanol and dH2O), 2 min at RT, and wash carefully with PBS twice.
- Count blue colonies present in each well under a microscope (magnification X4). No colonies should be observed in the negative control well.
- Calculate the total numbers of TU/mL considering the dilution factor.
2. Expansion and maintenance of 32D/G-CSF-R cells
If 32D/G-CSF-R cells are frozen, take an aliquot and thaw cells in a 37 °C water bath for 1 min and pour cells from the vial directly into 10 mL of pre-warmed RPMI 1640 medium supplemented with 10% FBS in a 15-mL tube. Invert the tube 3 times and spin the cells down at 400 × g. Remove supernatant and resuspend cells in IL-3 containing culture medium at a concentration of 0.3 × 106 cells/mL.
Optimal cell growth concentration is 0.25-0.5 × 106 cells/mL. Split cells every 2 days bringing them to a concentration of 0.1-0.2 × 106 cells/mL. NOTE: (Important) 32D/G-CSF-R cells may partially lose their ability to differentiate if grown to concentrations higher than 1 × 106 cells/mL. Therefore, it is very important to split 32D/G-CSF-R cells on time.
As needed, freeze and store cell aliquots for long periods of time at -80 °C or in liquid nitrogen. Spin down 3 × 106 cells, remove supernatant, and resuspend in 1 mL of freezing medium. Place tube in a freezing container at -80 °C. For long term storage, reposition cells to liquid nitrogen.
3. Transduction of 32D/G-CSF-R cells
Plate 32D/G-CSF-R cells into 6-well plate at a concentration of 0.3 × 106 cells/mL.
Add appropriate amount of virus to reach a MOI (multiplicity of infection) between 10 and 40. NOTE: Higher MOI will result in increased percentage of GFP+ cells as well as higher levels of expression of the gene of interest. Example: To infect 0.3 × 106 cells with a MOI of 10; 3 × 106 TU will need to be added to the 32D/G-CSF-R cells.
Add polybrene to final concentration 8 µg/mL and incubate for 6 h at 37 °C. Alternatively, perform a spin-inoculation procedure: centrifuge at 1200 × g for 90 min at 30 °C in a culturing plate using a swing-bucket rotor, and incubate for 3 h at 37 °C.
Collect cells, transfer to a 15-mL tube, and spin at 450 × g for 5 min (4 °C).
Discard the supernatant, resuspend pelleted cells in 6 mL of culture medium, place in a T25 cell culture flask, and culture at 37 °C.
48 h later, harvest cells and spin them at 450 × g for 5 min (4 °C). Discard the supernatant.
In case of a GFP reporter: place cells in 500 µL of PBS and sort GFP+ cells using a flow cytometry sorter. In case of a puromycin reporter containing vector: place cells in culture medium containing 2 µg/mL of puromycin.
Expand cells as needed for further analysis and experiments. NOTE: (Optional) Transduced cells can be frozen in freezing media in a freezing container at -80 °C, and further stored in liquid nitrogen.
4. 32D/G-CSF-R cell proliferation assay
To assess proliferation rate place 0.2 × 106 cells in 1 mL of culture medium and incubate overnight at 37 °C.
Next day count number of cells and dilute them back to a concentration of 0.2 × 106 cells/mL using culture medium. Incubate overnight at 37 °C. Keep records of cell concentrations and dilutions applied to the daily cultures using Table 3.
Repeat step 4.2 for as many days as proliferation needs to be assessed. NOTE: The length of the proliferation assays will depend on the effect of the gene of interest. In case of a strong phenotype, 8-9 days might be sufficient to observe a significant effect (See Figure 3A). However, in case of a mild phenotype, longer periods of time might be required.
Assess cell growth by making a proliferation curve as indicated in Table 3.
5. 32D/G-CSF-R cell differentiation assay
Wash 0.2 × 106 32D/G-CSF-R cells twice with RPMI medium without cytokines. NOTE: The washing steps before addition of G-CSF into the culture are important, since presence of residual IL-3 may inhibit differentiation.
Place cells in 1 mL differentiation medium (containing 100 ng/mL G-CSF) in a 24 well/plate, resulting in a cell concentration of 0.2 × 106 cells/mL (Day 0). Culture overnight at 37 °C.
Count number of cells/mL as indicated above (step 1.2.1).
Cytospin 2-5 × 104 cells on a glass slide (76 mm X 26 mm) using a centrifuge with necessary adaptors at 20 × g.
Refresh differentiation medium and plate cells at a concentration of 0.2 × 105 cells/mL on a 24 well/plate (Day 1). Discard the old plate, and proceed next day with step 5.6 with the new plate.
On days 2 to 7, repeat steps 5.3 to 5.5. Assess cell proliferation in the presence of G-CSF as described in step 4. NOTE: (Important) During differentiation, cell proliferation slows down, therefore after 3 to 4 days in the presence of G-CSF it is adequate to culture cells at higher concentrations.
- Assess differentiation state of 32D/G-CSF-R cells based on cell morphology.
- Fix cells from step 5.4 in methanol for 5 min at RT and leave slides to air-dry. NOTE: Slides from Day 0 to 7 can be stored at RT, and fixed simultaneously. Similarly, they can also be kept at RT for longer periods after fixation.
- Stain cells using May-Grünwald Giemsa staining following manufacturer's protocol.
- Visualize stained cells using a microscope and magnification X40.
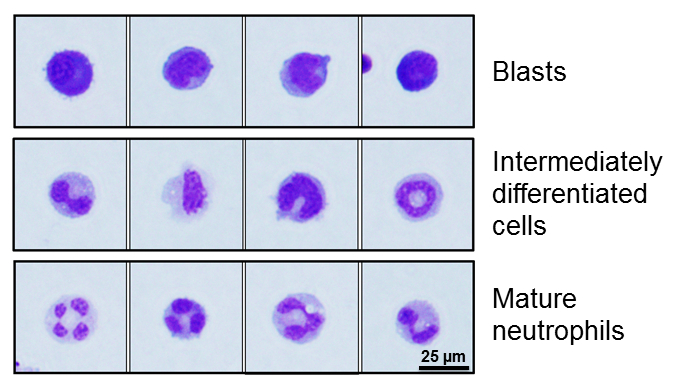
- Quantify number of blasts, intermediately differentiated cells, and mature granulocytes using illustrative pictures on Figure 1 and description in Table 4.
Representative Results
Proliferation and differentiation of 32D/G-CSF-R cells
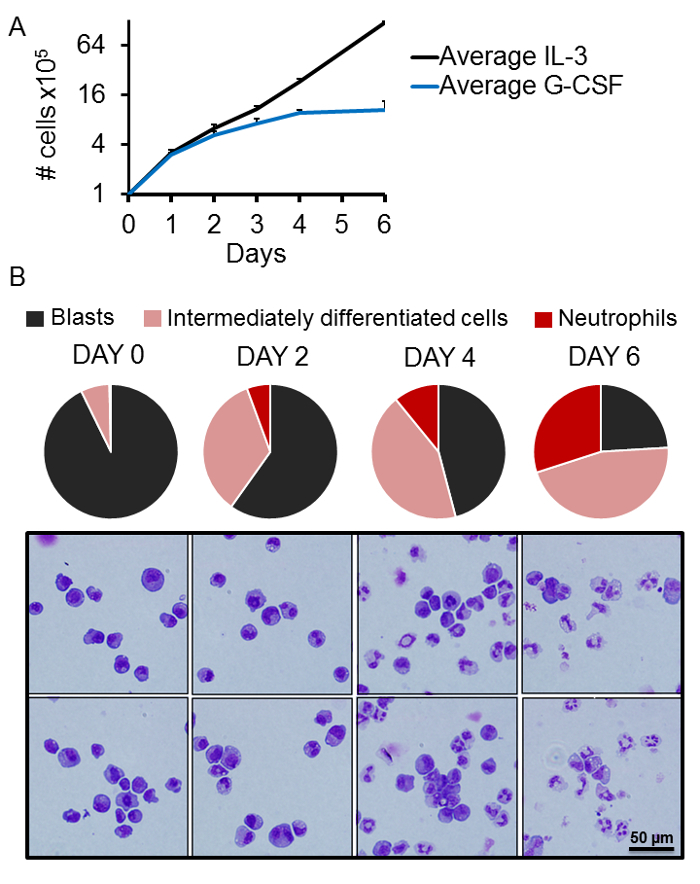
To assess proliferation of 32D/G-CSF-R cells under pro-proliferative and pro-differentiation conditions, 32D/G-CSF-R cells were cultured in media containing IL-3 and G-CSF, respectively. It was observed that cells cultured in IL-3 containing medium (10 ng/mL) divide approximately every 24 h (Figure 2A). Upon replacement of IL-3 with G-CSF (100 ng/mL) proliferation gradually slowed down and stopped after 4 days (Figure 2A). Further, the differentiation state of cells cultured in the presence of G-CSF using May-Grünwald Giemsa-stained cytospun cells was assessed. It was shown that on day 0 (before the start of G-CSF treatment), cells present an immature myeloblast-like morphology, characterized by a large nucleus and a dark cytoplasm (Figure 2B). Over the course of treatment, the nuclear size is reduced and the shape of the nucleus changes to a moon-shaped or donut-shape. Further, the cytoplasm is enlarged and loses the dark violet color. After 6 days of treatment with G-CSF, cells present signs of full neutrophilic differentiation, characterized by a lobulated nucleus and a light violet cytoplasm (Figure 2B). The differentiation of 32D/G-CSF-R cells is a gradual process, where not all cells evolve towards mature neutrophils at the exact same speed. Quantification of cells at different stages over the course of differentiation (namely blast, intermediately differentiated cell, and neutrophil) is shown in Figure 2B.
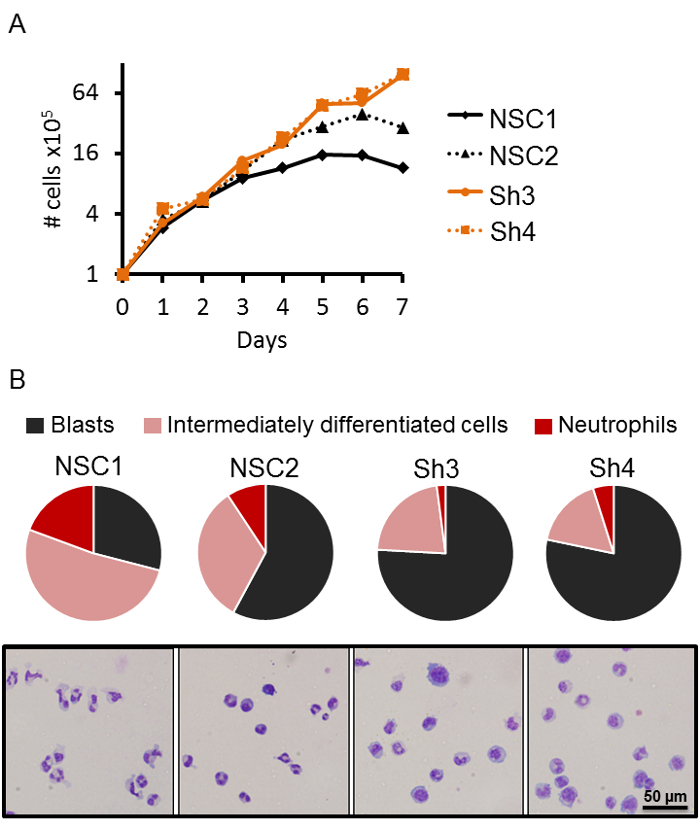
Murine Evi2b knockdown blocks neutrophilic differentiation in 32D/G-CSF-R cells
For downregulation of EVI2B (Ecotropic Viral Integration Site 2B) in 32D/G-CSF-R cells, 3 Evi2b-targeting (Sh2, Sh3, Sh4) and 1 non-targeting non-silencing control (NSC1) shRNAs were designed; these were cloned into a lentiviral vector carrying a GFP reporter16. 32D/G-CSF-R cells were transduced with control and Evi2b-silencing shRNAs using a MOI of 10. Two days after transduction, GFP+ (transduced) cells were sorted, and expanded for further experiments. In case of Sh3 and Sh4, EVI2B depletion reached 80%, however Sh2 was unable to efficiently downregulate EVI2B protein levels, and therefore was used as an additional control referred here as non-silencing control 2 (NSC2). Four days after transduction, differentiation and proliferation assays were performed in the presence of G-CSF, and the effects of Evi2b downregulation in these processes were evaluated. It was observed that Evi2b-depleted cells (Sh2, Sh3) sustained cell proliferation in the presence of GCSF, whereas the control cells (NSC1 and NSC2) reduced proliferation around day 4 (Figure 3A). Further, Evi2b-silenced 32D/G-CSF-R cells produced less intermediate and mature neutrophils compared to control cells (Figure 3B). Remarkably, cells transduced with NSC2, which demonstrated poor Evi2b knockdown efficiency, also showed slight reduction in the number of mature neutrophils (Figure 3B). These data were recently published16.
BCR-ABL fusion protein impairs neutrophilic differentiation in 32D/G-CSF-R cells
It has been shown that BCR-ABL fusion protein impairs myeloid differentiation, causing an extensive expansion of myeloid progenitors which results in hematopoietic failure during the acute phase of chronic myeloid leukemia25,26. Previous studies demonstrated that enforced expression of BCR-ABL in 32D cl-3 cells resulted in a block of neutrophil differentiation27,28. Therefore, we investigated whether similar results could be obtained with the 32D/G-CSF-R cell line. 32D/G-CSF-R cells were transduced with BCR-ABL or control MSCV retroviral vector carrying a GFP reporter (MOI = 20). 2 days after transduction, GFP+ cells were sorted and expanded for 2 weeks in IL-3 containing medium. Next, differentiation assays were performed in the presence of G-CSF. We observed that at day 0 (before transferring the cells to G-CSF containing medium), MSCV control and BCR-ABL expressing 32D/G-CSF-R cells presented similar morphology, mainly representing immature myeloid blast cells (Figure 4). However, we demonstrated that while empty vector transduced control cells underwent neutrophilic differentiation after 6 days of G-CSF treatment (producing 11.5 % of mature neutrophils and 56.4 % of intermediately differentiated cells), no mature neutrophils were generated from BCR-ABL-transduced 32D/G-CSF-R cells (Figure 4). Consistently, the percentage of immature blast in BCR-ABL expressing cells in G-CSF remained similar to the percentage of blast in IL-3 conditions.
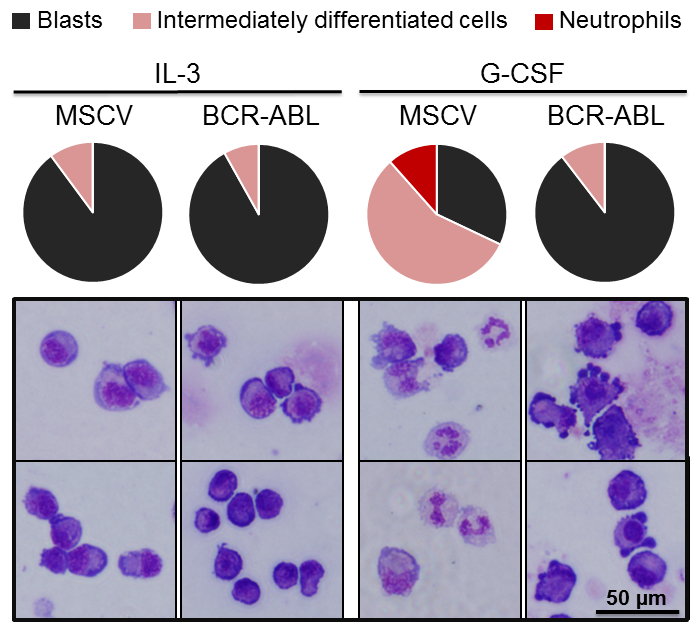
Figure 1. Representative images of distinct 32D/G-CSF-R cell morphology. 32D/G-CSF-R cells can be morphologically classified as blast, intermediately differentiated cells, and mature neutrophils. See Table 4 for description.
Figure 2. Proliferation and differentiation of 32D/G-CSF-R cells.(A) Proliferation of 32D/GCSFR cells in 10 ng/mL IL-3 (black line) or 100 ng/mL G-CSF (blue line) containing medium. The X axis represents days of treatment. The Y axis is shown in logarithmic scale (log2) and indicates the number of cells × 105. Results represent average of 3 independent experiments. Error bars indicate standard deviation. (B) Differentiation of 32D/G-CSF-R cells in G-CSF-containing medium (100 ng/mL). In the upper panel, the pie-plots demonstrate percentage of blasts (black), intermediately differentiated cells (pink), and neutrophils (red) in culture after 0, 2, 4, and 6 days of G-CSF treatment. The lower panel contains representative images of cytospun cells from the respective time points stained with May-Grünwald Giemsa. A minimum of 100 cells were evaluated for each time point.
Figure 3. Evi2b silencing blocks neutrophilic differentiation in 32D-G-CSF-R cells. (A) Proliferation of Evi2b-silenced (orange lines) and control (black lines) 32D/G-CSF-R cells in 100 ng/mL G-CSF containing medium. The X axis represents days of treatment. The Y axis is shown in logarithmic scale (log2) and indicates the number of cells × 105. Results demonstrate a representative result of 3 independent experiments. (B) Assessment of differentiation of 32D/G-CSF-R cells transduced with Evi2b-silencing and control shRNAs in G-CSF-containing medium (100 ng/mL) using May-Grünwald Giemsa staining of cytospun cells. In upper panel, the pie-plots demonstrate percentage of blasts (black), intermediately differentiated cells (pink), and neutrophils (red) in culture. The lower panels contain representative pictures of cytospun cells. Differentiation was assessed on day 7 of differentiation. At least 100 cells were evaluated for each condition.
Figure 4. Overexpression of BCR-ABL fusion protein impairs differentiation of 32D/G-CSF-R cells. Assessment of differentiation of 32D/G-CSF-R cells transduced either with control MSCV or BCR-ABL fusion gene in G-CSF-containing medium (100 ng/mL). Upper pie-plots demonstrate proportion of blasts (black), intermediately differentiated cells (pink), and neutrophils (red) on day 6 of differentiation. The lower panels show representative pictures of cytospun cells stained with May-Grünwald Giemsa. At least 100 cells were evaluated for each time point.
| Virus V [µL] | Number of plated cells | % GFP+ | GFP+ cell count | Linear? | TU/mL | Average (TU/mL) |
| 1 | 100 000 | 1.83 | 1 830 | Yes | 1 830 000 | 2 350 000 |
| 5 | 100 000 | 12.9 | 12 900 | Yes | 2 580 000 | |
| 10 | 100 000 | 26.4 | 26 400 | Yes | 2 640 000 | |
| 50 | 100 000 | 71.4 | 71 400 | No | ||
| 100 | 100 000 | 85.1 | 85 100 | No | ||
| 500 | 100 000 | 85.6 | 85 600 | No |
Table 1. Viral titer determination for GFP reporter containing vectors. Example of data obtained with the MSCV retrovirus and calculation performed to estimate the viral titer. Viral titer was calculated based on number of GFP+ cells acquired when certain amount of virus was applied. The amount of virus employed for infection should be in a linear correlation with the percentage of GFP+ cells measured. TU: transforming units.
| Tube | Tube description | DMEM+10% FBS | Virus | Total volume | Dilution factor |
| (µL) | (µL) | (µL) | |||
| 1 | dilution 1 | 1485 | 15 µL viral stock | 1500 | 1 x 102 |
| 2 | dilution 2 | 1350 | 150 µL tube 1 | 1500 | 1 x 103 |
| 3 | dilution 3 | 1350 | 150 µL tube 2 | 1500 | 1 x 104 |
| 4 | dilution 4 | 1350 | 150 µL tube 3 | 1500 | 1 x 105 |
| 5 | dilution 5 | 1350 | 150 µL tube 4 | 1500 | 1 x 106 |
Table 2. Viral titer determination for puromycin reporter containing vectors. Viral dilution strategy to determine the viral titer in puromycin containing vectors. Table indicates how to prepare 5 tubes (1-5) containing a serial dilution of the viral stock. Tube 1 contains 1485 µL DMEM + 10 % FBS and 15 µL of the produced virus, providing a 1 × 102 diluted virus. Tube 2 contains 1350 µL DMEM + 10% FBS and 150 µL of the 1 × 102 diluted virus (Tube 1), providing a 1 × 103 diluted virus. Tube 3 contains 1350 µL DMEM + 10% FBS and 150 µL of the 1 × 103 diluted virus (Tube 2), providing a 1 × 104 diluted virus. Tube 4 contains 1350 µL DMEM + 10% FBS and 150 µL of the 1 × 104 diluted virus (Tube 3), providing a 1 × 105 diluted virus. Tube 5 contains 1350 µL DMEM + 10% FBS and 150 µL of the 1 × 105 diluted virus (Tube 4), providing a 1 × 106 diluted virus. 1 ml from tubes 1-5 is used to titer the virus.
| Cell counts (x = number of cells per ml) | |||||||
| day 0 | day 1 | day 2 | day 3 | day 4 | day 5 | day 6 | |
| Sample 1 | x0 | x1 | x2 | x3 | x4 | x5 | x6 |
| Dilution (d) (y = volume of cells used for replating [mL]; z = final volume [mL] ) | |||||||
| day 0 | day 1 | day 2 | day 3 | day 4 | day 5 | day 6 | |
| Sample 1 | d0=1 | d1=y1/z1 | d2=y2/z2 | d3=y3/z3 | d4=y4/z4 | d5=y5/z5 | |
| Total cell count | |||||||
| day 0 | day 1 | day 2 | day 3 | day 4 | day 5 | day 6 | |
| Sample 1 | x0 | x1 /d0 | x2/d1 | x3/d2/d1 | x4/d3/d2/d1 | x5/d4/d3/d2/d1 | x6/d5/d4/d3/d2/d1 |
Table 3. Growth curve generation. The table describes equations necessary for assessment of proliferation rate of 32D/G-CSF-R cells.
| Nucleus | Cytoplasm | |
| Blasts | Dark, rounded | Dark, almost indistinguishable from nucleus |
| Intermediately differentiated cells | Pronounced changes in nuclear shape, often donut-like or moon-like shaped | Lighter, distinguishable cytoplasm |
| Mature neutrophils | Lobulated nucleus; almost no connections between the lobules | Light cytoplasm |
Table 4. 32D/G-CSF-R cell morphology during neutrophilic differentiation. The table summarizes major morphological features enabling distinction of immature blasts, intermediately differentiated cells, and mature neutrophils. Nuclear and cytoplasmic characteristics are described. See Figure 1 for representative images.
Discussion
The choice of an experimental model is one of the main issues in research. Though primary animal and human cells are believed to produce the most biologically relevant data, these models may involve ethical concerns and are often associated with expensive and/or sophisticated isolation/culturing procedures. Primary cells are limited in numbers and it is hard to genetically manipulate them. In addition, primary cells represent a heterogeneous population composed of various cell types that may complicate data interpretation29. In contrast, cell lines provide a virtually unlimited source of biological material, are cost effective, and allow to bypass the ethical issues. However, it is important to take into account that cell lines are considered to be an artificial experimental system, which may genetically and phenotypically differ from their tissue of origin. Nevertheless, cell lines when combined with other experimental approaches, provide a valuable model for the generation of reproducible and biologically relevant data.
The 32D/G-CSF-R cell line, as well as 32D cl-3 cells, are well-established and widely used cell culture models of murine neutrophilic differentiation14,15,24,30. Several research groups have used these cells to assess the role of particular genes in myelopoiesis, and obtained results that correlated with data obtained using in vivo models and primary cells16,31. Here we provide a protocol for handling the 32D/G-CSF-R cell line, however, similar procedures can be applied for culturing and manipulating 32D cl-3 cells. It is important to point out that different batches of the 32D cl-3 cells used in different laboratories present differences in karyotype, suggesting that different groups might be working with their own subclones32. Based on this observation, we hypothesize that genetic variability might similarly be present in 32D/G-CSF-R cells, and this needs to be taken into consideration when comparing results obtained by different research groups. Alternative cell culture models to 32D/G-CSF-R cells are immortalized hematopoietic progenitor lines22,33,34,35,36. This approach is useful when large-scale progenitor expansion is required, for instance in order to study protein interactions22. An advantage to 32D/G-CSF-R cells is that immortalized progenitors can be generated from any genetically modified mouse, although the time to establish the line is considerably long37. In addition, handling and manipulation of bone marrow immortalized progenitors requires more expertise than 32D/G-CSF-R cells.
To familiarize the reader with the 32D/G-CSF-R model, in the first part of the representative results we demonstrated proliferation and differentiation kinetics of 32D/G-CSF-R cells in the presence of IL-3 and G-CSF. Representative images demonstrating the morphology of the cells in IL-3 proliferating conditions as well as at different time points of GCSF-induced differentiation were presented. We assessed neutrophilic differentiation by morphological analysis, however it has been reported that 32D cl-3 cell differentiation can be determined by flow cytometric analysis using the CD11b cell surface marker27,31,38,39. To our knowledge and expertise, CD11b is not upregulated during neutrophilic differentiation of 32D/G-CSF-R cells. For standardization, in the 32D/G-CSF-R proliferation assays we employed commercially available IL-3. However, we observed that cells proliferate well in home-made IL-3, produced as described by Drobek and colleagues22. To obtain a good degree of G-CSF-induced neutrophilic differentiation, it is important to remember that the differentiation ability of 32D/G-CSF-R cells can be impaired if cells are cultured above a concentration of 1 × 106 cells/mL. In addition, the degree and speed of differentiation may be affected by the serum composition. Therefore, it is recommended to test the differentiation ability of 32D/G-CSF-R cells in several FBS batches, and select the one that better supports the development of mature neutrophils upon 6 to 9 days of culture in the presence of G-CSF.
We provided two representative experiments demonstrating possible ways to study the role of particular proteins in myeloid differentiation (Figure 3 and Figure 4). First, we showed that downregulation of EVI2B, a transmembrane protein expressed in hematopoietic cells, leads to a block of myeloid differentiation in 32D/G-CSF-R cells. These data were recently published by our group, in combination with assays performed using primary cells and Evi2b knockout mice16. Second, we investigated the effect of BCR-ABL, a fusion protein resulting from the genetic translocation t(9,22) (q34;q11), on neutrophilic differentiation of 32D/G-CSF-R cells. This translocation was originally identified in patients suffering from chronic myeloid leukemia40,41. It was previously shown that expression of BCR-ABL fusion oncoprotein in 32D cl-3 cells leads to a myeloid differentiation arrest27,28. Here we demonstrated that BCR-ABL overexpression has similar effects on 32D/G-CSF-R cells. In both sets of experiments presented here (involving Evi2b downregulation and BCR-ABL overexpression), genetic modification of 32D/G-CSF-R cells was performed through viral transduction, which results in stable gene expression. The choice of retroviral versus lentiviral delivery does not affect proliferation or differentiation of 32D/G-CSF-R cells. However, lentiviral infection is more efficient than retroviral transduction due to the presence of endogenous retrovirus in 32D/G-CSF-R cells. According to our experience, transfection of these cells using standard lipophilic reagents is not efficient. However, if transient construct expression is needed, transfection by electroporation is a viable approach42,43,44. Here, we presented genetic manipulation of 32D/G-CSF-R cells followed by GFP sorting and bulk analysis of the infected cells. However, if necessary, single cell sorting could be employed to generate single cell clones as previously reported12,45.
In addition to proliferation and differentiation assays, 32D/G-CSF-R cells have been employed to successfully determine the role of certain proteins in cell migration and apoptosis13,46,47,48. Further, the 32D/G-CSF-R line has been used to determine cytokine independent growth mediated by oncoproteins19,20. Another line frequently used to study cytokine independency is Ba/F3 cells21,49. Ba/F3 is an IL-3 dependent murine cell line derived from C3H mouse strain, similarly to 32D/G-CSF-R cells. Although both systems can be employed to study cytokine independent proliferation, Ba/F3 cells poorly differentiate in the presence of G-CSF.
Altogether, we suggest that 32D/G-CSF-R cells, though being less preferred than primary cells, offer several advantages including unlimited proliferation capacity and simple handling. We believe that for the generation of reliable data, experiments performed in 32D/G-CSF-R cells should be complemented with data acquired using alternative experimental approaches such as murine models and primary cultures.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank Prof. Ruud Delwel and Prof. Ivo Touw for providing us with the 32D/G-CSF-R cell line, and Prof. Daniel G. Tenen for providing us with the Bosc23 cell line. This work was supported by grants of the Grant Agency of the Czech Republic (GACR 15-03796S and GACR 17-02177S) to MA-J, support from the Institute of Molecular Genetics of the Czech Academy of Sciences (RVO 68378050) to MA-J, a GA UK fellowship (project No. 341015) from Charles University in Prague to MK, and a GA UK fellowship (project No. 1278217) from Charles University in Prague to PD.
References
- Bonilla MA, et al. Effects of recombinant human granulocyte colony-stimulating factor on neutropenia in patients with congenital agranulocytosis. N Engl J Med. 1989;320(24):1574–1580. doi: 10.1056/NEJM198906153202402. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Djulbegovic B, Norris LB, Armitage JO. Colony-stimulating factors for febrile neutropenia during cancer therapy. N Engl J Med. 2013;368(12):1131–1139. doi: 10.1056/NEJMct1210890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341(14):1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- Dong F, et al. Identification of a nonsense mutation in the granulocyte-colony-stimulating factor receptor in severe congenital neutropenia. Proc Natl Acad Sci U S A. 1994;91(10):4480–4484. doi: 10.1073/pnas.91.10.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol. 2007;7(2):105–117. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- Kota J, Caceres N, Constantinescu SN. Aberrant signal transduction pathways in myeloproliferative neoplasms. Leukemia. 2008;22(10):1828–1840. doi: 10.1038/leu.2008.236. [DOI] [PubMed] [Google Scholar]
- Kvinlaug BT, et al. Common and overlapping oncogenic pathways contribute to the evolution of acute myeloid leukemias. Cancer Res. 2011;71(12):4117–4129. doi: 10.1158/0008-5472.CAN-11-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene. 2002;21(21):3475–3495. doi: 10.1038/sj.onc.1205322. [DOI] [PubMed] [Google Scholar]
- Greenberger JS, Sakakeeny MA, Humphries RK, Eaves CJ, Eckner RJ. Demonstration of permanent factor-dependent multipotential (erythroid/neutrophil/basophil) hematopoietic progenitor cell lines. Proc Natl Acad Sci U S A. 1983;80(10):2931–2935. doi: 10.1073/pnas.80.10.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtieri M, et al. Cytokine-dependent granulocytic differentiation. Regulation of proliferative and differentiative responses in a murine progenitor cell line. J Immunol. 1987;138(11):3829–3835. [PubMed] [Google Scholar]
- Dong F, et al. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333(8):487–493. doi: 10.1056/NEJM199508243330804. [DOI] [PubMed] [Google Scholar]
- Jorda MA, Lowenberg B, Delwel R. The peripheral cannabinoid receptor Cb2, a novel oncoprotein, induces a reversible block in neutrophilic differentiation. Blood. 2003;101(4):1336–1343. doi: 10.1182/blood-2002-07-2034. [DOI] [PubMed] [Google Scholar]
- Jorda MA, et al. Hematopoietic cells expressing the peripheral cannabinoid receptor migrate in response to the endocannabinoid 2-arachidonoylglycerol. Blood. 2002;99(8):2786–2793. doi: 10.1182/blood.v99.8.2786. [DOI] [PubMed] [Google Scholar]
- Abbas S, et al. Integrated genome-wide genotyping and gene expression profiling reveals BCL11B as a putative oncogene in acute myeloid leukemia with 14q32 aberrations. Haematologica. 2014;99(5):848–857. doi: 10.3324/haematol.2013.095604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang D, Qiu Y, Kogan SC, Dong F. Increased CCAAT enhancer-binding protein epsilon (C/EBPepsilon) expression and premature apoptosis in myeloid cells expressing Gfi-1 N382S mutant associated with severe congenital neutropenia. J Biol Chem. 2006;281(16):10745–10751. doi: 10.1074/jbc.M510924200. [DOI] [PubMed] [Google Scholar]
- Zjablovskaja P, et al. EVI2B is a C/EBPalpha target gene required for granulocytic differentiation and functionality of hematopoietic progenitors. Cell Death Differ. 2017;24(4):705–716. doi: 10.1038/cdd.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini V, et al. The carboxy-terminal region of the granulocyte colony-stimulating factor receptor transduces a phagocytic signal. Blood. 2003;101(11):4615–4622. doi: 10.1182/blood-2002-07-2271. [DOI] [PubMed] [Google Scholar]
- Liu H, Qiu Y, Xiao L, Dong F. Involvement of protein kinase Cepsilon in the negative regulation of Akt activation stimulated by granulocyte colony-stimulating factor. J Immunol. 2006;176(4):2407–2413. doi: 10.4049/jimmunol.176.4.2407. [DOI] [PubMed] [Google Scholar]
- Kelly LM, et al. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99(1):310–318. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- Pikman Y, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller J, et al. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J. 1998;17(18):5321–5333. doi: 10.1093/emboj/17.18.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobek A, et al. PSTPIP2, a Protein Associated with Autoinflammatory Disease, Interacts with Inhibitory Enzymes SHIP1 and Csk. J Immunol. 2015;195(7):3416–3426. doi: 10.4049/jimmunol.1401494. [DOI] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70(8):5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberich-Jorda M, et al. C/EBPgamma deregulation results in differentiation arrest in acute myeloid leukemia. J Clin Invest. 2012;122(12):4490–4504. doi: 10.1172/JCI65102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103(11):4010–4022. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5(3):172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- Schuster C, et al. The effects of Bcr-Abl on C/EBP transcription-factor regulation and neutrophilic differentiation are reversed by the Abl kinase inhibitor imatinib mesylate. Blood. 2003;101(2):655–663. doi: 10.1182/blood-2002-01-0043. [DOI] [PubMed] [Google Scholar]
- Chang JS, et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2 dependent suppression of C/EBPalpha-driven myeloid differentiation. Blood. 2007;110(3):994–1003. doi: 10.1182/blood-2007-03-078303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velten L, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19(4):271–281. doi: 10.1038/ncb3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorda MA, Rayman N, Valk P, De Wee E, Delwel R. Identification, characterization, and function of a novel oncogene: the peripheral cannabinoid receptor Cb2. Ann N Y Acad Sci. 2003;996:10–16. doi: 10.1111/j.1749-6632.2003.tb03227.x. [DOI] [PubMed] [Google Scholar]
- Wurm AA, et al. Disruption of the C/EBPalpha-miR-182 balance impairs granulocytic differentiation. Nat Commun. 2017;8(1):46. doi: 10.1038/s41467-017-00032-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agliano AM, et al. On chromosomal instability: what is the karyotype of your 32D CI3 cell line. Blood. 2000;95(11):3636–3637. [PubMed] [Google Scholar]
- Wang GG, et al. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods. 2006;3(4):287–293. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- Houston IB, Huang KJ, Jennings SR, DeKoter RP. PU.1 immortalizes hematopoietic progenitors in a GM-CSF-dependent manner. Exp Hematol. 2007;35(3):374–384. doi: 10.1016/j.exphem.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Calvo KR, Sykes DB, Pasillas M, Kamps MP. Hoxa9 immortalizes a granulocyte-macrophage colony-stimulating factor-dependent promyelocyte capable of biphenotypic differentiation to neutrophils or macrophages, independent of enforced meis expression. Mol Cell Biol. 2000;20(9):3274–3285. doi: 10.1128/mcb.20.9.3274-3285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo KR, Sykes DB, Pasillas MP, Kamps MP. Nup98-HoxA9 immortalizes myeloid progenitors, enforces expression of Hoxa9, Hoxa7 and Meis1, and alters cytokine-specific responses in a manner similar to that induced by retroviral co-expression of Hoxa9 and Meis1. Oncogene. 2002;21(27):4247–4256. doi: 10.1038/sj.onc.1205516. [DOI] [PubMed] [Google Scholar]
- Fossati-Jimack L, et al. Phagocytosis is the main CR3-mediated function affected by the lupus-associated variant of CD11b in human myeloid cells. PLoS One. 2013;8(2):e57082. doi: 10.1371/journal.pone.0057082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwable J, et al. RGS2 is an important target gene of Flt3-ITD mutations in AML and functions in myeloid differentiation and leukemic transformation. Blood. 2005;105(5):2107–2114. doi: 10.1182/blood-2004-03-0940. [DOI] [PubMed] [Google Scholar]
- Worch J, et al. The serine-threonine kinase MNK1 is post-translationally stabilized by PML-RARalpha and regulates differentiation of hematopoietic cells. Oncogene. 2004;23(57):9162–9172. doi: 10.1038/sj.onc.1208164. [DOI] [PubMed] [Google Scholar]
- Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- Stam K, et al. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N Engl J Med. 1985;313(23):1429–1433. doi: 10.1056/NEJM198512053132301. [DOI] [PubMed] [Google Scholar]
- Holly SP, Larson MK, Parise LV. The unique N-terminus of R-ras is required for Rac activation and precise regulation of cell migration. Mol Biol Cell. 2005;16(5):2458–2469. doi: 10.1091/mbc.E03-12-0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JH, et al. Macrophage-colony-stimulating factor (CSF-1) induces proliferation, chemotaxis, and reversible monocytic differentiation in myeloid progenitor cells transfected with the human c-fms/CSF-1 receptor cDNA. Proc Natl Acad Sci U S A. 1990;87(15):5613–5617. doi: 10.1073/pnas.87.15.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JH, et al. Signal transduction through the EGF receptor transfected in IL-3-dependent hematopoietic cells. Science. 1988;239(4840):628–631. doi: 10.1126/science.3257584. [DOI] [PubMed] [Google Scholar]
- Oomen SP, et al. Somatostatin modulates G-CSF-induced but not interleukin-3-induced proliferative responses in myeloid 32D cells via activation of somatostatin receptor subtype 2. Hematol J. 2001;2(5):322–329. doi: 10.1038/sj.thj.6200121. [DOI] [PubMed] [Google Scholar]
- Oomen SP, et al. Somatostatin is a selective chemoattractant for primitive (CD34(+)) hematopoietic progenitor cells. Exp Hematol. 2002;30(2):116–125. doi: 10.1016/s0301-472x(01)00772-x. [DOI] [PubMed] [Google Scholar]
- Nogami A, et al. FLT3-ITD confers resistance to the PI3K/Akt pathway inhibitors by protecting the mTOR/4EBP1/Mcl-1 pathway through STAT5 activation in acute myeloid leukemia. Oncotarget. 2015;6(11):9189–9205. doi: 10.18632/oncotarget.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodel JE, Link DC. Suppression of apoptosis during cytokine deprivation of 32D cells is not sufficient to induce complete granulocytic differentiation. Blood. 1996;87(3):858–864. [PubMed] [Google Scholar]
- Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci U S A. 1988;85(23):9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]