

Abstract
Notch transcription complexes (NTCs) drive target gene expression by binding to two distinct types of genomic response elements, NTC monomer-binding sites and sequence-paired sites (SPSs) that bind NTC dimers. SPSs are conserved and are linked to the Notch-responsiveness of a few genes, but their overall contribution to Notch-dependent gene regulation is unknown. To address this issue, we determined the DNA sequence requirements for NTC dimerization using a fluorescence resonance energy transfer (FRET) assay, and applied insights from these in vitro studies to Notch-“addicted” leukemia cells. We find that SPSs contribute to the regulation of approximately a third of direct Notch target genes. While originally described in promoters, SPSs are present mainly in long-range enhancers, including an enhancer containing a newly described SPS that regulates HES5. Our work provides a general method for identifying sequence-paired sites in genome-wide data sets and highlights the widespread role of NTC dimerization in Notch-transformed leukemia cells.
Introduction
Notch receptors participate in a highly conserved signaling pathway that regulates many aspects of development and cellular homeostasis in metazoans (for review, see (1)). Mammals have genes encoding four Notch receptors (NOTCH1-4) and four functional ligands [delta-like 1 (DLL1), DLL4, jagged-1 (JAG1) and JAG2]. Receptor activation is initiated upon binding of ligands expressed on neighboring cells, an event that triggers successive cleavages of Notch by ADAM metalloproteases and γ-secretase. The latter event releases the Notch intracellular domain (NICD) from the cell membrane, allowing NICD to traffic to the nucleus and form a Notch transcription complex (NTC) with the DNA binding protein RBPJ (recombination signal binding protein for immunoglobulin κJ region) and coactivators of the mastermind-like protein (MAML) family. The NTC recruits other transcriptional regulators, such as p300 (2), lysine-specific demethylase 1A (LSD1) (3), and mediator (4), to increase the transcription of target genes. Although the canonical elements of the Notch pathway are highly conserved, outcomes vary widely across cellular contexts, and inappropriate increases or decreases in Notch signaling are associated with several developmental disorders and many human cancers (5, 6), emphasizing the importance of precise regulation of the timing and size of transcriptional responses to Notch activation.
NTCs can activate target genes by binding to two different types of Notch response elements (NREs), monomeric sites containing single RBPJ binding sites (7, 8), and sequence-paired sites (SPSs) containing two RBPJ binding sites (9, 10). The DNA sequence requirements for binding of NTCs to monomeric sites have been characterized in vitro by studying the binding of RBPJ to an array containing all possible 8 base pair double-stranded DNA sequences in the presence and absence of other NTC components (11). This study revealed that the binding affinity and sequence preference of RBPJ was unaffected by the presence of other NTC components, and yielded protein binding microarray “scores” for RBPJ binding to every possible 8-mer DNA sequence. By contrast, the DNA sequence requirements for binding of NTC dimers have been studied less extensively. DNA sequences containing two head-to-head RBPJ binding sites separated by spacer sequences of 15 to 17 base pairs are competent for binding of NTC dimers (9). The crystal structure of a NTC dimer bound to a SPS showed that RBPJ makes the same contacts with DNA in dimeric complexes as in monomeric complexes, and that NTC dimers are stabilized by homotypic contacts between residues Lys1945, Glu1949, and Arg1984 on the convex face of the NOTCH1 ankyrin repeat (ANK) domain (9).
SPSs were described originally in promoter elements within the Drosophila enhancer of split [E(Spl)] locus and also are found in the promoters of vertebrate E(spl) homologs, such as Hes1 (12), Hes4, and Hes5 (9). The cooperative binding of NTCs to paired sites suggests that such elements serve to “tune” or sharpen transcriptional responses within a subset of Notch target genes. Indeed, prior work in Drosophila cell lines has shown that genes in the E(spl) locus respond very rapidly to a pulse of activated Notch (13). We previously identified a set of NTC dimer-dependent target genes that contribute to the development of Notch-driven T-ALL in the mouse (14), and Castel et al. reported that 14% (22/158) of high confidence RBPJ sites identified by ChIP-Seq in murine C2C12 myoblasts have associated motifs resembling SPSs. These studies suggest that SPSs have a substantial role in regulating gene expression in mammalian cells, but to date genome-wide studies linking putative functional SPSs to gene expression have been lacking. Moreover, the SPS in the Hes5 promoter is “cryptic”, consisting of a high affinity RBPJ site paired with a site that fails to match the RBPJ binding consensus sequence (9), suggesting that SPSs may not be identified reliably by “sequence-gazing” alone.
To fill these gaps in current knowledge, we first developed a fluorescence resonance energy transfer (FRET)-based assay using purified NTC components to quantitatively measure NTC dimerization in the presence of defined DNA sequences. We determined that dimer formation on any particular DNA sequence is strongly correlated with the product of the PBM scores for the two potential RBPJ binding sites, providing a means to identify possible cryptic SPSs, and then applied this metric to identify high confidence SPSs in T-ALL cells. To evaluate our predictions, we introduced a dimerization defective form of NICD, and noted a strong correlation between the presence (or absence) of SPSs in functional NREs and Notch dimerization-dependent (or independent) expression of flanking target genes. Altogether, we conservatively estimate that approximately 15% to 20% of functional NTC binding sites in T-ALL cells are SPSs, an estimate in line with experimental perturbations showing that approximately a third of genes that respond to Notch activation do so in a NTC dimer-dependent fashion. Relative to Drosophila, human leukemia cells contain a broader diversity of NREs, including enhancers with multiple SPSs or mixtures of SPSs and monomeric NREs. These observations greatly expand the known role of SPSs in mammalian gene regulation and reveal the presence of diverse NRE architectures that contribute to the regulation of Notch target genes.
Results
Identification of DNA sequences that support NTC dimerization using a fluorescent resonance energy transfer (FRET) assay
Using the structure of an NTC dimer on the Hes1 proximal promoter SPS as a model (Fig. 1A), we designed a FRET assay to monitor assembly of purified NTC dimer components on 40 base pair duplex DNA oligonucleotides in solution (Fig. 1B; see Materials and Methods). A FRET signal was generated only when DNA containing an SPS and all critical NTC components (key portions of NOTCH1, MAML1, and RBPJ) were present (Fig. 1, C and D). Furthermore, the mutation R1984A in NOTCH1 (Fig. 1A inset) abrogated the FRET signal and prevented both NTC dimerization (Fig. S1A) and activation of transcription from reporter genes containing SPSs (Fig. S1B), yet it had no effect on NICD1 activation of reporter genes containing monomeric NREs (Fig. S1C), findings in line with prior reports (9, 10).
Figure 1. Detection of dimerization of Notch transcription complexes on DNA by fluorescence resonance energy transfer (FRET) assay.
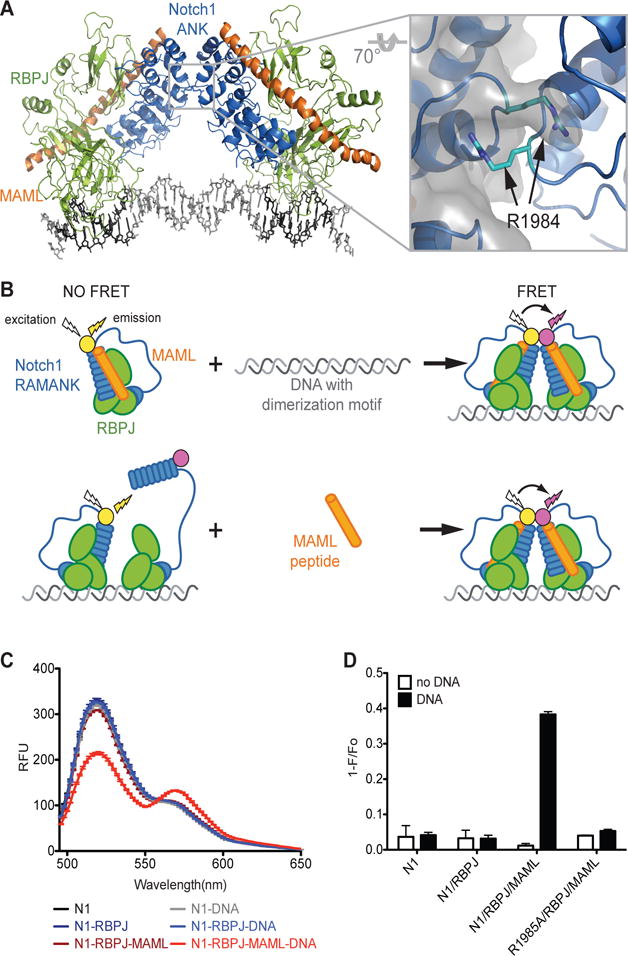
(A) Structure of a Notch transcription complex (NTC) dimer bound to the Hes1 proximal promoter sequence paired site (SPS). The inset shows the key ANK-ANK interface involving the residue R1984A. (B) Cartoon demonstrating the structural basis of the FRET-based NTC dimerization assay. (C) Wavelength scans for labeled NOTCH1 (N1) RAMANK (50:50 mix of RAMANK labeled with AlexaFluor-488 or AlexaFluor-546) in various combinations with purified RBPJ, MAML1 (MAML1, residues 13-74), and DNA containing the Hes1 SPS. (D) Quantitation of FRET from data represented in (C). FRET is expressed as 1−F/Fo, where Fo is the fluorescence intensity in the absence of SPS DNA at the emission maxima for AlexaFluor-488. Data are mean ± SEM of 3 independent measurements.
Using this FRET assay, we next tested a variety of DNA sequences for their ability to support NTC dimerization (Fig. 2A). First, we assessed the effect of DNA spacer length on NTC dimerization and observed that dimerization occurs only with DNAs containing spacer segments of 15-17 base pairs (Fig. 2B, and Fig. S2). We then determined the effect of RBPJ binding site sequence variants on NTC dimerization using with identical 5′ (site A) and 3′ (site B) sites (Fig. 2C) and distinct 5′ and 3′ sites (Fig. 2D). Introduction of non-consensus base pairs into RBPJ binding sites produced a broad range of FRET signals (Fig. 2, C and D), suggesting that NTC dimerization on any particular DNA sequence differs as a continuous function of overall site affinity. Notably, the weak RBPJ binding site within the cryptic human HES5 promoter SPS (sequence 13) facilitated NTC dimerization when paired with a strong RBPJ binding site (Fig. 2D), but failed to support dimerization when scored as a palindromic sequence (Fig. 2C). We also tested a variety of naturally occurring SPSs. We observed that the cryptic murine Hes5 promoter site supported the loading of NTC dimers, in line with prior functional studies (9), whereas mutated versions of this site did not (Fig. S3). Similarly, the murine Myc Notch-dependent enhancer site supported NTC dimerization (Fig. S3), in line with the dimer-dependency of Myc regulation by Notch in murine T-ALL cells (15).
Figure 2. Quantitation of NTC dimerization on various DNAs using FRET.
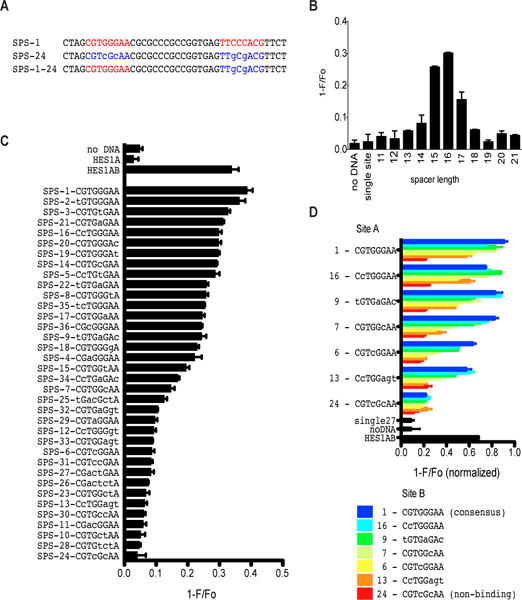
(A) Example of DNA sequences used in dimerization assays. SPS-1 has paired head-to-head consensus RBPJ motifs separated by a spacer of 16 base pairs. SPS1-24 is an example of a DNA with a non-consensus site B sequence, while SPS-24 has non-consensus substitutions in both sites A and B. Non-consensus base substitutions are shown in lower case. (B) Effect of spacer length on NTC dimerization. Each DNA contains the two consensus RBPJ binding sites shown in SPS-1. (C) NTC dimerization on the indicated palindromic DNA sequences with 16 base pair spacers. (D) NTC dimerization on the indicated non-palindromic DNA sequences with 16 base pair spacers. In (C) and (D), non-consensus base substitutions are shown in lower case, and sequence 13 corresponds to a low affinity RBPJ site found in an SPS located in the promoter of human HES5. Data are mean ± SEM of 3 independent measurements.
To study the relationship between RBPJ binding affinity and dimerization in greater detail, we used biolayer inferometry to determine the affinity of RBPJ binding to a series of increasingly divergent RBPJ binding motifs, including the weak RBPJ site in the HES5 promoter SPS (Fig. S4A). Of note, the measured binding affinities correlated well with affinities determined using isothermal calorimetry (16). NTC dimerization on DNAs containing pairs of these sequences aligned head-to-head and separated by a 16-base pair spacer also tightly correlated with the log(Kd) value for RBPJ binding to each sequence as a monomer (Fig. S4B), highlighting the cooperative nature of NTC dimerization on DNA.
SPSs are enriched in functional Notch response elements in human T-ALL cells
Functional NREs are characterized by dynamism, defined as the rapid unloading and reloading of NTCs as cells are “toggled” between the Notch-off and Notch–on states (17). Dynamic RBPJ/NOTCH1 binding sites are strongly associated with chromatin regions bearing acetylated histone H3K27 (H3K27ac) marks, are located predominantly in enhancers, and are spatially associated with genes whose expression is Notch-sensitive (17). The genomes of human T-ALL cell lines such as CUTLL1 also contain non-dynamic “static” RBPJ/NOTCH1 sites of uncertain function that show little change in occupancy when cells are cycled between the Notch-on and -off state and are associated with Notch-insensitive genes (17). To determine the frequency of SPSs among RBPJ/NOTCH1 binding sites and test the idea that dynamic RBPJ/NOTCH1 sites are enriched for SPSs relative to static sites, 30 base pair sequences immediately flanking dynamic and static RBPJ/NOTCH1 sites were surveyed for secondary RBPJ motifs using a position-weighted-matrix (PWM) motif score (18) (Fig. 3A). This revealed that secondary RBPJ sequence motifs are uniquely enriched adjacent to dynamic RBPJ/NOTCH1 binding sites (Fig. 3B). Moreover, the identified motifs possess the two key characteristics of SPSs, head-to-head orientation and separation by a 15-17 base pair spacer (Fig. 3B), in agreement with prior studies (9) and our FRET assay results (Figs. 1 and 2). K-means clustering of sequences flanking dynamic RBPJ sites also identified a cluster defined by adjacent RBPJ sites in a head-to-head orientation separated by spacers with a preferred spacer length of 16 base pairs (Fig. 3C).
Figure 3. Identification of sequence-paired site motifs in the genomes of human T-ALL cells.
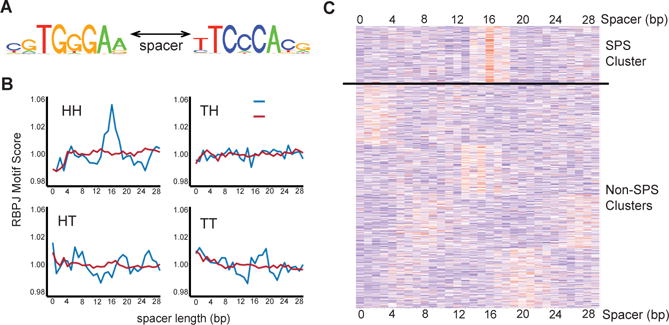
(A) Position Weighted Matrix (PWM) logo in the head to head orientation used for subsequent calculations. (B) Motif matching scores for secondary RBPJ sites oriented head-to-head (HH), head-to-tail (HT), tail-to-head (TH), and tail-to-tail (TT) calculated for all possible spacer lengths across a 30bp region flanking the highest scoring RBPJ consensus sequence associated with dynamic and non-dynamic NOTCH1 binding sites. *, 16 base pair spacer. (C) Heat map showing K-means clustering of RBPJ sequence motif matching scores generated for sequences flanking dynamic RBPJ binding sites in CUTLL1 cells. Each row represents a single high confidence dynamic RBPJ site, and each column represents the sequence motif matching score in flanking DNA for positions 1 to 30 base pairs from the primary RBPJ site.
Predicting SPSs in functional Notch response elements in T-ALL cells
Although sequence scanning indicated that functional NREs are enriched for likely SPSs, the ability of DNA sequences to support NTC dimerization forms a continuum, without any clear cut-off to assign sequences to SPS and non-SPS classes (Fig. 2). We therefore explored two different metrics that take into account the DNA sequence content of both 8-mers within potential SPSs. We first tested a PWM product score metric that was generated by multiplying the PWM score of site A times the PWM score of site B, but observed that this score was poorly correlated with NTC dimerization in solution (Fig. 4A). As an alternative approach, we used data from studies of NTC “capture” onto a protein-binding oligonucleotide microarray (PBM) to generate a PBM score for all possible 8-mer DNA sequences (11). Unlike the PWM product score, the PBM product score [PBM(site A) × PBM(site B)] was highly correlated with FRET signal (Fig. 4B), indicating that this metric can be used to identify DNA sequences that are likely to support NTC dimerization.
Figure 4. Generation and application of a metric for identifying genomic sequence-paired sites.
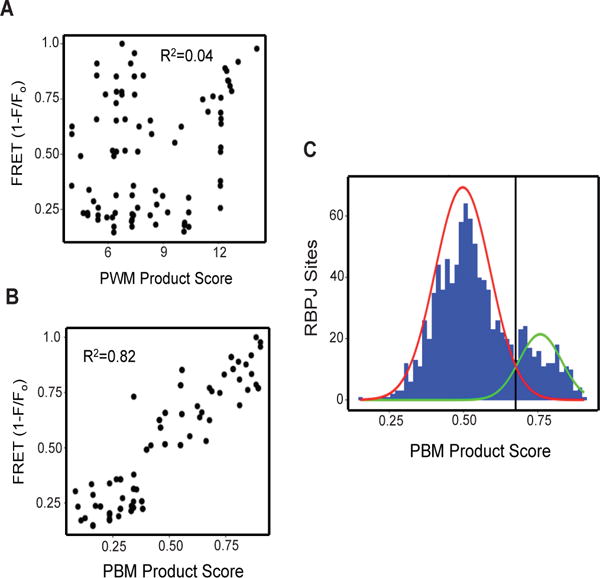
(A) Relationship between NTC dimerization measured by FRET and the sums of PWM scores for RBPJ sites A and B. (B) Relationship between NTC dimerization measured by FRET and protein binding microarray (PBM) product scores [PBM(A) × PBM(B)] for RBPJ sites A and B. (C) Bimodal distribution of PBM product scores of dynamic RBPJ/NOTCH1 binding sites in CUTLL1 genomes. Based on mixed Gaussian modeling, the red line corresponds to the distribution of PBM product scores corresponding to monomeric RBPJ/NOTCH1 sites and the green line corresponds to the distribution of PBM product scores for SPSs. Sites with PBM product scores of >0.67 (corresponding to the black line) were designated likely SPSs for purposes of further analysis.
We then calculated the PBM product scores of all dynamic RBPJ/NOTCH1 genomic binding sites in CUTLL1 cells, assuming possible spacer lengths of 15 to 17 base pair spacers. Using a Gaussian mixed model, we identified two Gaussian distributions in the data, one corresponding to probable monomeric sites and one corresponding to probable SPSs, defined as sites that are statistically likely to fall under the SPS Gaussian distribution (Fig. 4C; see Materials and Methods for details). Of the 1182 dynamic RBPJ/NOTCH1 sites identified in our reanalyzed ChIP-Seq data sets from CUTLL1 cells, 996 are associated with high confidence RBPJ binding motifs (p<0.05), of which 197 (20%) contain high-confidence SPSs (summarized in table S1). Among these 197 sites, maximal PBM product scores were observed with spacers of 15 (n=78, 40%), 16 (n=60, 30%), and 17 base pairs (n=60, 30%), which correspond to spacer lengths that support NTC dimerization in vitro. As might be expected of dimeric complexes, the average NOTCH1 and RBPJ ChIP-Seq signals for predicted SPSs were greater than the signals seen at predicted monomeric sites. By contrast, the lowest signals were seen at dynamic sites that lack RBPJ motifs (Fig. 5, A and B), which may be enriched for sites involved in looping interactions with bona fide RBPJ/NOTCH1 binding sites. Mapping of likely SPSs showed that (as is generally true of dynamic RBPJ/NOTCH1 binding sites in CUTLL1 cells (17)), most dynamic sites containing probable SPSs (86%) were found within distal enhancers (Fig. 5, C and D).
Figure 5. Characteristics of high confidence sequence-paired sites in human T-ALL cell genomes.
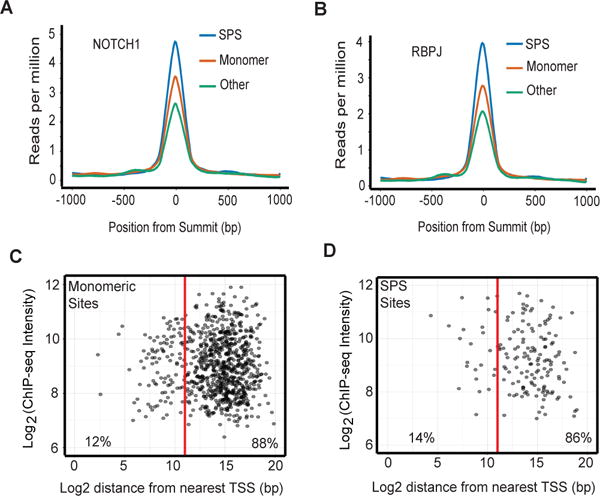
(A) NOTCH1 and (B) RBPJ ChIP-seq signals for predicted SPSs, monomeric RBPJ sites, and dynamic ChIP-seq peaks lacking RBPJ consensus sequences. (C, D) Location of predicted dynamic RBPJ/NOTCH1 monomeric sites (C) and SPSs (D) relative to transcription start sites (TSS) and RBPJ and NOTCH1 ChIP-seq signal intensity.
Contribution of dimeric NTC complexes to gene expression in T-ALL cells
Of the homotypic NICD1-NICD1 contacts that are required for NTC dimerization on SPSs, R1984 is particularly important, as an R1984A substitution severely impairs NTC dimerization on DNA and abrogates activation of transcription from genes containing SPSs (Fig. S1A,B) (9). Because the R1984A mutant retains the ability to activate genes containing monomeric RBPJ sites (Fig. S1A,B) (9), it can be used to selectively perturb the expression of genes that depend on NTC binding to SPSs. Thus, to determine the contribution of NTC dimerization to gene expression in T-ALL cells, RNA-seq was performed on CUTLL1 cells transduced with: GFP, in the presence (i) or absence (ii) of GSI; iii) wild type NICD1 in the presence of GSI; or iv) R1984A NICD1 in the presence of GSI, which inhibits the activity of endogenous NOTCH1 in CUTLL1 cells. Importantly, wild type NICD1 and R1984A mutant NICD1 were expressed at comparable levels in transduced cells (Fig. 6A). We observed that 93 of 281 genes “rescued” from GSI by wild type NICD1 failed to be rescued by R1984A NICD1 (Fig. 6B, summarized in table S2). To narrow our focus to genes that are likely to be directly regulated by Notch, we relied on previous observations showing that direct Notch target genes tend to be found within the same CTCF-bounded domain as one or more dynamic RBPJ-NOTCH1 binding sites (17). We observed that the expression of 36% (38/107) of these high probability direct Notch target genes depended on NTC dimerization (Fig. 6C; summarized in table S2), and that the home CTCF domains of dimer-dependent genes were enriched for predicted SPSs, whereas the home CTCF domains of dimer-independent genes were enriched for predicted monomeric NTC binding sites (Fig. 6D,). These relationships also are evident in a waterfall plot showing that the more sensitive a gene is to the R1984A mutant, the more likely it is to be associated with a predicted SPS (Fig. 6E. Of further interest, local ChIP analyses showed that R1984A mutant NICD1 failed to bind stably to known SPSs (such as those in the promoters of HES1 and HES4), whereas association with predicted monomeric RBPJ/NOTCH1 binding sites (such as those in the IL7R and MYC enhancers) was comparable to that of wild type NICD1 (Fig. S1, D-G). This result is consistent with experiments in purified systems showing that (in the presence of RBPJ and MAML1) the R1984A NICD1 mutant forms stable complexes on monomeric RBPJ sites, but fails to form higher order complexes on SPSs (10).
Figure 6. Association of predicted sequence-paired sites and monomeric Notch response elements with dimer-dependent and -independent Notch target genes in human T-ALL cells.
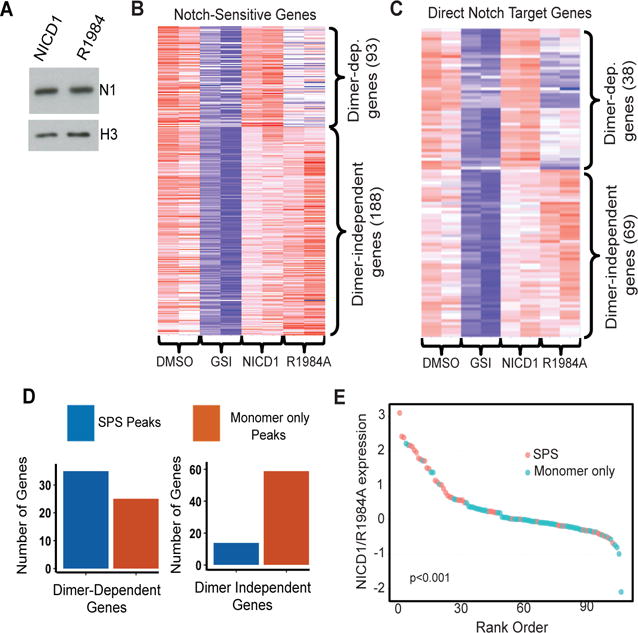
(A) Abundance of wild type NICD1 and R1984A mutant NICD1 in transduced CUTLL1 cells. (B and C) Heat maps of Notch-dependent RNA expression (B) and high confidence Notch target gene expression (C) in cells treated with vehicle (DMSO), GSI (1μM DBZ), or GSI following transduction with wild type or R1984A mutant NICD1. Expression was determined by sequencing RNA obtained from two independent experiments. (D) Notch dimer-dependent and –independent genes are associated with predicted sequence-paired sites and monomeric RBPJ/NOTCH1 sites, respectively. Notch regulated genes were paired with nearby predicted SPS or monomeric RBPJ/NOTCH1 binding sites as described (17). Genes rescued by R1984A expression in the presence of GSI are preferentially associated with monomeric RBPJ/NOTCH1 binding sites than with SPSs (p < 1 × 10−5, Fisher Exact Test). (E) Waterfall plot showing the association between the dimer-dependency of likely direct Notch target genes and the presence of one or more nearby SPSs (p<1 × 10−3, Wilcoxon one-sided signed-rank test). The Y-axis corresponds to the log2 expression ratio of individual Notch target genes in GSI treated cells transduced with wild type or R1984A mutant NICD1.
Discrepant associations (dimer-independent genes associated with SPSs and dimer-dependent genes associated with monomeric sites) likely have several bases. Most notably, some of the most robust Notch target genes that score as NTC dimer-independent are associated with a mixture of predicted SPS and monomeric sites. Included among such genes is NRARP, which is regulated by a large 5′ enhancer element that contains 5 dynamic RBPJ/NOTCH1 sites (17), of which 3 are predicted to be monomeric sites and 2 are predicted to be SPSs. Presumably, in CUTLL1 cells NRARP expression can be stimulated maximally without a need for dimerization on SPSs. Our results are similar to those obtained by Hass et al. (19), who used a split DNA adenine methylase fused to NTC components to show that the 5′ Nrarp enhancer in murine mK4 kidney also contains several SPSs, yet behaves like a gene that does not require NTC dimerization to respond to Notch activation. Overall, 7 of 11 dimer-independent Notch target genes associated with SPSs (Fig. 6D) also are associated with monomeric RBPJ/NOTCH1 sites. Other discrepant associations may stem from the experimental system used, as stable expression of wild type or R1984A mutant NICD1 undoubtedly produces secondary effects on gene expression. Nevertheless, despite these limitations, our findings show that NTC dimerization on SPSs contributes significantly to the Notch-dependent program of gene expression in human T-ALL cells.
To further test the ability of our approach to identify genomic SPSs, we analyzed previously collected gene expression and ChIP-Seq data sets obtained from the murine T-ALL cell line T6E (14, 20), which expresses a membrane-tethered form of Notch1 that is GSI-sensitive (21). PBM product scores for all RBPJ/NOTCH1 binding sites associated with a RBPJ motif again showed two Gaussian distributions within the data set, with 241 of 1654 high confidence sites (14.6%) falling into the distribution of predicted SPSs (Fig. S5A). Of the top 50 Notch-sensitive genes in T6E cells that are associated with a flanking Rbpj/Notch1 binding site, 21 (42%) were dimerization-dependent, defined by restoration of expression in GSI treated cells by transduced wild type NICD1 and not by the R1984A NICD1 mutant (Fig. S5B). As in human T-ALL cells, dimerization-dependent genes in murine T-ALL cells were highly associated with sites predicted to be SPSs based on their PBM product scores (Fig. S5C).
Identification of SPSs involved in Notch-dependent regulation of HES5 and LUNAR1
One observation emerging from our analyses of CUTLL1 cells was the absence of NTC binding in the HES5 promoter region and the presence of two NTC binding sites (E1 and E2) within an enhancer element 5′ of the HES5 gene body (Fig. 7A). Based on PBM product scores for E1 and E2 (0.497 and 0.892, respectively), the E1 site is a monomeric RBPJ/NOTCH1 binding site and the E2 site is an SPS; thus, like NRARP, in T-ALL cells HES5 is a Notch target gene that is predicted to be regulated by a mixture of SPS and monomeric response elements.
Figure 7. Co-regulation of HES5 and HANR1 by enhancer RBPJ/NOTCH1 binding sites.
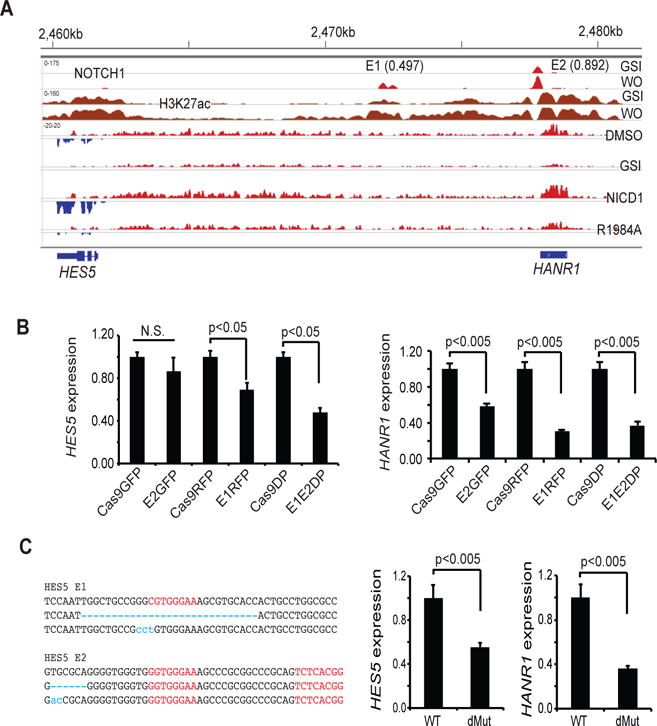
(A) HES5/HANR1 co-regulation. Upper panels show NOTCH1 and H3K27ac aligned read from ChIP-Seq data for CUTLL1 cells in the Notch-off (GSI) and Notch-on (GSI washout [WO]). Dynamic NOTCH1 binding sites designated E1 and E2 have PBM product scores of 0.497 and 0.892, respectively. Lower panels show stranded RNA-seq data obtained from CUTLL1 cells treated with vehicle, GSI (1μM DBZ), or DBZ following transducing of wild type NICD1 or the R1984A NICD1 mutant. HANR1 and HES5 aligned reads from opposite DNA strands are shown in red and blue, respectively. (B, C) Effect of CRISPR/Cas9 editing of E1 and E2 on HES5 and HANR1 expression, as determined by RT-PCR. In (B), HES1 and HANR1 expression was determined in bulk CUTLL1 cells transduced singly or in combination with empty lentiviruses (Cas9GFP, Cas9RFP) or lentiviruses carrying guide RNAs specific for E1 (E1RFP) or E2 (E2GFP). In the left panel in (C), RBPJ motifs are in red and mutated sequences identified by Sanger sequencing of single PCR products are in blue. Data are mean ± SEM of independent determinations done in triplicate. Representative results are shown.
To assess transcripts within the HES5 region for dimer-dependency, we reanalyzed our strand-specific RNA-seq data using Trinity, a de novo transcriptome assembly method (22). Alignment of RNA-seq reads demonstrated that expression of both HES5 and an enhancer-associated transcript encoding a predicted non-coding RNA (termed HES5-associated non-coding RNA1, or HANR1) was suppressed in the presence of the R1984A NICD1 mutant, indicating that both are dimer-dependent transcripts (Fig. 7A). Dependency of HES5 and HANR1 expression on NTC dimerization was independently confirmed by RT-PCR analyses (Fig. S6). To link the E1 and E2 SPSs to HES5 and HANR1 expression, CRISPR/Cas9 gene editing was performed with gRNAs designed to target the E1 or E2 sites or immediately flanking sequences, alone and in combination. Initial studies performed on bulk transduced cells showed that co-targeting of both sites was more effective at suppressing HES5 and HANR1 expression than targeting of either site alone (Fig. 7B). T7E1 nuclease analysis confirmed targeting of both E1 and E2 (Fig. S7). A single cell clone with biallelic mutations within or immediately adjacent to sites E1 and E2 also showed impaired expression of HES5 and HANR1 (Fig. 7C). Taken together, these studies show that the E1 and E2 RBPJ/NOTCH1 binding sites located in the HES5 5′ enhancer both contribute to Notch-dependent regulation of HES5 and HANR1 in CUTLL1 cells.
Co-regulation of HES5 and noncoding HANR1 transcripts in CUTLL1 cells by Notch provoked us to ask if these transcripts also were Notch-dependent in other T-ALL cells lines. Prior expression profiling carried out in a panel of Notch-dependent T-ALL cell lines failed to identify HES5 as a general target of Notch (23), suggesting that CUTLL1 is an outlier with respect to Notch regulation of HES5. In line with these observations, H3K27ac landscapes in two other Notch-dependent human T-ALL cell lines, KOPTK1 and DND41, were devoid of H3K27ac in the region of the HES5 gene body (Fig. S8). Notably, H3K27ac in the region of RBPJ/NOTCH1 binding site E1 was restricted to CUTLL1 cells, whereas H3K27ac was observed in the region of HANR1 and site E2 in all three T-ALL lines (Fig. S8), suggesting that the activity of these two elements is dissociable.
We also scanned our data set for evidence of SPS-dependent co-regulation of other coding genes and noncoding RNAs. This resulted in identification of NTC dimer-dependent regulation of the IGF-1 receptor gene (IGF1R) and LUNAR1, a noncoding RNA proposed to play a structural role in looping interactions between an intronic Notch-sensitive IGF1R enhancer and the IGF1R proximal promoter (24). The intronic IGF1R RBPJ/NOTCH1 binding site has a PBM product score of 0.862, consistent with an ability to function as an SPS. In line with the model proposed by Trimarchi et al. (24), expression of the NICD1 R1984A mutant led to loss of expression of both IGFR1 and LUNAR1, despite the absence of RBPJ/NOTCH1 binding signals near the LUNAR1 transcriptional start site (Fig. S9). Together, these analyses suggest that SPSs within distal enhancers can co-regulate associated genes and enhancer lncRNAs, findings that are of interest given the existence of numerous Notch-regulated long intervening non-coding RNAs in T-ALL cells (24, 25).
Discussion
We and others have hypothesized that cooperative dimerization of NTCs on SPSs serves to properly “tune” transcriptional responses to Notch signals (10, 13), but understanding the role of SPSs has been limited by the lack of a robust method to identify SPSs genome-wide. Here, building on direct measures of binding of NTC dimers to DNA in purified systems, we provide a method for identifying likely SPSs in genome-wide ChIP-Seq data sets and show that SPSs are often found in long-range enhancers and exist both alone and in combination with monomeric NTC binding sites, and regulate subsets of both coding and non-coding RNAs. Using a sensitive method employing NTC components fused to complementary portions of DNA adenine methyltransferase, Hass et al. also have suggested that SPSs are prevalent with the genomes of mammalian cells (19); however, while highly sensitive, this method labels genomic DNA over regions of approximately 5Kb, precluding precise SPS localization. Thus, our findings further extend the known role of SPSs in regulation of mammalian gene expression, laying the groundwork for assessment of the role of this conserved class of Notch response element in other Notch-dependent systems.
What remains to be determined is precisely how SPSs contribute to regulation of transcriptional responses to Notch. Prior work in fly cells has shown that E(spl) genes associated with promoter-localized SPSs respond very rapidly to Notch activation (13), leading to the suggestion that SPS-regulated genes are early responders, possibly at low Notch doses because of the cooperative binding of NTC dimers on SPSs. Here, we also note that SPSs on average have stronger RBPJ/NOTCH1 ChIP-Seq signals, suggesting that SPS-associated genes also have larger or more sustained responses to Notch activation. However, linking specific response element architectures to the kinetics and size of response to Notch activation is complicated by the diversity of NREs that are found in mammalian cells. Most prior work has focused on the existence of genes with promoters containing one of two types of Notch response elements, SPSs and monomeric response elements. Our genome-wide analyses revealed many variations on these themes, including genes associated with multiple monomeric response elements (for example, IL4), and mixtures of SPS and monomeric response elements, often within long-range enhancers (such as NRARP, DTX1, IL7R, MYC, HES5). Additional variation in response to a particular dose of Notch may be created by: (i) differing affinities of individual RBPJ binding sites; (ii) differences in the number of functional NTC binding sites; and (iii) the presence of nearby cis-regulatory elements that cooperate with NTCs to drive transcription, such as regulatory lncRNAs. These variables are likely to yield a large range of responsiveness to Notch signals, which are normally balanced by feedback controls involving HES family repressors (13, 26) and Notch inhibitors such as NRARP (27, 28) to enable precise regulation of the strength and duration of Notch responses. Of note, while SPSs are present in flies, they appear to be absent from worms, suggesting that SPSs arose to permit more precise and varied responses to Notch signals in complex multicellular animals.
Other levels of emerging complexity are reflected in cross-cell line, cross-lineage, and cross-species variation in the regulation of Notch target genes. We previously identified a cryptic dimeric SPS in the HES5 promoter (9), yet our analyses indicate that in CUTLL1 T-ALL cells HES5 is regulated by two enhancer-based 5′ RBPJ/NOTCH1 binding sites, one of which is an SPS. However, HES5 is not a Notch-sensitive gene in other Notch-dependent human T-ALL cell lines (23), apparently because they possess different 5′ enhancer states (Fig. S8A). CUTLL1 cells are unusual in having a genomic rearrangement involving NOTCH1 and the TCRB locus that creates a mutant NOTCH1 allele that drives high levels of NICD1 (29), raising the possibility that this might lead to “capture” of genes that are not normally regulated by Notch in T lineage cells.
Other complexity is evidence in Notch-regulation of MYC. Our group and Ferrando’s group recently reported that in T-ALL cells and normal developing thymocytes Notch regulates MYC through a enhancer element located ~1.3Mb 3′ of the MYC gene body (15, 30), whereas Kieff’s group has shown that EBV-mediated upregulation of MYC in B cells appears to occur via a RBPJ binding site located ~500kb 5′ of the MYC gene body (31). In the mouse the T-cell specific 3′ Notch-dependent MYC enhancer contains a SPS that is absolutely required for Notch to regulate MYC transcription in T-ALL cells (15), and the dimer-defective R1984A NICD1 mutant cannot support murine T-ALL development or sustain the growth of murine T-ALL cells (14). However, the dimer-defective R1984A NICD1 mutant drives MYC expression in human T-ALL cells and rescues these cells from blockade of endogenous NOTCH1 with GSI. This distinction apparently stems from the presence of several monomeric RBPJ binding sites in human MYC 3′ enhancer that are absent from the murine Myc 3′ enhancer. How these varied regulatory architectures contribute to physiologic Notch function in distinct cell contexts remains to be determined, but their importance in regulation of transcriptional outputs is emphasized by a report showing that approximately 5% of human T-ALLs harbor duplications of the 3′ Notch-dependent MYC enhancer (30). It will be of interest to determine if alterations in Notch response elements underlie other human disease states.
Materials and Methods
Protein expression and purification
Parent and R1984A mutant polypeptides consisting of the RAM and ANK domains of human NOTCH1 (RAMANK, residues 1760-2126) were made cysteine-free by introducing the mutations C1871A and C1890A and were prepared as described (32). In control electrophoretic mobility shift experiments, the C1817A/C1890A mutants were equivalent to wild type RAMANK and the R1984A RAMANK mutant in terms of assembly into NTC complexes on DNA. Single cysteine substitutions (D1942C and H2018C) that permit incorporation of fluorophores were made in the C1871A/C1890A RAMANK mutants by Quick Change mutagenesis. RAMANK proteins were fluorescently labeled using AlexaFluor 546 C5-maleimide or AlexaFluor 488 C5-maleimide. RAMANK proteins (25μM) in 20mM HEPES, pH 7.5, containing 150mM NaCl and 1mM TCEP were incubated with each fluor (1mM) overnight at 4° C in the dark. Reactions were quenched with β-mercaptoethanol. Excess fluor was removed using a 30KDa centrifugal filter followed by size-exclusion chromatography. RBPJ (residues 9-435) and MAML1 (residues 13-74) proteins were expressed and purified as described (33).
FRET Assay
Assay design was based on the X-ray crystal structure of an NTC dimer bound to the SPS in the Hes1 promoter in which the DNA is bent and undertwisted compared to B-form DNA. A model created by superimposing two NTCs on ideal B-form DNA was used as a hypothetical “open” conformation in which the two NTCs are not on the same DNA face. Two surface residues of RAMANK, D1942 and H2018, were chosen that are predicted: i) to allow mutation to cysteine and fluorophore attachment without blocking complex assembly; and ii) to be close together in the fully assembled NTC dimers and far apart in the theoretical “open” conformation. Specifically, the two D1942 residues are 14Å apart in the dimeric structure and 96 Å apart in the “open” model and are located on the surface of the ANK domain distal to the DNA-binding region of the NTC complex. The two copies of H2018 are 14Å apart in the dimeric X-ray structure and 61Å apart in the “open” conformation on B-form DNA, and are located on the surface of the ANK domain in the cavity between the ANK-ANK dimer interface and the DNA. Duplex DNA was generated by annealing complementary oligonucleotides at 100°C for 5 min and cooling slowly to room temperature. Complexes were assembled by combining 150-300nM fluorescently labeled RAMANK with untagged 600nM RBPJ, 2μM MAML1 and 150-200nM DNA in 20mM Tris pH 8.5, containing 150mM NaCl and 5mM EDTA. Assays were performed using a 1:1 mixture of D1942C-Alexa488 and D1942C-Alexa546, or with H2018-Alexa546 only. In mixtures of D1942C-Alexa488 and D1942C-Alexa546, loss of Alexa488 and gain of Alexa546 signal were coincident and energy transfer was measured as loss of Alexa488 signal (E= 1−F/Fo, where Fo is the fluorescence in the absence of DNA). In experiments using H2018-Alexa546, loss of Alexa 546 signal due to self-quenching was measured (E= 1−F/Fo).
Biolayer Interferometry
Dissociation constants for RBPJ and DNA were determined using a ForteBio Octet RED384 instrument at 30°C in a binding buffer of 25mM HEPES pH 7.9, containing 150mM NaCl, 5mM DTT and 0.1% Tween 20. Pairs of biotinylated 27-mer complementary oligonucleotides containing the sequence (CTAGXXXXXXXXCGCGCGGTGAGTTCT), where 8X corresponds to a variant candidate RBPJ binding site sequence, were annealed at 100°C for 5 min and cooled slowly to room temperature. Biotinylated DNA duplexes (2 μM) were immobilized onto Streptavidin (SA) biosensors and RBPJ binding was measured at a range of concentrations between 0.01μM and 10μM. Kd values were determined using steady-state analysis and fit to the binding isotherm, R = Rmax*[RBPJ]/(Kd + [RBPJ]).
Cell culture
Human CUTLL1 cells and murine T6E cells were maintained in RPMI1640 medium containing 10% fetal bovine serum and 1X penicillin-streptomycin-glutamine (PSG). Cells transduced with NICD1 and R1984A were maintained in the presence of the γ-secretase inhibitors 1μM dibenzazepine (DBZ) or 1μM compound E.
Retroviral packaging and transduction
Wild type NICD1 and dimer-defective R1984A mutant NICD1 cDNAs were cloned into the retrovirus vector MigR1 (14), packaged into pseudotyped retrovius (34), and transduced into CUTLL1 and T6E cells by spinoculation as described (34).
ChIPseq analysis
Prior NOTCH1, RPBJ, and H3K27Ac data sets ((17), (GEO accession GSE72175) were reanalyzed using BWA (35) after alignment to human genome build hg38 and uniquely mapped, non-redundant reads were retained. Genomic wiggle traces were generated using MACS2 (36) and were normalized by the total non-redundant read count of each dataset. ChIP-seq data processing was performed in R. Peaks with multiple summits greater than 150 base pairs apart were split in R, and dynamic peaks were determined as described (17). All new ChIPseq and RNAseq data are deposited in the Gene Expression Omnibus (GEO) under the accession numbers GSE51800 and GSE72175, respectively.
RBPJ sequence motif analysis
RBPJ sequence motif hits (RBPJ consensus sequences) near dynamic NOTCH1/RBPJ binding sites were identified using FIMO (37). A 300 base pair region centered at the summit of each RBPJ/NOTCH1 ChIP-seq peak was screened and RBPJ sequence motif hits with an adjusted p value<0.05 were identified. A RBPJ sequence-motif scan was then performed in the flanking 30 base pair region in each of 4 potential RBPJ-RBPJ site orientations, head-to-head (HH), head-to-tail (HT), tail-to-head (TH), and tail-to-tail (TT). The RBPJ motif used for the screening and motif matching calculation was the position-weighted matrix (PWM) calculated using RBPJ protein binding microarray data in Del Bianco et al. (11).
Calculation of PBM product scores
Protein binding microarray data for RBPJ from Del Bianco et al. (11) was downloaded and enrichment scores were transformed from values of −0.5 to 0.5 to values of 0.0 to 1.0 by adding 0.5 to all scores. PBM product scores were then generated for all 996 dynamic RBPJ/NOTCH1 sites bearing RBPJ motifs in CUTLL1 cells. The highest ranked RBPJ site in each dynamic RBPJ/NOTCH1 ChIP-Seq peaks was determined by FIMO (37) and was designated site A. PBM product scores were then generated by multiplying the adjusted scores for each site A and each potential site B, assuming that endogenous SPSs have 15-17 bp spacers.
ChIP-seq peak to gene assignment
The ENCODE union of CTCF sites (as described in the methods of (17)) was used to associated ChIP-seq peaks and genes. If a peak was in the same CTCF domain as a gene, it was assigned to that gene.
Local ChIP
Local ChIP with a polyclonal antibody against NOTCH1 was carried out in triplicate as described (17). Sequences of primers used to quantify NOTCH1 binding to sites near HES1, HES4, IL7R, and MYC, as well as a negative control region that is devoid of NOTCH1 binding near MYC, are listed in Table S3.
RNAseq analysis
GFP-positive NICD1-expressing or R1984A mutant NICD1-expressing CUTLL1 cells with equivalent GFP fluorescent intensities were isolated 2 days post-transduction using on a BD FACSaria flow-sorting instrument. Total RNA was extracted with the RNAeasy kit. After ribosomal RNA depletion, stranded RNA libraries were prepared following the TruSeq library preparation guide (Illunima # 15031048). Multiplexed RNAseq was performed using a HiSeq2000 instrument (Illumina). RNA-seq data were processed using TopHat and Cufflinks (Version 2.0.2) (40) with default parameters. RPKM levels for all annotated genes with official gene symbols were obtained from the Cufflinks output for each sample. Genes with RPKM > 0.5 in at least one of the 8 samples were used in differential expression analysis. A total of 10495 expressed genes were retained. Notch-dependent genes were defined by i) higher expression in DMSO than in GSI, ii) higher expression in NICD1-transduced cells treated with GSI than in GSI-treated cells transduced with empty virus. Notch dimer-dependent genes were defined by higher expression in NICD1-transduced cells treated with GSI than in R1984A mutant NICD1-transduced cells treated with GSI. The significance of concordant high-ranked differential expression was determined based on the null hypothesis that the sum of the ranks of differential expression log fold-change follows the Irwin-Hall distribution. Notch-dependent genes were identified under a false discovery rate (FDR) cutoff of 0.01 and dimer-dependent genes were identified under a FDR cutoff of 0.05, both determined by the Benjamini-Hochberg procedure.
To identify expressed long intervening non-coding RNA (lncRNA) transcripts, RNA-seq data from all samples were pooled together and processed using Trinity (41) with default parameters. Candidate lncRNAs were defined as transcripts of ≥200 base pairs in length that do not overlap with any annotated coding exon in the human genome and that have low coding potential based on CPAT (42).
Gene expression profiling
Gene expression profiling data were obtained from murine T6E cells transduced with empty MigRI viruses or viruses expressing NICD1 or the R1984A NOTCH1 mutant. Approximately 15 hours after transduction, transduced MigRI cells were treated with GSI (compound E, 1 uM) or DMSO, whereas NICD1 and R1984A transduced cells were treated with GSI only. After 6 h of treatment, GFP+ cells were isolated by FACS sorting. Total RNA was prepared using TRIzol (Invitrogen), processed to fluorescently labeled cRNAs according to manufacturer’s instructions (Affymetrix), and hybridized to Affymetrix GeneChip Mouse Genome 430 2.0 arrays. The .CEL files were imported into R using the oligo package (43). Data were analyzed in R and batch effects were corrected with ComBat (44). Differential expression was assessed in R using the limma package (45). Affymetrix gene expression data are deposited in GEO under the accession number GSE72175.
CRISPR-Cas9-mediated targeted genome editing
Lentiviral vectors capable of co-expressing guide RNA and Cas9-eGFP or Cas9-RFP657 (38) were used to target enhancer regions HES5. Lentivirus production and transduction procedure was as described (38). Transduced cells were sorted by flow cytometry. Mutation of targeted regions in bulk population of cells was assessed by T7E1 assay as described (39). Briefly, 100-200ng of PCR product containing the targeted genomic region was denatured, annealed, treated with T7E1 endonuclease I, and visualized on an agarose gel. To isolate individual clones, CRISPR-Cas9-sgRNA expressing cells were sorted into 96-well plates at a density of 1 cell/well and cultured for 3-4 weeks. Mutation of particular regions of the genome was assessed by PCR amplification of the targeted region, cloning with the blunt-end Topo cloning kit (Life Technologies), and Sanger sequencing of DNA obtained from individual bacterial colonies.
Real-time (RT)-quantitative PCR
RNA was extracted with the RNeasy kit (Qiagen) and cDNA was synthesized with a cDNA synthesis kit (BioRad). RT-qPCR was performed in triplicate using CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). Expression of HES5 and HANR1 was analyzed with CFX manager software (BioRad). PCR primer sequences used for RT-PCR are listed in Table S3.
Reporter Gene Assays
Notch reporter gene assays were carried out in transiently transfected U2OS cells as described (46).
Supplementary Material
Figure S1: Characterization of the dimerization-defective NOTCH1 mutant R1984A in FRET, reporter gene, and local ChIP assays.
Figure S2: Sequences used to assess the effect of spacer length on NTC dimerization.
Figure S3: Endogenous Hes5 promoter and Notch-dependent Myc enhancer sequence paired sites bind Notch transcription complex dimers.
Figure S4: RBPJ binding affinity for DNA correlates with FRET efficiency.
Figure S5: Identification of sequence paired sites and Notch dimer-dependent target genes in murine T6E T-ALL cells.
Figure S6: HES5 and the HANR1 long noncoding RNA expression depend on NTC dimerization.
Figure S7: Co-targeting of the HES5 E1 and E2 elements with Cas9/CRISPR.
Figure S8: Chromatin landscapes near HES5 in T-ALL cell lines.
Figure S9: IGF1R/LUNAR1 co-regulation.
Table S3: RT-qPCR primers.
Table S1: Dynamic NOTCH1 binding sites in the genome of CUTLL1 T-ALL cells
Table S2: Notch-sensitive genes in human CUTLL1 T-ALL cells.
Acknowledgments
Funding: This work was supported by grants from the NIH (5P01 CA119070 and 5R01 CA092433) and the Leukemia and Lymphoma Society.
Footnotes
Author contributions: J.C.A., S.C.B., X.L., and W.S.P. designed the project; K.A., H.W., and H.L. performed the experiments; E.S., K.A., C.Z., and L.T. analyzed the data; J.C.A., S.C.B., and W.S.P. wrote the paper.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All new ChIPseq and RNAseq data are deposited in the Gene Expression Omnibus (GEO) under the accession numbers GSE51800 and GSE72175, respectively, while gene expression profiling data are deposited the accession number GSE72175. Plasmids and other genomic and transcriptomic sequence information are available upon request.
References
- 1.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oswald F, et al. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol Cell Biol. 2001;21:7761–7774. doi: 10.1128/MCB.21.22.7761-7774.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mulligan P, et al. A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Mol Cell. 2011;42:689–699. doi: 10.1016/j.molcel.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. 2004;16:509–520. doi: 10.1016/j.molcel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 5.Aster JC. In brief: Notch signalling in health and disease. J Pathol. 2014;232:1–3. doi: 10.1002/path.4291. [DOI] [PubMed] [Google Scholar]
- 6.Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res. 2015;21:955–961. doi: 10.1158/1078-0432.CCR-14-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi SH, et al. Conformational locking upon cooperative assembly of notch transcription complexes. Structure. 2012;20:340–349. doi: 10.1016/j.str.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nam Y, Sliz P, Song L, Aster JC, Blacklow SC. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell. 2006;124:973–983. doi: 10.1016/j.cell.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 9.Arnett KL, et al. Structural and mechanistic insights into cooperative assembly of dimeric Notch transcription complexes. Nat Struct Mol Biol. 2010;17:1312–1317. doi: 10.1038/nsmb.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nam Y, Sliz P, Pear WS, Aster JC, Blacklow SC. Cooperative assembly of higher-order Notch complexes functions as a switch to induce transcription. Proc Natl Acad Sci U S A. 2007;104:2103–2108. doi: 10.1073/pnas.0611092104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Del Bianco C, et al. Notch and MAML-1 complexation do not detectably alter the dna binding specificity of the transcription factor CSL. PLoS One. 2010;5:e15034. doi: 10.1371/journal.pone.0015034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jarriault S, et al. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 13.Housden BE, et al. Transcriptional dynamics elicited by a short pulse of notch activation involves feed-forward regulation by E(spl)/Hes genes. PLoS Genet. 2013;9:e1003162. doi: 10.1371/journal.pgen.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H, et al. Notch dimerization is required for leukemogenesis and T-cell development. Genes Dev. 2010;24:2395–2407. doi: 10.1101/gad.1975210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yashiro-Ohtani Y, et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc Natl Acad Sci U S A. 2014;111:E4946–4953. doi: 10.1073/pnas.1407079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedmann DR, Kovall RA. Thermodynamic and structural insights into CSL-DNA complexes. Protein Sci. 2010;19:34–46. doi: 10.1002/pro.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, et al. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A. 2014;111:705–710. doi: 10.1073/pnas.1315023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 19.Hass MR, et al. SpDamID: Marking DNA Bound by Protein Complexes Identifies Notch-Dimer Responsive Enhancers. Mol Cell. 2015;59:685–697. doi: 10.1016/j.molcel.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci U S A. 2011;108:14908–14913. doi: 10.1073/pnas.1109023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weng AP, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20:2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palomero T, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trimarchi T, et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. 2014;158:593–606. doi: 10.1016/j.cell.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durinck K, et al. The Notch driven long non-coding RNA repertoire in T-cell acute lymphoblastic leukemia. Haematologica. 2014;99:1808–1816. doi: 10.3324/haematol.2014.115683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krejci A, Bernard F, Housden BE, Collins S, Bray SJ. Direct response to Notch activation: signaling crosstalk and incoherent logic. Sci Signal. 2009;2:ra1. doi: 10.1126/scisignal.2000140. [DOI] [PubMed] [Google Scholar]
- 27.Lamar E, et al. Nrarp is a novel intracellular component of the Notch signaling pathway. Genes Dev. 2001;15:1885–1899. doi: 10.1101/gad.908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yun TJ, Bevan MJ. Notch-regulated ankyrin-repeat protein inhibits Notch1 signaling: multiple Notch1 signaling pathways involved in T cell development. J Immunol. 2003;170:5834–5841. doi: 10.4049/jimmunol.170.12.5834. [DOI] [PubMed] [Google Scholar]
- 29.Palomero T, et al. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to gamma-secretase inhibitors. Leukemia. 2006;20:1279–1287. doi: 10.1038/sj.leu.2404258. [DOI] [PubMed] [Google Scholar]
- 30.Herranz D, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20:1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B, et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc Natl Acad Sci U S A. 2011;108:14902–14907. doi: 10.1073/pnas.1108892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Del Bianco C, Aster JC, Blacklow SC. Mutational and energetic studies of Notch 1 transcription complexes. J Mol Biol. 2008;376:131–140. doi: 10.1016/j.jmb.2007.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnett KL, Blacklow SC. Analyzing the nuclear complexes of Notch signaling by electrophoretic mobility shift assay. Methods Mol Biol. 2014;1187:231–245. doi: 10.1007/978-1-4939-1139-4_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weng AP, et al. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol. 2003;23:655–664. doi: 10.1128/MCB.23.2.655-664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heckl D, et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol. 2014;32:941–946. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reyon D, et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 45.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aster JC, et al. Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol Cell Biol. 2000;20:7505–7515. doi: 10.1128/mcb.20.20.7505-7515.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knoechel B, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–370. doi: 10.1038/ng.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Characterization of the dimerization-defective NOTCH1 mutant R1984A in FRET, reporter gene, and local ChIP assays.
Figure S2: Sequences used to assess the effect of spacer length on NTC dimerization.
Figure S3: Endogenous Hes5 promoter and Notch-dependent Myc enhancer sequence paired sites bind Notch transcription complex dimers.
Figure S4: RBPJ binding affinity for DNA correlates with FRET efficiency.
Figure S5: Identification of sequence paired sites and Notch dimer-dependent target genes in murine T6E T-ALL cells.
Figure S6: HES5 and the HANR1 long noncoding RNA expression depend on NTC dimerization.
Figure S7: Co-targeting of the HES5 E1 and E2 elements with Cas9/CRISPR.
Figure S8: Chromatin landscapes near HES5 in T-ALL cell lines.
Figure S9: IGF1R/LUNAR1 co-regulation.
Table S3: RT-qPCR primers.
Table S1: Dynamic NOTCH1 binding sites in the genome of CUTLL1 T-ALL cells
Table S2: Notch-sensitive genes in human CUTLL1 T-ALL cells.