

Abstract
This protocol is a practical guide to the N-methyl-D-glucamine (NMDG) protective recovery method of brain slice preparation. Numerous recent studies have validated the utility of this method for enhancing neuronal preservation and overall brain slice viability. The implementation of this technique by early adopters has facilitated detailed investigations into brain function using diverse experimental applications and spanning a wide range of animal ages, brain regions, and cell types. Steps are outlined for carrying out the protective recovery brain slice technique using an optimized NMDG artificial cerebrospinal fluid (aCSF) media formulation and enhanced procedure to reliably obtain healthy brain slices for patch clamp electrophysiology. With this updated approach, a substantial improvement is observed in the speed and reliability of gigaohm seal formation during targeted patch clamp recording experiments while maintaining excellent neuronal preservation, thereby facilitating challenging experimental applications. Representative results are provided from multi-neuron patch clamp recording experiments to assay synaptic connectivity in neocortical brain slices prepared from young adult transgenic mice and mature adult human neurosurgical specimens. Furthermore, the optimized NMDG protective recovery method of brain slicing is compatible with both juvenile and adult animals, thus resolving a limitation of the original methodology. In summary, a single media formulation and brain slicing procedure can be implemented across various species and ages to achieve excellent viability and tissue preservation.
Keywords: Neuroscience, Issue 132, Brain slice, patch clamp, electrophysiology, NMDG, protective recovery method, synaptic connectivity, neocortex, Na spike-in, multi-neuron recording
Introduction
The acute brain slice preparation is an essential experimental model system in neuroscience. For roughly half of a century, this platform has enabled dynamic functional studies of the living brain across a wide variety of anatomical brain regions and animal species. Whether the intended application is biochemistry, functional imaging, morphology, or electrophysiology, it is of the utmost importance to ensure optimal integrity and viability of the sliced tissue. It is for this reason that the highly resilient juvenile rodent brain slice preparation (i.e., younger than postnatal day 30 for mice) has been the most preferred to date. The difficulty in obtaining sufficiently healthy brain slices from mature adult and aging animals has proven to be a formidable challenge for most and has imposed severe limitations for studying the functional architecture of the mature brain. This is particularly true for patch clamp recording, a technique that requires excellent morphological and functional preservation and is indispensable for characterizing detailed intrinsic and synaptic properties of identified single neurons. For the past several decades, the vast majority of patch clamp electrophysiologists have relied on a 'protective cutting' method using sucrose-substituted low Na+ aCSF1 for preparing healthy brain slices from juvenile, and to a far lesser extent, young adult animals. This method is based on the premise that passive Na+ influx and subsequent water entry and cell swelling during the slice cutting step is the predominant insult that leads to poor survival of neurons, particularly for those neurons located in the superficial layers that are most likely to sustain direct trauma from the blade movement. However, the protective cutting method still leaves much to be desired for brain slice preparation from mature adult animals regardless of the particular aCSF formulation implemented.
A simple but effective solution to this problem has been described2,3,4,5,6 and termed the 'protective recovery' brain slice method. The original version of this method uses an NMDG-substituted aCSF, as NMDG was identified as the most versatile and effective among various other candidate sodium ion substitutes (including sucrose, glycerol, choline, and Tris). The media formulation was further enhanced by addition of HEPES to resist brain slice edema and provide stronger pH buffering7, as well as the addition of supplements to counteract the detrimental effects of oxidative stress (Table 1). It was empirically determined that an initial recovery incubation step in low Na+, low Ca2+, and high Mg2+ NMDG aCSF immediately after adult brain tissue slicing was both necessary and sufficient for improved neuronal preservation over a wide range of brain regions, cell types, and animal ages3,5,6.
Notably, earlier incarnations of what is now dubbed the protective recovery method can be found in the literature1,8,9,10,11,12,13, although the full potential for mature adult and aging animal brain slice and patch clamp recording was not recognized or demonstrated in these earlier works. In addition, nuanced procedural variations continue to emerge in support of specific experimental applications4,14,15,16. The collective body of work of these numerous research groups imparts high confidence in the robustness of the protective recovery method for improved tissue preservation. The NMDG protective recovery method has now been widely adopted and implemented in numerous published research studies utilizing adult animal brain slice preparations. These acute slice studies span neocortical3,17,18, hippocampal15,19,20,21, striatal22,23,24, midbrain25,26,27,28,29, and hindbrain30,31,32,33,34 regions, and a variety of neurotransmitter and neuromodulator types including glutamatergic4,30, GABAergic18,20,31,35,36, dopaminergic24,29,37,38, cholinergic14,37,38,39, noradrenergic40, and serotonergic27,28 neurotransmission. The method is also well suited for optogenetic control of neuronal activity in slices derived from transgenic animals3,39 or following in vivo viral injections17,27,28,40,41,42,43, as well as functional Ca2+ imaging of neuronal activity2,44,45,46. Analyses of both short term plasticity4,47,48 and diverse forms of long-term plasticity16,35,48 have been reported. A recent study applied the NMDG protective recovery method to facilitate extensive and systematic probing of synaptic connectivity in the visual cortex in mature adult mouse brain slices using the octopatch recording configuration49 — a powerful demonstration of the utility and robustness of this method. The protective recovery method has even been applied successfully in previously unforeseen experimental contexts, such as, improved preservation of vasculature and pericytes in adult cortical brain slices50, patch clamp recording from transplanted interneuron populations in 1–1.5 year old Alzheimer's Disease mouse models20, and an adult brain slice receptor trafficking assay51.
The following protocol describes step-by-step procedures for implementing an optimized NMDG protective recovery method of brain slice preparation to improve the viability of acute brain slices. The principles for improved neuronal preservation are discussed, as well as demonstration of the clear benefits of this methodology for complex multi-neuron patch clamp recording experiments in both young adult transgenic mouse brain slices and mature adult human neurosurgical brain slices. The following protocol has been validated for mice from 21 days old to more than one years old, as well as for human neurosurgical specimens derived from adult patients.
Protocol
Procedures involving transgenic mice have been approved by the Institutional Animal Care and Use Committee (IACUC) at the Allen Institute for Brain Science. Both male and female C57BL/6 mice (weight range 10-30 g) were used in these experiments. Some of the representative results describe data collected from living human brain slices. Neocortical tissue specimens were obtained during neurosurgeries for tumor removal. It was necessary to remove the overlying neocortical tissue to gain access to the diseased tissue. Informed patient consent was obtained in all cases for the use of neurosurgical tissue for research purposes under a protocol approved by the institutional review board of Swedish Medical Center.
1. Preparation of Media and Reagents (Table 1)
NOTE: Solutions should be made up in purified water that is free of trace metals and other impurities. It is recommended that solutions be made freshly on the day of the experiment, although unused solutions may be stored at 4 °C for up to 1 week, if desired. 1 L of each formulation above is sufficient for 1–2 slicing procedures. All aCSF solutions must be saturated with carbogen (95% O2/5% CO2) prior to use to ensure stable pH buffering and adequate oxygenation. The pH of all solutions should be adjusted to 7.3-7.4 and osmolality measured and adjusted to 300-310 mOsmol/kg.
Prepare NMDG-HEPES aCSF (in mM): 92 NMDG, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2·2H2O, and 10 MgSO4·7H2O. Titrate pH to 7.3–7.4 with 17 mL +/- 0.5 mL of 5 M hydrochloric acid. NOTE: This titration step should ideally be performed prior to the addition of divalent cations to avoid precipitation; however, the precipitation can be reversed upon adjustment of the pH to the physiological range.
Prepare HEPES holding aCSF (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2·2H2O, and 2 MgSO4·7H2O. Titrate pH to 7.3–7.4 with several drops of concentrated 10 N NaOH.
Prepare recording aCSF (in mM): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 12.5 glucose, 5 HEPES, 2 CaCl2·2H2O, and 2 MgSO4·7H2O. Titrate pH to 7.3-7.4 with a few drops of concentrated 10 N NaOH.
Prepare Na+ spike-in solution (2 M): 580 mg of NaCl dissolved in 5 mL of freshly prepared NMDG-HEPES aCSF. This is enough solution for one brain slice prep.
Prepare 2% agarose to be used for tissue embedding. Dissolve 2 g of agarose type 1B (see Table of Materials) in 100 mL of 1x PBS and microwave until just boiling. Swirl to mix, then pour the mixture into a sterile 10 cm Petri dishes and allow to solidify. Store the agarose plate in a sealed plastic bag at 4 °C until use.
Prepare injectable anesthetic working stock solution. Mix 2.5 g of 2,2,2-Tribromoethanol with 5 mL of 2-methyl-2-butanol and then gradually dissolve in 200 mL PBS, pH 7.0–7.3. Filter the stock solution with a 0.22 µm filter before use and store at 4 °C protected from light. NOTE: Consult respective animal use committee guidelines and rules for determining the expiration and disposal procedure for the anesthetic working stock solution.
2. Setup of the Slicing Station
Set up the slicing station with the tissue slicer machine and surgical instruments (see Table of Materials). To calibrate the slicer machine, attach a zirconium ceramic injector blade to the blade arm using fast-adhesive glue, then insert the specimen holder and align the leading edge of the blade to the specimen holder rim leaving a tiny gap to ensure the blade does not scrape the metal. NOTE: If the blade edge is not physically damaged it can be reused for many weeks or even months without replacement.Various tissue slicer models are commercially available, many of which can provide excellent performance when optimally calibrated. The ideal instrument should have minimal z-axis deflection, either directly measured and tuned or empirically observed.
Set up a 250 mL beaker filled with 200 mL of NMDG-HEPES aCSF and pre-chill on ice with constant carbogenation (applied via a gas diffuser stone immersed in the media) for >10 min. NOTE: This solution will be used for transcardial perfusion and for filling the slicing machine reservoir during sectioning.
Set up the initial brain slice recovery chamber filled with 150 mL of NMDG-HEPES aCSF (maintain constant carbogenation) and place the chamber in a heated water bath maintained at 32–34 °C. NOTE: A slice chamber after the design of Edwards and Konnerth (1992)52 is recommended for this step. These chambers can be made with readily available laboratory items (250 mL beaker, nylon mesh netting, 50 mL conical tube, 35 mm plastic round dish). Care must be taken to ensure that the slice netting remains free of air bubbles, particularly those continuously produced by the carbogen gas bubble stones as these can cause slices to float up and become damaged. The netting should be submerged roughly 1 cm under the liquid surface.
Set up a brain slice holding chamber; a design with multiple independent wells in a larger reservoir is recommended (see Table of Materials). Fill the reservoir with 450 mL of HEPES aCSF and warm to room temperature under constant carbogenation until use. NOTE: The brain slices will be transferred from the initial recovery chamber to this chamber for long-term storage before electrophysiological recording. Care must be taken to ensure that the slice netting remains free of air bubbles at all times.
Prepare molten agarose for tissue embedding. Use the open end of a 50 mL conical vial like a cookie cutter to cut out a block of 2% agarose from the previously prepared dish. Loosely cap the conical vial, then microwave for 10–30 s until the agarose is just melted. Do not overheat.
Pour the molten agarose into 1.5 mL tubes. Maintain the agarose in the molten state using a thermomixer set to 42 °C with vigorous shaking. Carefully ensure that the molten agarose does not solidify prematurely.
Place the accessory chilling block for the slicer on ice to pre-cool at this time.
3. Transcardial Perfusion
NOTE: The transcardial perfusion procedure is an important step when working with adult animals and is important to achieve rapid cooling of the brain and slowed metabolism via brain infusion of low Na+, low Ca2+/high Mg2+ aCSF solution. The transcardial perfusion also serves to clear red blood cells from brain vasculature, which reduces autofluorescence that might interfere with visualization and targeting of fluorescently labeled cell populations in transgenic lines. It is not advisable to omit transcardial perfusion.
Deeply anesthetize mice by intraperitoneal injection of anesthetic working stock solution (250 mg/kg: 0.2 mL of 1.25% anesthetic working stock solution per 10 g body weight, see the Table of Materials). After ~2–3 min, verify sufficient depth of anesthesia by assessing toe pinch reflex. If required, inject an additional volume of anesthetic working stock and reassess toe pinch reflex after another 2-3 min.
Load a 30 mL syringe with 25 mL of carbogenated NMDG-HEPES aCSF from the pre-chilled 250 mL beaker (2–4 °C is optimal, as opposed to slushy or frozen solution). Attach a 25 5/8 gauge needle.
With the mouse on its back, pin down the forepaws and hind paws for stability. A 15 cm glass Petri dish filled with hardened silicone works well as the base.
Using a scalpel, make a lateral incision to open the thoracic cavity at the level of the diaphragm. Use fine scissors to cut through the rib cage on either side taking care to avoid clipping the heart and lungs.
Pin back the center portion of the rib cage to expose the heart. Insert the needle of the 30 mL syringe into the left ventricle and cut the right atrium with fine scissors to allow blood to exit the heart.
Depress the syringe plunger using manual constant pressure and perfuse the animal with the chilled NMDG-HEPES aCSF at a rate of ~10 mL/min. NOTE: If the perfusion is successful the liver will change in color from deep red to pale yellow, and in some cases clear fluids can be observed exiting the nostrils towards the end of the procedure.
4. Brain Dissection and Slicing
Decapitate the animal. Use a scalpel to open the skin on the head and expose the skull cap.
Use fine super-cut scissors to cut away the skin over the skull cap and make small incisions laterally on either side at the caudal/ventral base of the skull. Make additional shallow cuts starting at the caudal/dorsal aspect of the skull moving in the rostral direction up the dorsal midline taking care not to damage the underlying brain. Make a final 'T' cut perpendicular to the midline at the level of the olfactory bulbs. NOTE: Care must be taken to ensure no damage is done to the brain region(s) of interest. In particular, at no time should there be any compressive force applied to the brain itself.
Use the round-tip forceps to grasp the skull starting at the rostral-medial aspect and peel back towards the caudal-lateral direction. Repeat for both sides to crack open and remove the dorsal halves of the skull cap to expose the brain. Gently scoop out the intact brain into the beaker of pre-chilled NMDG-HEPES aCSF. Allow the brain to uniformly cool for ~1 min.
Use the large spatula to lift the brain out of the beaker and onto the Petri dish covered with filter paper. Trim and block the brain according to the preferred angle of slicing and desired brain region of interest. Work quickly to avoid prolonged oxygen deprivation during handling. NOTE: Many slicing angles are possible. The exact blocking method and slicing angle will depend on the exact brain region, cell type, and circuit to be studied.
Affix the brain block to the specimen holder using adhesive glue. Retract the inner piece of the specimen holder enough to withdraw the brain block fully inside. Pour the molten agarose directly into the holder until the brain block is fully covered in agarose. Clamp the pre-cooled accessory chilling block around the specimen holder for ~10 s until the agarose has solidified.
Insert the specimen holder into the receptacle on the slicer machine and verify proper alignment. Fill the reservoir with remaining pre-chilled, oxygenated NMDG-HEPES aCSF from the 250 mL beaker and move a bubble stone into the reservoir for the duration of slicing to ensure adequate oxygenation.
Adjust the micrometer to begin advancing the agarose-embedded brain specimen. Start the slicer and empirically adjust the advance speed and oscillation frequency to the desired level. NOTE: Both settings should be in the low range. For best results, a single pass of the blade arm should take approximately 20 s and the oscillation should produce a very smooth and gentle humming noise with no overt buzzing.
Continue advancing and slicing the tissue in 300 µm increments (or other preferred thickness) until the brain region of interest is fully sectioned; the total time for the slicing procedure should be less than 15 min.
5. Optimized NMDG Protective Recovery Procedure
Initial NMDG recovery step (critical step): Upon completion of the sectioning procedure, collect up all of the slices using a cut-off plastic Pasteur pipe t and transfer them into a pre-warmed (34 °C) initial recovery chamber filled with 150 mL of NMDG-HEPES aCSF. Transfer all slices in short succession and start a timer as soon as all slices are moved into the recovery chamber.
Consult Table 2 to determine the optimal Na+ spike-in schedule according to mouse age. NOTE: This procedure is a practical method to achieve a controlled rate of reintroduction of Na+ into the brain slice chamber and is optimized for a specific brain slice chamber geometry and reservoir volume and type (see Table of Materials).
Carry out the stepwise Na+ spike-in procedure by adding the indicated volumes of Na+ spike-in solution at the indicated times. Add the Na+ spike-in solution directly into the bubbler chimney of the initial recovery chamber to facilitate rapid mixing.
Transfer all slices to the HEPES aCSF long-term holding chamber maintained at room temperature. Allow slices to recover for an additional 1 h in the HEPES holding chamber prior to initiating patch clamp recording experiments.
6. Patch Clamp Recording
NOTE: The following basic procedures merely provide some practical considerations and are not intended to represent detailed protocols for patch clamp recordings, as these can be found elsewhere53,54. A patch clamp electrophysiology rig is required for this application. This will generally be composed of an upright microscope equipped with infrared differential interference contrast (IR-DIC) optics and a fluorescence illumination system, a patch clamp amplifier and data digitizer, motorized micromanipulator and microscope platform, vibration isolation table, Faraday cage, and solution heating and perfusion system. The sample chamber and platform should be designed for submerged slice recording. For multi-neuron patch clamp recordings, a rig equipped with multiple amplifiers, head stages, and high quality micromanipulators is required. In addition, for best results, a rig equipped with a 900 nm IR band pass filter and matching optical components is highly recommended to ensure adequate visualization of cells located >50 µm deep in the brain slices. Proper alignment for Kӧhler illumination is also important for clear visualization.
Prepare intracellular pipette solution (in mM): 130 K-Gluconate, 4 KCl, 10 HEPES, 0.3 EGTA, 10 phosphocreatine-Na2, 4 MgATP, 0.3 Na2-GTP, and 13.4 biocytin. Adjust the pH to 7.35 with 1 M KOH and the osmolality to 285–290 mOsmol/kg using sucrose as needed.
Prepare patch clamp electrodes from thick-walled borosilicate glass capillaries; the ideal patch clamp electrode has a relatively short and stubby taper with a ~3–6 MOhm filled tip resistance in the bath.
Ensure that the silver electrode wires are properly chlorided in order to ensure stable recordings. Do this (typical) by submerging the last 3-4 mm of the silver wire in liquid bleach for approximately 30 min or until the wire turns black.
Establish solution perfusion using a peristaltic pump set to 3–4 mL/min. Circulate carbogenated recording aCSF through the recording chamber taking care to match inflow and outflow to avoid overflow.
Transfer a single brain slice into the submersion recording chamber and secure in place using a U-shaped slice anchor with nylon cross strings. Identify the target brain region using a 4X air objective before switching to a higher power objective (e.g., high numerical aperture 40X or 60X water immersion objectives).
Visually identify a healthy target cell. The appearance of the neuronal membrane at the soma, as visualized by IR-DIC microscopy, is used to judge the suitability of a candidate cell for patch clamp recording. NOTE: Healthy neurons typically exhibit the following features: neither shrunken nor swollen somata, soft contrast of membrane edges, and smooth membrane appearance. In addition, the majority of healthy cells are located >30 µm deep in the slice, as superficial neurons are likely to be damaged with severed dendritic processes. Neurons that exhibit a crinkled appearance, clearly visible nuclei or 'fried egg' appearance, or very dark or high contrast membrane edges should be avoided.
Back-fill the patch pipette with ~5 µL of intracellular solution and load it onto the electrode holder. Move the pipette into the recording bath above the brain slice and apply light positive pressure to clear any obstructions in the tip. Zero the pipette offset and monitor the tip resistance using a membrane test function.
Move the pipette tip into contact with the target neuron's cell body; a small dimple should form on the membrane surface due to the light positive pressure.
As soon as a membrane dimple is observed, swiftly remove the positive pressure and apply gentle suction to facilitate seal formation. Once the pipette resistance increases to >100 MOhm, turn on a holding command to a level that matches the anticipated resting membrane potential for the targeted cell type (-70 mV is a good starting point).
Once the tip resistance reaches ≥1 gigaohm, attempt to break into the cell by rupturing the membrane underneath the patch pipette using brief bouts of sharp suction; the 'zap' feature may be utilized to facilitate break-in as needed. NOTE: A holding potential of -70 mV is suggested for voltage clamp experiments on cortical neurons. Healthy neurons will have a leak current no more negative than -100 pA for the duration of the experiment, but this is in part dependent on the cell type. A neuron would be excluded from analysis if the resting membrane potential was more depolarized than -50 mV, or if the access resistance changed by more than 20%.
- Once a stable whole-cell patch clamp recording is obtained, target additional neurons for recording by repeating steps 6.6–6.10. Take care to avoid mechanical disturbances that would result in losing the first recording.
- Select additional neurons within 100 µm from the first neuron to ensure a reasonable probability of finding synaptically-coupled neurons. NOTE: It may be helpful in some cases to identify multiple candidate neurons up front and to pre-load and pre-position all pipettes nearby to the various targeted cells before establishing the first patch clamp recording.
Representative Results
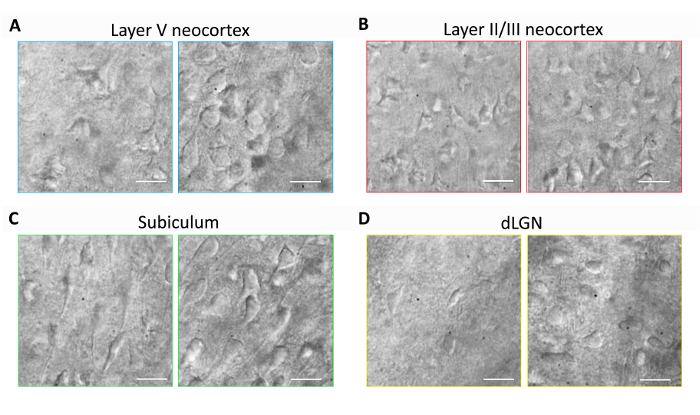
This section provides representative results for routine brain slice preparation and patch clamp electrophysiology experiments using the optimized NMDG protective recovery method (i.e., NMDG protective recovery combined with gradual Na+ spike-in procedure). First, morphological preservation of neurons was evaluated in various brain regions of brain slices prepared with or without implementing the optimized NMDG protective recovery method (Figure 1). Three month old adult mice were selected for these experiments, and we used IR-DIC microscopy to determine neuron health based on the shape and overall appearance of the somata and proximal dendrites. Note the shriveled, pyknotic appearance of most neurons in the representative images of brain slices prepared without the protective recovery method (all images were obtained 1–2 h after slice preparation). These control slices were prepared using NMDG aCSF for transcardial perfusion and slicing steps but were initially recovered in high Na+-containing HEPES aCSF. In contrast, the representative images from the slices prepared using the optimized NMDG protective recovery method reveal neurons with improved morphologies (smoother, fuller, less crinkled appearance) that are suitable for patch clamp recording (Figure 1). The improved neuronal preservation was observed across multiple brain regions including neocortical layers II/III and V, subiculum, and dorsal lateral geniculate nucleus (dLGN).
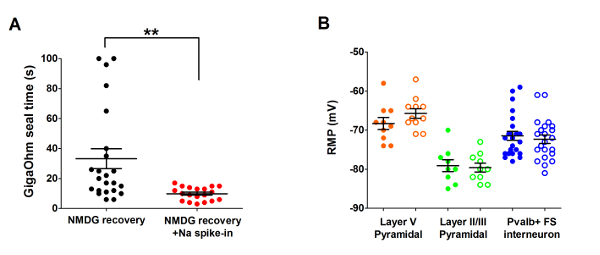
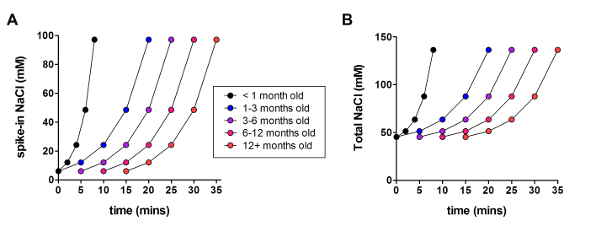
Next, the optimized NMDG protective recovery method was compared with the original NMDG protective recovery method (i.e., without the gradual Na+ spike-in procedure). The average time for gigaohm seal formation in patch clamp recording attempts was dramatically and significantly reduced (9.9 s versus 33.3 s, **p <0.005, paired t-test) when the gradual Na+ spike-in procedure was applied together with the NMDG protective recovery step (Figure 2). The faster and more reliable membrane sealing times greatly improved the throughput of patch clamp recording in young adult brain slices. The optimal Na+ spike-in schedule was further modified according to animal age (Table 2) and was beneficial for all ages tested (3 weeks to 1 year old mice). The profile of gradual sodium ion concentration elevation throughout the course of the spike-in procedure is provided (Figure 3) to accompany the schedules shown in Table 2.
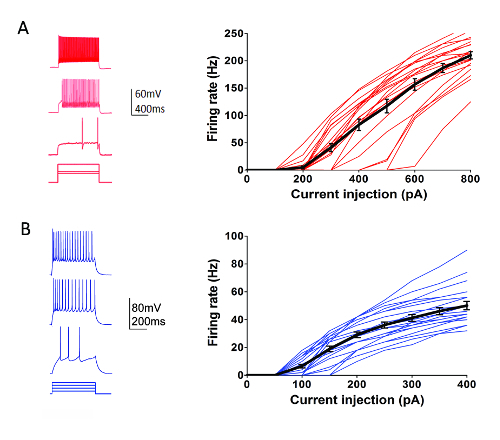
As part of the Allen Institute Cell Types Program (http://celltypes.brain-map.org/) a large scale effort is underway to systematically characterize the intrinsic electrophysiological properties of individual neurons in young adult (postnatal day 40-80) mouse visual cortical brain slices derived from transgenic lines with cell type-specific fluorescent marker expression in genetically-defined neuronal populations (cortical layer and cell type specific Cre driver lines crossed to a Cre-dependent fluorescent reporter line55). Figure 4 shows example traces of the firing patterns recorded from Parvalbumin (Pvalb)-expressing cortical fast-spiking (FS) interneurons (Pvalb-IRES-Cre/Ai14 mice) in response to a series of 1 s current injection steps that cover the dynamic range of neuron firing. The F-I curve for a dataset of 22 cortical FS interneurons is shown at right. Similar targeted patch clamp recording experiments were performed to characterize 23 Rorb-expressing excitatory neurons in layer IV from Rorb-IRES-Cre/Ai14 mice (Figure 4). Diverse healthy neuron types including FS interneurons and pyramidal neurons across cortical regions and layers can routinely and reliably be targeted for patch clamp recording for at least 6-8 h after slice preparation using this optimized protocol.
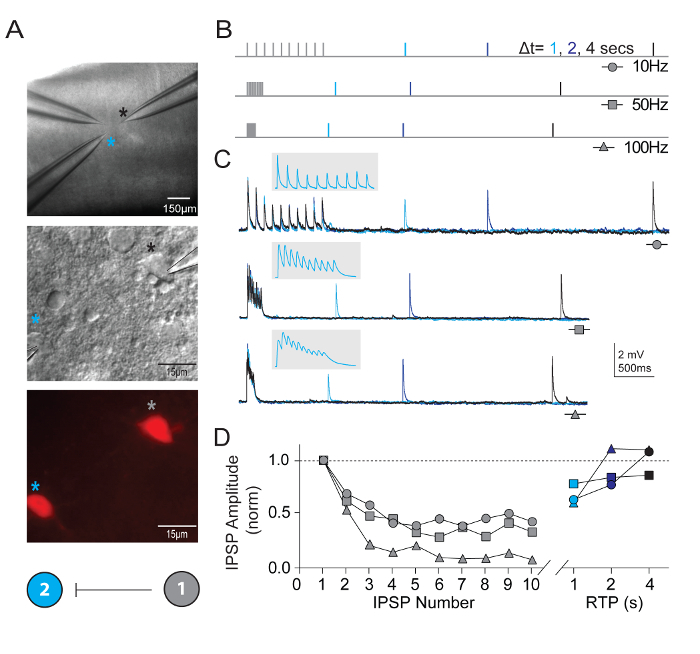
In addition to measuring intrinsic neuronal properties, synaptic connectivity was probed between multiple simultaneously recorded neurons of defined types in visual cortical microcircuits. The multi-neuron patch clamp recording technique is exceptionally demanding, as numerous healthy candidate neurons of defined types must be present within a relatively small field of the brain slice in order to ensure a reasonable chance of obtaining high quality simultaneous recordings and identifying bona fide synaptic connections. Figure 5 shows paired recording of two tdTomato+ FS interneurons in the visual cortex of brain slices derived from young adult Pvalb-IRES-Cre/Ai14 mice. A strong unidirectional inhibitory synaptic connection was detected (recorded with high chloride internal pipette solution). Example recordings and protocols for measuring properties of short-term synaptic plasticity are presented. Bouts of high frequency train stimulation (10 pulses each at 10, 50, and 100 Hz) were followed by single recovery test pulses at various time intervals (1, 2, or 4 s) to measure the time course of recovery from synaptic depression.
Excellent success has also been obtained for human neocortical neurons in mature adult ex vivo brain slices. Neurosurgical specimens are obtained from patients undergoing scheduled surgeries for tumor removal at local hospitals. The procedures for human neurosurgical tissue collection and brain slice preparation differ from the mouse brain slice procedures in a few practical ways. In brief, the resected neocortical tissue (distal to the site of pathology) is collected from the operating room and immersed into ice-cold oxygenated NMDG-HEPES aCSF and transported with continuous chilling and oxygenation from the operating room to the laboratory within 30 min or less. The brain slices are prepared using the NMDG protective recovery procedure and allowed to recover for an extended time of approximately 3 h before initiating patch clamp recordings. Figure 6 shows a successful paired recording experiment and a successful quadruple patch clamp recording experiment from human ex vivo brain slices prepared in this manner from the frontal cortex region. The paired recording demonstrates unidirectional excitatory synaptic input from a cortical pyramidal neuron onto a cortical interneuron (recorded as excitatory postsynaptic currents). In the quad patch experiment two excitatory and two inhibitory neurons were recorded simultaneously, and three inhibitory synaptic connections were detected (recorded as inhibitory postsynaptic potentials) out of twelve total connections probed. Thus, this optimized brain slice methodology allows reliable experimental success in the most challenging of brain slice applications, including multi-neuron patch clamp experiments to study circuit connectivity in acutely resected mature adult human brain tissue.
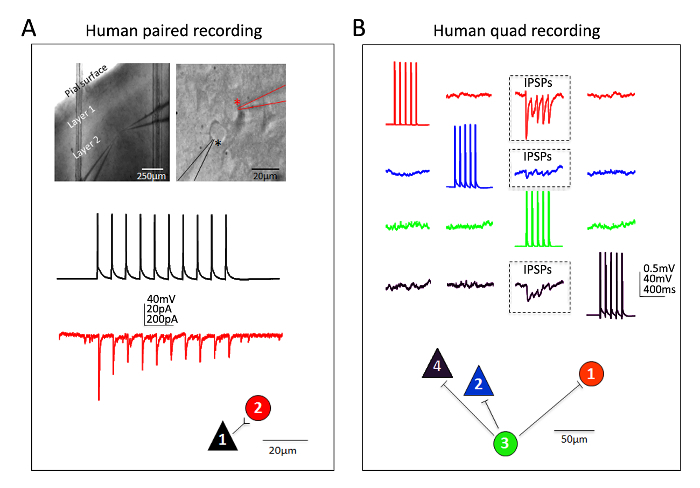
Figure 1: Improved neuronal preservation with the optimized NMDG protective recovery method of brain slice preparation. Representative IR-DIC images were acquired from diverse brain regions in acute slices from a three month old mouse. Control NMDG protective cutting method without a protective recovery step (left panels) versus optimized NMDG protective recovery method (right panels). (A) Layer V of neocortex, (B) Layer II/III of neocortex, (C) subiculum, and (D) dorsal lateral geniculate nucleus (dLGN). Scale bars in all panels are 20 µm. Please click here to view a larger version of this figure.
Figure 2: Accelerated speed and improved reliability of gigaohm seal formation in patch clamp recording experiments using the NMDG protective recovery method with Na+ spike-in procedure. (A) Plot of gigaohm seal formation times for NMDG recovery alone (black data points, n = 19) versus NMDG recovery plus Na+ spike-in procedure (red data points, n = 23). All recorded cells were pyramidal neurons in layer II/III or V of visual cortex. Note, the maximal time is capped at 100 s to account for cells that never formed gigaohm seals. Paired t-test, **p <0.005. (B) Plot of resting membrane potential (RMP) for layer V pyramidal neurons (orange data points, n = 10/11), II/III pyramidal neurons (green data points, n = 9/10), or cortical Pvalb+ FS interneurons (blue data points, n = 23/22). Solid circles denote NMDG recovery condition and open circles NMDG recovery plus Na+ spike-in procedure. Each data point represents one neuron and both the mean and +/-SEM are displayed. There are no significant differences in RMPs comparing brain slice preparation conditions for all three cell types (paired t-test). Please click here to view a larger version of this figure.
Figure 3: Profile of gradual sodium ion concentration elevation throughout the course of the spike-in procedure. (A) Plot of spike-in NaCl concentration versus time. (B) Plot of total extracellular NaCl concentration versus time. Please click here to view a larger version of this figure.
Figure 4: Intrinsic electrophysiological properties of genetically-defined cortical cell types. (A) Example traces of neuronal firing in response to current injection steps for Pvalb+ cortical FS interneurons (left panel). tdTomato+ neurons were targeted for recordings in brain slices from Pvalb-IRES-Cre/Ai14 mice. Summary data for firing rate-current injection relationship (F-I curve) are shown at right (n = 22). (B) Example traces of neuronal firing in response to current injection steps for Rorb-expressing cortical layer IV excitatory neurons (left panel). tdTomato+ neurons were targeted for recordings in brain slices from Rorb-IRES-Cre/Ai14 mice. Summary data for firing rate-current injection relationship (F-I curve) are shown at right (n = 23). Each thin colored line represents the F-I curve for a single neuron; whereas, the thick black lines represents the average for each group +/-SEM. Please click here to view a larger version of this figure.
Figure 5: Short-term plasticity in synaptically connected Pvalb-expressing cortical FS interneurons. (A) Differential interference contrast and epifluorescence were used to target tdTomato-positive, Pvalb-expressing FS interneurons of mouse primary visual cortex. A color coded schematic connectivity diagram demonstrating a unilateral synaptic connection between the two interneurons. (B) Schematic of the stimulation protocols used to assess short-term dynamics of synaptically-connected neurons. Trains of 10 action potentials (10, 50, and 100 Hz) are evoked in the presynaptic neuron, followed by a single action potential (Recovery Test Pulse, RTP) delivered with different time delay (1, 2, and 4 s) after the train has terminated. Each RTP are color coded for clarity. (C) Average traces of corresponding unilateral inhibitory post-synaptic potentials (uIPSPs) in cell #2 in response to trains of action potentials evoked in cell #1. Gray inset has expanded time scale to show delineation of uIPSP train. (D) Normalized uIPSP amplitudes are plotted as a function of their position during trains at varying frequencies. Net depression is clear across all input rates with a substantial recovery of uIPSP at 4 s. Please click here to view a larger version of this figure.
Figure 6: Multi-neuron patch clamp recordings in adult human neurosurgical brain slices. (A) Low and high magnification IR-DIC images indicating the location and identity of the recorded neurons (top panels). A pyramidal neuron (black asterisk) and neighboring interneuron (red asterisk) were recorded simultaneously. Example color coded traces of a unidirectional excitatory synaptic connection (ESPC) from pyramidal cell to interneuron (measured as EPSCs in voltage clamp) and corresponding physical connection map (bottom panels). (B) Quadruple patch clamp recording experiment in an adult human neurosurgical brain slice of dorsolateral prefrontal cortex. Two pyramidal neurons and two interneurons are recorded simultaneously, allowing for sequential probing of twelve possible synaptic connections. A train of evoked action potentials in cell #3 (green traces) led to detection of inhibitory post-synaptic potentials (IPSPs) in each of the other three simultaneously recorded neurons (three boxed regions, top panels). The responses recorded from each individual neuron are color coded for clarity. The physical connection map is shown in the bottom panel. Note the raw traces shown in (B) represent the averages of at least 20 consecutive raw data sweeps. Putative identification of cell type was based on soma shape, analysis of morphology by fluorescent dye filling during recordings, and intrinsic electrophysiological properties including the firing patterns in response to current injection steps. Please click here to view a larger version of this figure.
| NMDG-HEPES aCSF | HEPES holding aCSF | Recording aCSF | |||||||
| Component | mM | MW | g/Liter | mM | MW | g/Liter | mM | MW | g/Liter |
| NMDG | 92 | 195.22 | 17.96 | ||||||
| HCl | 92 | 36.46 | * | ||||||
| NaCl | 92 | 58.44 | 5.38 | 124 | 58.44 | 7.25 | |||
| KCl | 2.5 | 74.55 | 0.19 | 2.5 | 74.55 | 0.19 | 2.5 | 74.55 | 0.19 |
| NaH2PO4 | 1.2 | 138.00 | 0.17 | 1.2 | 138.00 | 0.17 | 1.2 | 138.00 | 0.17 |
| NaHCO3 | 30 | 84.01 | 2.52 | 30 | 84.01 | 2.52 | 24 | 84.01 | 2.02 |
| HEPES | 20 | 238.31 | 4.77 | 20 | 238.31 | 4.77 | 5 | 238.31 | 1.19 |
| Glucose | 25 | 180.20 | 4.51 | 25 | 180.20 | 4.51 | 12.5 | 180.20 | 2.25 |
| sodium ascorbate | 5 | 198.00 | 0.99 | 5 | 198.00 | 0.99 | 0 | 198.00 | 0.00 |
| Thiourea | 2 | 76.12 | 0.15 | 2 | 76.12 | 0.15 | 0 | 76.12 | 0.00 |
| sodium pyruvate | 3 | 110.04 | 0.33 | 3 | 110.04 | 0.33 | 0 | 110.04 | 0.00 |
| MgSO4.7H2O | 10 | 246.48 | 5 mL | 2 | 246.48 | 1 mL | 2 | 246.48 | 1 mL (2M Stock) |
| CaCl2.2H2O | 0.5 | 147.01 | 0.25 mL | 2 | 147.01 | 1 mL | 2 | 147.01 | 1 mL (2M stock) |
| * titrate pH of NMDG-HEPES aCSF to 7.3-7.4 using concentrated HCl | |||||||||
| All solutions should be in the range 300-310 mOsm/Kg |
Table 1: Media formulations.
| Animal Age | |||||
| Time (min)* | <1 month | 1-3 months | 3–6 months | 6–12 months | 12+ months |
| 0 | 250 µL | 250 µL | |||
| 1 | |||||
| 2 | 250 µL | ||||
| 3 | |||||
| 4 | 500 µL | ||||
| 5 | 250 µL | 250 µL | |||
| 6 | 1000 µL | ||||
| 7 | |||||
| 8 | 2000 µL | ||||
| 9 | |||||
| 10 | transfer | 500 µL | 250 µL | 250 µL | |
| 11 | |||||
| 12 | |||||
| 13 | |||||
| 14 | |||||
| 15 | 1000 µL | 500 µL | 250 µL | 250 µL | |
| 16 | |||||
| 17 | |||||
| 18 | |||||
| 19 | |||||
| 20 | 2000 µL | 1000 µL | 500 µL | 250 µL | |
| 21 | |||||
| 22 | |||||
| 23 | |||||
| 24 | |||||
| 25 | transfer | 2000 µL | 1000 µL | 500 µL | |
| 26 | |||||
| 27 | |||||
| 28 | |||||
| 29 | |||||
| 30 | transfer | 2,000 µL | 1,000 µL | ||
| 31 | |||||
| 32 | |||||
| 33 | |||||
| 34 | |||||
| 35 | transfer | 2,000 µL | |||
| 36 | |||||
| 37 | |||||
| 38 | |||||
| 39 | |||||
| 40 | transfer | ||||
| *Time zero is the moment slices are transferred into the initial recovery chamber |
Table 2: Recommended schedule for gradual Na + spike-in procedure according to mouse age.
Discussion
Na+ Spike-in Improves Gigaohm Seal Formation and Patch Clamp Recording Success The initial version of the NMDG protective recovery method was specifically designed for adult and aging animals2,5. Some early adopters have also sought to apply this methodology to juvenile animal brain slicing (i.e., mice <30 days old). However, it has been noted that in contrast to the outstanding visually-confirmed neuronal preservation with the NMDG protective recovery method in this age range, gigaohm seal formation can frequently stall out, leading to failed patch clamp recording attempts. One hypothesis is that NMDG cations are more readily trapped in juvenile brain slices relative to adult brain slices and can impede seal formation; however, gigaohm seals can readily form while juvenile brain slices are fully submerged in NMDG aCSF (data not shown), thus indicating that NMDG aCSF per se is not impeding gigaohm seal formation.
The rapid transition from low-to-high Na+ solution at the completion of the initial brain slice recovery step causes damage to neuronal membranes and perturbs the seal formation process. This is intuitive given that the transition from low-to-high Na+, cold-to-warm temperature, and dramatic elevation of the Ca2+ to Mg2+ ratio collectively lead to a massive resurgence of spontaneous synaptic activity. This inhibitory rebound phase in the brain slicing procedure is likely to mirror reperfusion injury following an ischemic insult. Thus, to further mitigate neuronal membrane damage in the initial recovery phase a gradual Na+ spike-in procedure has been incorporated in which the elevation of Na+ concentration in the NMDG protective recovery incubation chamber is slowly and reproducibly elevated with precise timing. As in the original protective recovery procedure, the temporal dissociation of Na+ elevation from temperature and Ca2+/Mg2+ ratio elevation is beneficial. But additionally, the Na+ spike-in procedure leads to small incremental increases in extracellular Na+ concentration over the early time points and large increases towards the late time points, thereby affording the brain tissue an opportunity to better accommodate to the rising Na+ levels. This procedure is an alternative to gradual solution exchange controlled by a perfusion pump or gravity drip lines which lead to constant increases in Na+ levels and require attention to both inflow and outflow to avoid overflow of the slice chamber. Notably, in this Na+ spike-in procedure the osmolality of the solution in the slice chamber gradually rises over a period of several minutes before the slices are returned to normal osmolality solution, but this did not adversely affect the slice health or patch clamp recording success. A high osmolality cutting solution has previously been used for midbrain slice preparations in order to better preserve dopamine neurons for patch clamp recordings57,58, thus demonstrating that this temporary hyperosmolality may be beneficial in some contexts.
By implementing an optimized procedure combining the NMDG protective recovery method and gradual Na+ spike-in step the utility of this brain slice methodology has been extended to cover juvenile through mature adult animal ages. This updated protocol is now suitable for a wide range of animal ages using a single optimal NMDG aCSF formulation and procedure. If necessary, the Na+ spike-in procedure can be applied with a progressively longer delay and/or slower time course to enhance the viability of brain slices from older animals, and we have provided a basic guide of recommended spike-in schedules according to animal age (see Table 2). While we have provided a basic framework suitable for a wide array of applications, additional advanced steps can be explored for further enhancing viability and longevity of brain slices from adult and aging animals. For example, glutathione restoration strategies are particularly effective in this respect and can be implemented as described elsewhere2,6.
Improving Throughput for Challenging Experiments The analysis of synaptic connectivity by patch clamp recording is a demanding application that requires excellent preservation of both neuronal structure and function in order to achieve a high reliability of success. As the number of neurons to be simultaneously recorded goes up linearly, the technical difficulty level goes up supra-linearly. There are numerous failure modes, and one of the most frequent causes of failures is the inability to form adequate gigaohm seals onto one or more of the targeted cells. This can dramatically slow progress, particularly when three or more neurons must be recorded simultaneously. Consistent with the finding of faster gigaohm seal formation time with the optimized NMDG protective recovery method, there was a marked improvement in the success rate and throughput of multi-neuron patch clamp recording experiments with both adult transgenic mouse brain slices and adult human neurosurgical brain slices. The improved efficiency is almost certainly attributable to both the more rapid and reliable gigaohm seal formation and the improved neuronal preservation of the slices with this protocol. Although this protocol focuses on the benefits explicitly for patch clamp recording applications, similar gains are anticipated for other challenging experimental applications where brain slice viability is paramount.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was funded by the Allen Institute for Brain Science. The authors wish to thank the Allen Institute founders, Paul G. Allen and Jody Allen, for their vision, encouragement, and support. We also thank the Allen Institute technical support staff for performing animal care, husbandry, and genotyping.
References
- Aghajanian GK, Rasmussen K. Intracellular studies in the facial nucleus illustrating a simple new method for obtaining viable motoneurons in adult rat brain slices. Synapse. 1989;3(4):331–338. doi: 10.1002/syn.890030406. [DOI] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol. 2014;1183:221–242. doi: 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, et al. Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods. 2011;8(9):745–752. doi: 10.1038/nmeth.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peca J, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472(7344):437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JTF, et al. Improved methods for acute brain slice preparation from adult and aging animals. Society for Neuroscience Abstracts. 2011;520 [Google Scholar]
- Ting JTF, et al. Improved methods for acute brain slice preparation from adult and aging animals (Part II): Glutathione depletion underlies rapid deterioration of adult brain slices. Society for Neuroscience Abstracts. 2012;505 [Google Scholar]
- MacGregor DG, Chesler M, Rice ME. HEPES prevents edema in rat brain slices. Neurosci Lett. 2001;303(3):141–144. doi: 10.1016/s0304-3940(01)01690-1. [DOI] [PubMed] [Google Scholar]
- Liu YB, Guo JZ, Chiappinelli VA. Nicotinic receptor-mediated biphasic effect on neuronal excitability in chick lateral spiriform neurons. Neuroscience. 2007;148(4):1004–1014. doi: 10.1016/j.neuroscience.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Huguenard JR, Prince DA. GABAA receptor-mediated currents in interneurons and pyramidal cells of rat visual cortex. J Physiol. 1998;506:715–730. doi: 10.1111/j.1469-7793.1998.715bv.x. (Pt 3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JR, Jonas P. Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc. 2006;1(4):2075–2081. doi: 10.1038/nprot.2006.312. [DOI] [PubMed] [Google Scholar]
- Contractor A, et al. Loss of kainate receptor-mediated heterosynaptic facilitation of mossy-fiber synapses in KA2-/- mice. J Neurosci. 2003;23(2):422–429. doi: 10.1523/JNEUROSCI.23-02-00422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pita-Almenar JD, Collado MS, Colbert CM, Eskin A. Different mechanisms exist for the plasticity of glutamate reuptake during early long-term potentiation (LTP) and late LTP. J Neurosci. 2006;26(41):10461–10471. doi: 10.1523/JNEUROSCI.2579-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Contractor A, Swanson GT. Attenuated plasticity of postsynaptic kainate receptors in hippocampal CA3 pyramidal neurons. J Neurosci. 2004;24(27):6228–6236. doi: 10.1523/JNEUROSCI.1302-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dort CJ, et al. Optogenetic activation of cholinergic neurons in the PPT or LDT induces REM sleep. Proc Natl Acad Sci U S A. 2015;112(2):584–589. doi: 10.1073/pnas.1423136112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferando I, Mody I. Altered gamma oscillations during pregnancy through loss of delta subunit-containing GABA(A) receptors on parvalbumin interneurons. Front Neural Circuits. 2013;7:144. doi: 10.3389/fncir.2013.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AG, et al. Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proc Natl Acad Sci U S A. 2015;112(4):1196–1201. doi: 10.1073/pnas.1416196112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi YK, et al. Neural estimates of imagined outcomes in the orbitofrontal cortex drive behavior and learning. Neuron. 2013;80(2):507–518. doi: 10.1016/j.neuron.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, et al. Preserving GABAergic interneurons in acute brain slices of mice using the N-methyl-D-glucamine-based artificial cerebrospinal fluid method. Neurosci Bull. 2015;31(2):265–270. doi: 10.1007/s12264-014-1497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaSarge CL, Santos VR, Danzer SC. PTEN deletion from adult-generated dentate granule cells disrupts granule cell mossy fiber axon structure. Neurobiol Dis. 2015;75:142–150. doi: 10.1016/j.nbd.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong LM, et al. Inhibitory interneuron progenitor transplantation restores normal learning and memory in ApoE4 knock-in mice without or with Abeta accumulation. J Neurosci. 2014;34(29):9506–9515. doi: 10.1523/JNEUROSCI.0693-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming RL, et al. Binge-pattern ethanol exposure during adolescence, but not adulthood, causes persistent changes in GABAA receptor-mediated tonic inhibition in dentate granule cells. Alcohol Clin Exp Res. 2013;37(7):1154–1160. doi: 10.1111/acer.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer KK, El Rawas R, Kress M, Saria A, Zernig G. Social interaction and cocaine conditioning in mice increase spontaneous spike frequency in the nucleus accumbens or septal nuclei as revealed by multielectrode array recordings. Pharmacology. 2015;95(1-2):42–49. doi: 10.1159/000370314. [DOI] [PubMed] [Google Scholar]
- Qi J, et al. A glutamatergic reward input from the dorsal raphe to ventral tegmental area dopamine neurons. Nat Commun. 2014;5:5390. doi: 10.1038/ncomms6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, et al. Modulation of dopamine release in the striatum by physiologically relevant levels of nicotine. Nat Commun. 2014;5:3925. doi: 10.1038/ncomms4925. [DOI] [PubMed] [Google Scholar]
- Siuda ER, et al. Spatiotemporal Control of Opioid Signaling and Behavior. Neuron. 2015. [DOI] [PMC free article] [PubMed]
- Tucker KR, Huertas MA, Horn JP, Canavier CC, Levitan ES. Pacemaker rate and depolarization block in nigral dopamine neurons: a somatic sodium channel balancing act. J Neurosci. 2012;32(42):14519–14531. doi: 10.1523/JNEUROSCI.1251-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt RA, et al. Serotonergic versus nonserotonergic dorsal raphe projection neurons: differential participation in reward circuitry. Cell Rep. 2014;8(6):1857–1869. doi: 10.1016/j.celrep.2014.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DV, et al. Mesopontine median raphe regulates hippocampal ripple oscillation and memory consolidation. Nat Neurosci. 2015;18(5):728–735. doi: 10.1038/nn.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, Shih PY, McIntosh JM, Drenan RM. alpha4alpha6beta2* nicotinic acetylcholine receptor activation on ventral tegmental area dopamine neurons is sufficient to stimulate a depolarizing conductance and enhance surface AMPA receptor function. Mol Pharmacol. 2013;84(3):393–406. doi: 10.1124/mol.113.087346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KM, Ribnicky DM, Rogers RC, Hermann GE. Artemisia santolinifolia enhances glutamatergic neurotransmission in the nucleus of the solitary tract. Neurosci Lett. 2014;582:115–119. doi: 10.1016/j.neulet.2014.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergacheva O, Boychuk CR, Mendelowitz D. Developmental changes in GABAergic neurotransmission to presympathetic and cardiac parasympathetic neurons in the brainstem. J Neurophysiol. 2013;110(3):672–679. doi: 10.1152/jn.01054.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergacheva O. Chronic intermittent hypoxia alters neurotransmission from lateral paragigantocellular nucleus to parasympathetic cardiac neurons in the brain stem. J Neurophysiol. 2015;113(1):380–389. doi: 10.1152/jn.00302.2014. [DOI] [PubMed] [Google Scholar]
- Dyavanapalli J, et al. Chronic intermittent hypoxia-hypercapnia blunts heart rate responses and alters neurotransmission to cardiac vagal neurons. J Physiol. 2014;592:2799–2811. doi: 10.1113/jphysiol.2014.273482. (Pt 13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolides PF, Trussell LO. Regulation of interneuron excitability by gap junction coupling with principal cells. Nat Neurosci. 2013;16(12):1764–1772. doi: 10.1038/nn.3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA. Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron. 2013;77(5):942–954. doi: 10.1016/j.neuron.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferando I, Mody I. In vitro gamma oscillations following partial and complete ablation of delta subunit-containing GABAA receptors from parvalbumin interneurons. Neuropharmacology. 2015;88:91–98. doi: 10.1016/j.neuropharm.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DJ, et al. M5 receptor activation produces opposing physiological outcomes in dopamine neurons depending on the receptor's location. J Neurosci. 2014;34(9):3253–3262. doi: 10.1523/JNEUROSCI.4896-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, Broderick HJ, Drenan RM. Local application of drugs to study nicotinic acetylcholine receptor function in mouse brain slices. J Vis Exp. 2012. p. e50034. [DOI] [PMC free article] [PubMed]
- Wang L, et al. Temporal components of cholinergic terminal to dopaminergic terminal transmission in dorsal striatum slices of mice. J Physiol. 2014;592:3559–3576. doi: 10.1113/jphysiol.2014.271825. (Pt 16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway BB, et al. Monosynaptic glutamatergic activation of locus coeruleus and other lower brainstem noradrenergic neurons by the C1 cells in mice. J Neurosci. 2013;33(48):18792–18805. doi: 10.1523/JNEUROSCI.2916-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePuy SD, et al. Glutamatergic neurotransmission between the C1 neurons and the parasympathetic preganglionic neurons of the dorsal motor nucleus of the vagus. J Neurosci. 2013;33(4):1486–1497. doi: 10.1523/JNEUROSCI.4269-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott SB, Holloway BB, Viar KE, Guyenet PG. Vesicular glutamate transporter 2 is required for the respiratory and parasympathetic activation produced by optogenetic stimulation of catecholaminergic neurons in the rostral ventrolateral medulla of mice in vivo. Eur J Neurosci. 2014;39(1):98–106. doi: 10.1111/ejn.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergacheva O, Dyavanapalli J, Pinol RA, Mendelowitz D. Chronic intermittent hypoxia and hypercapnia inhibit the hypothalamic paraventricular nucleus neurotransmission to parasympathetic cardiac neurons in the brain stem. Hypertension. 2014;64(3):597–603. doi: 10.1161/HYPERTENSIONAHA.114.03603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, et al. Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron. 2012;76(2):297–308. doi: 10.1016/j.neuron.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo FH, Horn ME. Sohal V.S. Putative microcircuit-level substrates for attention are disrupted in mouse models of autism. Biological Psychiatry. 2016;79(8):667–675. doi: 10.1016/j.biopsych.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KM, Rogers RC, Hermann GE. PAR1-activated astrocytes in the nucleus of the solitary tract stimulate adjacent neurons via NMDA receptors. J Neurosci. 2015;35(2):776–785. doi: 10.1523/JNEUROSCI.3105-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano P, Andrade R, Bykhovskaia M. Synapsin II and Rab3a cooperate in the regulation of epileptic and synaptic activity in the CA1 region of the hippocampus. J Neurosci. 2013;33(46):18319–18330. doi: 10.1523/JNEUROSCI.5293-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomioka NH, et al. Elfn1 recruits presynaptic mGluR7 in trans and its loss results in seizures. Nat Commun. 2014;5:4501. doi: 10.1038/ncomms5501. [DOI] [PubMed] [Google Scholar]
- Jiang X, et al. Principles of connectivity among morphologically defined cell types in adult neocortex. Science. 2015;350(6264):9462. doi: 10.1126/science.aac9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, et al. Imaging pericytes and capillary diameter in brain slices and isolated retinae. Nat Protoc. 2014;9(2):323–336. doi: 10.1038/nprot.2014.019. [DOI] [PubMed] [Google Scholar]
- Gabriel LR, Wu S, Melikian HE. Brain slice biotinylation: an ex vivo approach to measure region-specific plasma membrane protein trafficking in adult neurons. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed]
- Edwards FA, Konnerth A. Patch-clamping cells in sliced tissue preparations. Methods Enzymol. 1992;207:208–222. doi: 10.1016/0076-6879(92)07015-g. [DOI] [PubMed] [Google Scholar]
- Qi G, Radnikow G, Feldmeyer D. Electrophysiological and morphological characterization of neuronal microcircuits in acute brain slices using paired patch-clamp recordings. J Vis Exp. 2015. p. e52358. [DOI] [PMC free article] [PubMed]
- Booker SA, Song J, Vida I. Whole-cell patch-clamp recordings from morphologically- and neurochemically-identified hippocampal interneurons. J Vis Exp. 2014. p. e51706. [DOI] [PMC free article] [PubMed]
- Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. The permeability of the sodium channel to organic cations in myelinated nerve. J Gen Physiol. 1971;58(6):599–619. doi: 10.1085/jgp.58.6.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9(7):868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- Chen BT, et al. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59(2):288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]