

Abstract
Ex vivo normothermic machine perfusion (NMP) is a new clinical strategy to assess and resuscitate organs likely to be declined for transplantation, thereby increasing the number of viable organs available. Short periods of NMP provide a window of opportunity to deliver therapeutics directly to the organ and, in particular, to the vascular endothelial cells (ECs) that constitute the first point of contact with the recipient’s immune system. ECs are the primary targets of both ischemia-reperfusion injury and damage from preformed antidonor antibodies, and reduction of perioperative EC injury could have long-term benefits by reducing the intensity of the host’s alloimmune response. Using NMP to administer therapeutics directly to the graft avoids many of the limitations associated with systemic drug delivery. We have previously shown that polymeric nanoparticles (NPs) can serve as depots for long-term drug release, but ensuring robust NP accumulation within a target cell type (graft ECs in this case) remains a fundamental challenge of nanomedicine. We show that surface conjugation of an anti-CD31 antibody enhances targeting of NPs to graft ECs of human kidneys undergoing NMP. Using a two-color quantitative microscopy approach, we demonstrate that targeting can enhance EC accumulation by about 5-to 10-fold or higher in discrete regions of the renal vasculature. In addition, our studies reveal that NPs can also non-specifically accumulate within obstructed regions of the vasculature that are poorly perfused. These quantitative preclinical human studies demonstrate the therapeutic potential for targeted nanomedicines delivered during ex vivo NMP.
INTRODUCTION
Kidney transplantation is the most effective treatment for end-stage renal failure with 1-year graft survival rates that exceed 90%. However, two considerable challenges remain: the shortage of suitable donor organs and the rate of late graft failure (1). In the United States, about 100,000 patients currently await kidney transplants, and only about 18,000 transplantations are conducted annually, resulting in a median wait time of 3.6 years (1). A recent advance can potentially address the first challenge by expanding the pool of available kidneys: resuscitation of marginal organs via short periods (typically 1 hour) of ex vivo normothermic machine perfusion (NMP) (2–5). The second challenge of late graft loss primarily occurs as a result of chronic rejection (6). Notably, the rate of late graft loss has not substantially improved despite large gains in reducing acute graft rejection through new drugs and better immunosuppressive regimens (6, 7). Perioperative events, such as ischemia-reperfusion injury resulting in delayed graft function or perioperative injuries caused by preformed host antidonor-specific antibodies (Abs), pose an additional risk for late graft loss, likely because inflammation at the time of organ implantation primes a more vigorous host antigraft adaptive immune response (8).
NMP itself may provide a therapeutic benefit by reducing the severity of ischemia-reperfusion injury to graft vasculature (9). In addition, NMP also creates a window of opportunity for the ex vivo delivery of therapeutic agents to further alter the graft microenvironment directly in the transplanted organ. In particular, the endothelial cell (EC) lining of the graft vasculature—the primary target of graft injury due to ischemia reperfusion or preformed anti-donor Abs (10–14)—is directly accessible to the perfusate during NMP. Reduction of preformed Abs by plasmapheresis or immunoadsorption, sometimes in combination with administration of intravenous immunoglobulin, may partially mitigate early Ab-mediated rejection (15). However, these approaches are expensive, and neither strategy has been fully successful on its own. Ex vivo treatment of the vascular EC targets in the isolated graft (rather than the organ recipient) may represent a complementary strategy to enhance efficacy and minimize adverse systemic effects.
Because the nature of the host’s adaptive immune response can evolve over the first few postoperative weeks, a prolonged therapeutic response over this time period will likely be needed. We have previously shown that drugs encapsulated within polymeric nanoparticles (NPs) can be internalized by human ECs and that these NPs can serve as a slow release depot for drug delivery because the NPs slowly degrade by hydrolysis (16). In the case of a small interfering RNA (siRNA), the therapeutic effect may persist 6 weeks or longer (17). The challenge we address in this study is how to best ensure that NPs introduced during NMP are efficiently delivered to graft ECs.
Targeted NPs have thus far had only limited success in clinical medicine because of several challenges associated with systemic administration (18, 19). First and foremost, NPs typically cannot efficiently escape from the vasculature to reach extravascular targets (20). This results in marked accumulation within phagocytes of the liver and spleen that compete nonspecifically with the intended target cell (21). In addition, surface adsorption of serum proteins can create a so-called “protein corona” that can mask targeting ligands and abolish targeting benefits in vivo (22–24). Targeting graft ECs during NMP, which uses a serum-free perfusate, can potentially circumvent these challenges.
Anti-CD31 Abs conjugated either to NPs or directly to therapeutic molecules have been previously used to target ECs in vivo in animal models, including in the context of organ transplantation. In one study, a conjugate of anti-CD31 and catalase enzyme accumulated in lung ECs after intravenous administration in mice and was subsequently able to reduce ischemia-reperfusion injury after lung transplant into a second animal, thereby mitigating effects of cold ischemia (25). ECs of other organs, such as the heart and the brain, have also been successfully targeted by anti-CD31–therapeutic conjugates when administered in specific anatomic locations to enhance the first-pass rapid accumulation in the organ of interest (26, 27). However, it was unclear how these results would translate into targeting of NPs to the ECs of the human kidney, the most commonly transplanted organ in clinical practice.
Here, we report that conjugating anti-human CD31 Abs to poly(lactic acid)–poly(ethylene) glycol (PLA-PEG) NPs loaded with a fluorescent dye and administering them to isolated human kidneys during ex vivo NMP can lead to enhanced vascular retention compared to nontargeted NPs. Using two-color quantitative microscopy on cryosectioned biopsies, we observed the time-dependent accumulation of CD31-targeted and nontargeted NPs simultaneously in physiologically viable human kidneys during a period of NMP. This approach showed that Ab targeting to ECs can produce enrichment of accumulation in both renal glomerular and interstitial microvascular ECs in regions that were well perfused.
RESULTS
CD31 expression in the human renal cortical vasculature
To evaluate the potential use of molecularly targeted NPs for use during ex vivo perfusion of the human kidney, we first sought to identify a suitable surface protein on ECs to target. CD31 [also known as platelet EC adhesion molecule–1 (PECAM-1)] is a pan-EC marker typically expressed at relatively high quantities (28). Although it is also expressed on platelets and new thymically released T cells, neither of these potentially competing cell types are present in isolated kidneys that are perfused with leucocyte-depleted and washed red blood cell (RBC) suspensions. CD31 has been shown to be a viable target to facilitate binding and subsequent internalization of Ab-conjugated NPs within ECs (29–31), provided that an appropriate epitope is recognized by the conjugated Ab (29). These features suggest that CD31 may be well suited as a receptor for targeting of Ab-NPs uniformly to human renal ECs. However, the renal cortex contains a variety of vascular beds—including arteries, afferent and efferent arterioles, glomerular capillaries, peritubular capillaries, venules, and veins—that are molecularly distinct and may vary in the quantity of CD31 expressed (Fig. 1A). To confirm the appropriateness of CD31 as our target, we probed for the expression of CD31 throughout the renal cortex of three separate human donor kidneys using fluorescence immunohistochemistry. CD31 staining was consistently present throughout the vascular beds of the renal cortex, suggesting that CD31 is an EC receptor with the potential to induce widespread accumulation of targeted NPs (Fig. 1, B and C).
Fig. 1. CD31 is present throughout the human renal vasculature.
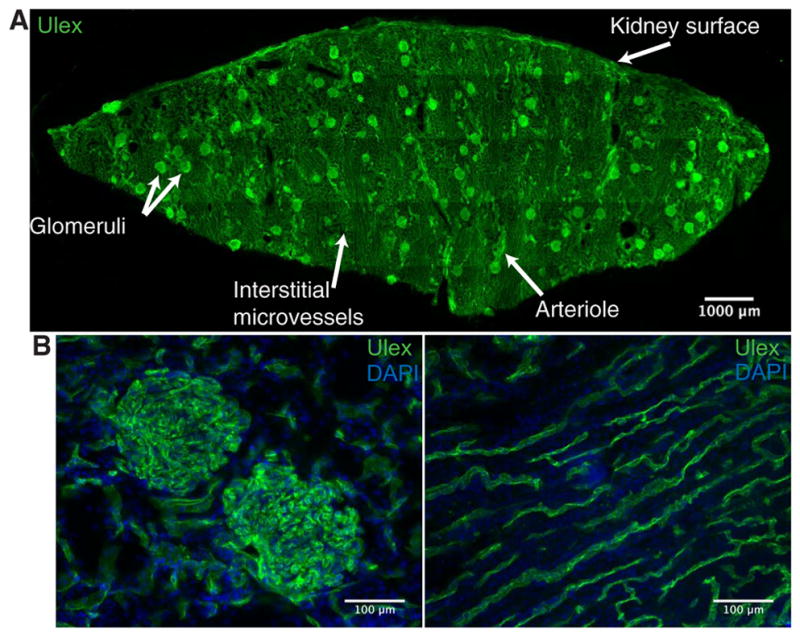
(A) Representative fluorescent immunohistochemistry staining image of a cryosection taken from a wedge biopsy of a human kidney undergoing normothermic machine perfusion (NMP). Section was stained for Ulex to depict vascular endothelium (green). White arrows label different vascular beds and the kidney surface. Multiple 20× images were tiled to reconstruct the entire cryosection. (B) Representative images for CD31 (green) and nuclear 4′,6-diamidino-2-phenylindole (DAPI; blue) staining of glomerular capillaries (left) and interstitial microvessels (right). Representative staining from three different donor kidneys.
Specific anti-CD31 NP binding to human endothelium in cell culture
We used nanoprecipitation to assemble NPs from a block copolymer of PLA-PEG (Fig. 2A). This approach produces monodisperse NPs with an inert surface coating that reduces NP clumping (32) and thereby decreases the likelihood that aggregates of NPs might plug vessels and interfere with graft function. In addition, the PEGylated surface allows Abs to be conjugated using simple standard approaches (33). NPs encapsulating either 1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) or 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) dye were about 165 to 170 nm in diameter with a polydispersity index (PDI) of <0.15 by dynamic light scattering (DLS; Fig. 2B). Dye-loaded NPs were conjugated to either an anti-CD31 (CD31-NPs) or a nonbinding isotype-matched Ab (Control-NPs) via covalent coupling to terminal COOH end groups on the PEG. This procedure yielded about 100 Abs per NP, as determined using I125-labeled Ab. DLS measurements after the conjugation procedure displayed an about 20-nm increase in diameter, consistent with the addition of an Ab layer to the NP surface (fig. S1A). NP-binding specificity was confirmed by fluorescence microscopy after a 2-hour treatment of human umbilical vein ECs (HUVECs) with 100 μg/ml of either CD31-NPs or Control-NPs. CD31-NPs strongly associated with HUVECs, whereas Control-NPs showed minimal association (Fig. 2C). Quantification of this association via flow cytometry demonstrated that CD31-NPs enhanced accumulation by about 80× relative to Control-NPs under these static incubation conditions (Fig. 2, D and E). In addition, a quantification of the number of NPs per cell after 30 min of static incubation suggested that our CD31-NP formulation is capable of accumulating at an amount commensurate with previously validated CD31-targeted NPs evaluated under similar conditions (fig. S1, B and C) (30, 31, 34). Combined treatment of red CD31-NP and green Control-NP for 2 hours demonstrated that simultaneous treatment of CD31-NP and Control-NP did not have an appreciable impact on either specific or non-specific NP accumulation (fig. S1D). A dose titration of CD31-NPs, followed by a 24-hour incubation period, show no adverse effects on EC viability (fig. S1E).
Fig. 2. CD31 provides robust NP targeting in vitro but slow kinetics.

(A) Representative transmission electron microscopy image of poly(lactic acid)–poly(ethylene) glycol NPs. Scale bar, 200 nm. (B) Quantification of NP size distribution and polydispersity index as measured by dynamic light scattering for both 1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)– and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO)–loaded nanoparticles (NPs; n = 3; a t test was used for statistical analysis). N.S., not statistically significant. (C) Representative 20× images of CD31-NPs versus Control-NPs after a 2-hour incubation with human umbilical vein endothelial cells (HUVECs) in static culture. DiO-loaded NPs are shown in green (right column) overlaid with brightfield images (left column). Scale bars, 150 μm. n = 3. (D) Representative fluorescence-activated cell sorting (FACS) histograms for HUVECs treated identically to (C). (E) Quantification of triplicate FACS experiments as shown in (D). n = 3. MFI, mean fluorescence intensity; a.u., arbitrary units. (F) Representative images of accumulation of DiI NPs as a function of time on HUVECs in a microfluidic cell culture during perfusion with CD31-NPs (100 μg/ml; top) or Control-NPs (100 μg/ml; bottom) at 2 dyne/cm2. Each image is 300 μm wide, representing the full width of the microfluidic channel. NPs are pseudocolored green for better visualization. (G) Quantification of mean area of positive fluorescence for flow experiments as performed in (F) at a range of concentrations (n = 3 independent imaging regions for each dose and time point). Lines through data represent fit to a rate equation. (H) Plot of rates as a function of NP concentration as extracted from quantification in (G). Line through data represents linear fit. Rates are normalized to a concentration of 10 μg/ml (dashed line, 1). All error bars represent 1 SD.
Although targeted NP association in static culture is frequently used as a measure of potential for targeting in vivo (16), its relevance is unclear because association of NPs with ECs in the vasculature will occur under flow. The kinetics of targeted NP association with ECs are likely influenced by both the presence of flow and NP concentration. These features are particularly relevant for clinical NMP, given the relatively short perfusion period (typically about 1 hour). Thus, we quantified the targeting kinetics of CD31-NPs under physiologically relevant flow conditions and as a function of NP concentration. Previous studies have evaluated the kinetics of NP association with surface-immobilized target receptors via surface plasmon resonance (35); however, to our knowledge, quantitative studies evaluating the kinetics of NP association with live cells have not been reported.
Using microfluidic flow chambers (shear of 2 dyne/cm2) and live-cell imaging, we measured the association of CD31-NPs to HUVEC monolayers over an NP concentration range of 10 to 100 μg/ml. CD31-NPs accumulated on the EC monolayer, whereas Control-NP at 100 μg/ml did not (Fig. 2F). CD31-NP binding did not fully saturate the surface of the cultured HUVECs over a 2-hour observation period even at the highest NP concentration tested (100 μg/ml; Fig. 2, F and G). As predicted for first-order kinetics, the rate of CD31-NP association scaled linearly with NP concentration (Fig. 2H). Collectively, these cell culture data demonstrate both that our CD31-NPs bind ECs with robust specificity and that targeting is occurring under non-saturating kinetics. This finding of nonsaturating kinetics is consistent with previous examples of CD31 targeting to cultured ECs under flow, which showed apparent linear increases in NP accumulation with increasing NP dose, but a direct comparison of our results with previous studies is difficult because the previous studies did not directly examine the time dependence of NP accumulation (30, 31, 34). The critical point raised by the observation that targeting to ECs is a kinetic (time-dependent) process, rather than equilibrium process, is that achieving robust accumulation within human kidneys during a short period of ex vivo NMP will require the optimization of both NP concentration and duration of exposure.
Donor characteristics and experimental design for human kidney perfusion series
To determine whether CD31-NPs can provide enhanced accumulation during ex vivo NMP, eight experimentally viable human kidneys that had been declined for transplantation were perfused with NPs. Four of these kidneys were declined because there was a concern that the donor had a nonrenal malignancy, two kidneys were declined because of poor in situ cold perfusion characteristics in the donor during procurement, one kidney was declined on the basis of histological features consistent with diabetes, and the final kidney was declined because of a damaged ureter and artery (Table 1). Other donor demographics are presented in Table 1.
Table 1. Declined human kidney characteristics and experimental conditions.
Donor demographics, donor type; donation after brain death (DBD), donation after circulatory death (DCD), reason for decline, warm ischemic time, cold ischemic time, duration of ex vivo normothermic perfusion (NMP), mean renal blood flow (RBF), total urine output, macroscopic appearance, NPs, and time to administration of NPs.
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | |
|---|---|---|---|---|---|---|---|---|
| Donor age (years) | 57 | 25 | 44 | 68 | 45 | 64 | 65 | 57 |
| Donor type (DBD/DCD) | DCD | DBD | DCD | DBD | DBD | DCD | DCD | DCD |
| Left/right kidney | Left | Right | Right | Right | Left | Left | Left | Left |
| Reason for decline | Poor in situ perfusion | Malignancy | Suspected malignancy | Suspected malignancy | Poor in situ perfusion + prolonged agonal phase | Suspected malignancy | Histological changes consistent with diabetes | Damage to ureter and artery |
| Warm ischemic time (min) | 13 | — | 13 | — | — | 12 | 14 | 11 |
| Cold ischemic time (hour:min) | 13:13 | 17:17 | 34:37 | 16:03 | 22:38 | 27:05 | 18:20 | 25:58 |
| NMP duration (hour:min) | 4:30 | 6:50 | 5:45 | 5:00 | 5:00 | 8:30 | 8:30 | 8:30 |
| Mean RBF (ml/min per 100 g) | 88.0 ± 19.6 | 123.7 ± 35.2 | 91.6 ± 23.9 | 67.9 ± 31.7 | 83.5 ± 35.5 | 75.0 ± 22.1 | 77.3 ± 25.4 | 80.7 ± 37.7 |
| Total urine output (ml) | 177 | 730 | 107 | 1045 | 373 | 598 | 451 | 257 |
| Macroscopic appearance | Moderate | Moderate | Moderate | Moderate | Moderate | Excellent | Excellent | Moderate |
| NP type | CD31-NP | Control-NP | CD31-NP + Control-NP | CD31-NP + Control-NP | CD31-NP + Control-NP | CD31-NP + Control-NP | CD31-NP + Control-NP | CD31-NP + Control-NP |
| NP circulating concentration | 4 μg/ml | 4 μg/ml | 10 μg/ml | 50 μg/ml | 100 μg/ml | 50 μg/ml | 50 μg/ml | 50 μg/ml |
| NP administration time (from start of perfusion) | 30 min | 50 min | 45 min | 75 min | 60 min | 30 min | 30 min | 30 min |
Experiments with these eight human kidneys were divided into three stages: (i) Low-dose (4 μg/ml) CD31-NP or Control-NP were perfused in separate organs using a single-color NP to establish our experimental approach (two kidneys). (ii) A dose titration (10, 50, and 100 μg/ml) of simultaneously administered CD31-NP and Control-NP in two NP colors (red and green, respectively) was performed for the identification of optimal NP concentration and in situ discrimination of CD31-specific versus nonspecific NP accumulation (three kidneys). (iii) Comparison of CD31-targeting benefit in three separate kidneys using an optimized NP dose and sampling conditions (three kidneys). The results from each stage of the study are described below.
Single-color evaluation of separate kidneys by quantitative microscopy
We first examined accumulation of CD31-NPs versus Control-NPs in two separate kidneys using the same low NP dose (4 μg/ml). Note that this is the initial NP concentration in the perfusate reservoir; the actual concentration in the perfusate will change as fluid moves in and out of the intravascular space during normal kidney function and as NPs are depleted from the circulating perfusate. The goal of these experiments was to establish our methodology, including detection within tissue samples by quantitative fluorescence microscopy. On the basis of our cell culture–targeting studies (Fig. 2), we predicted that several hours of perfusion would be necessary for substantial NP accumulation at this dose. Therefore, using quantitative imaging of cryosections, we evaluated the accumulation of either CD31-NPs or Control-NPs in glomerular vessels and surrounding interstitial microvessels after 4 hours of NMP (Fig. 3, A and B). Although there was considerable variability, CD31-NPs accumulated about 2.5-fold more than Control-NPs [93.2 ± 23.2 μm−2 (CD31-NPs) versus 36.8 ± 15.4 μm−2 (Control-NP) normalized mean intensity; ± represent 95% confidence interval of the mean; Fig. 3B]. Notably, we did not find evidence for extravascular accumulation of NPs in either CD31-NP or Control-NP perfused kidneys, nor did we find NPs present in the urine during NMP. Accumulation within the vasculature was heterogeneous. For both CD31-NP and Control-NP, there were vessels that showed NP accumulation, whereas adjacent vessels appeared completely free of NP. Moreover, there was a statistically significant elevation of accumulation in glomeruli compared to surrounding interstitial microvasculature (P < 0.0001; Fig. 3B). Accumulation within glomeruli was notably heterogeneous with strong accumulation in discreet regions, whereas other vessel segments appeared devoid of any accumulation (Fig. 3A).
Fig. 3. CD31 targeting enhances NP accumulation in perfused human kidney.

(A) Representative 20× fluorescent images of glomeruli (top) and interstitial microvessels (bottom) for kidneys perfused with either CD31-NPs (left) or Control-NPs (right). Overlay images show vascular stain (Ulex; green), DAPI nuclear stain (blue), and NP accumulation (red). Scale bars, 100 μm. Images are cropped to provide finer details. (B) Quantification of individual image MFI after background subtraction. Each image was normalized to the area of Ulex vascular stain, and every dot represents an individual normalized image mean intensity. The red dots correspond to the images shown in (A). *P < 0.0001 using a Mann-Whitney t test. For the single CD31-NP–treated kidney, n = 35 for images of glomeruli and n = 59 for images of microvasculature; for the single Control-NP–treated kidney, n = 31 for images of glomeruli and n = 56 for images of microvasculature. (C) Time-dependent normalized mean intensity values for CD31-NP versus Control-NP in glomeruli with corresponding fits to a linear equation (CD31-NP, black line) or saturating kinetic equation (Control-NP, dashed gray line). Error bars refer to 95% confidence window of the mean. n for each condition is provided in Table 3.
Our cell culture results (Fig. 2) suggested that accumulation of NP in renal vascular ECs could increase over several hours. Quantification of CD31-NP association with glomerular ECs at early time points showed low NP signal that increased linearly with time (Fig. 3C). In contrast, the NP signal observed in the kidney treated with Control-NPs appeared to show a rapid accumulation to a low, saturated value within the first 30 min. The rapidity of these non-specific interactions, combined with their heterogeneous distribution throughout the vasculature, suggested that these NPs may be accumulating at specific sites due to discrete anatomical features of the tissue rather than binding to ECs. This raised the possibility that the difference observed between CD31-NPs and Control-NPs could have resulted from differences between the two kidneys rather than the nature of the NPs. Thus, although these results are consistent with an enhancement of accumulation of CD31-NP relative to Control-NP after 4 hours of perfusion, we next developed an approach to compare specific versus nonspecific accumulation simultaneously in the same kidney.
Dose escalation and two-color analysis of specific versus nonspecific NP accumulation
To determine the extent of specific versus nonspecific NP accumulation within an individual organ, we used a two-color NP approach where red-labeled (DiI dye) CD31-NPs and green-labeled (DiO dye) Control-NPs were simultaneously administered to each perfused organ; relative accumulation was then determined by quantifying the ratio of the two NP signals. The two-color NPs were matched for intensity under identical imaging and analysis conditions, as used for perfused kidney sections (fig. S2). The Control-NPs were specifically chosen for the green DiO channel to ensure that any tissue autofluorescence (which is predominately present in the green channel; fig. S3) did not artificially inflate the calculated degree of targeting specificity.
The analysis of kidney 1 confirmed that CD31-NP targeting of renal ECs during NMP is time-dependent. Because our in vitro studies showed that increasing NP concentration increased the rate of delivery to ECs, we next focused on optimizing the concentration of NPs in the perfusate fluid above the initial concentration tested (4 μg/ml). A mixture of red CD31-NP (DiI) and green Control-NP (DiO) were administered to three donor kidneys, each at matched doses of 10, 50, and 100 μg/ml. Individual images of either glomeruli (masked to exclude any surrounding microvessels) or interstitial microvasculature (masked to exclude any glomerular vessels if present) taken within cryosections cut from wedge biopsies collected at various time points after perfusion were evaluated by quantitative microscopy. Quantification of the mean intensity per unit vascular area of the CD31-NP within glomeruli for all four NP doses revealed an apparent linear dependence on NP concentration (R2 = 0.465, as determined by nonlinear regression analysis), consistent with NP delivery under non-saturating conditions (fig. S4). This linear dependence of mean NP intensity on NP dose demonstrates the reliability of our quantitative microscopy approach. The dose titration also revealed that, although all three doses provided sufficient signal for quantitative evaluation in both glomeruli and interstitial microvessels, the intensity at NP concentrations of 50 and 100 μg/ml allowed for optimal imaging conditions that maximized NP signal and minimized green-channel autofluorescent artifacts from the tissue (fig. S5).
On the basis of the presence of apparent nonspecific accumulation in the single-color Control-NP kidney, we hypothesized that such areas would accumulate roughly equal amounts of red (CD31-NP) and green (Control-NP). A salient feature observed in all human kidneys treated with two-color NPs were distinct regions of specific (red only) versus nonspecific (yellow, about equal amounts of red and green) accumulation (Fig. 4, A to C, and fig S5). Green-only areas, representing accumulation of Control-NPs without CD31-NPs, were rarely observed and occurred at a frequency commensurate with the green-channel tissue autofluorescence present in tissues collected before NP introduction (fig. S3). Quantitative evaluation of yellow regions, either by line scan or total region evaluation, confirmed the nonspecific (about 1:1) nature of NP accumulation in these regions (Fig. 4, D and E). Within the same field of view as these nonspecific regions, there were other regions with distinctly higher CD31-NP–specific accumulation (about five- to sevenfold enhancement of CD31-NP relative to Control-NP in the highlighted regions shown in Fig. 4). The presence of regions of about 1:1 CD31-NP/Control-NP accumulation in the same field of view and within the same cryosection as other vessels with five- to sevenfold higher values of CD31-NP–specific accumulation further validated our quantitative microscopy–based approach and supports the conclusion that CD31-NPs are capable of enhancing retention in regions of the kidney vasculature that do not otherwise accumulate NPs. In contrast, other regions of the same kidney accumulate NPs by a different mechanism that is not improved by targeting. In both cases, NP accumulation was exclusively intravascular.
Fig. 4. Specific versus nonspecific NP accumulation.

Representative thresholded 20× fluorescent images of (A) glomeruli from the NP kidney (10 μg/ml), (B) interstitial microvessels from the NP kidney (50 μg/ml), and (C) glomeruli and interstitial microvessels from the NP kidney (100 μg/ml) with white boxed regions highlighting areas of either specific or nonspecific accumulation within the same field of view or same section (DAPI, blue; CD31-NPs, red; Control-NPs, green; NP overlay, yellow; Ulex, white). The boxed regions are shown at a larger scale in the insets for both (A) and (B). The white dashed lines across the insets in (A) refer to the line scale analysis shown in (D) for nonspecific (region 1) and specific (region 2) accumulation. Scale bars, 50 μm. (D) Line scan plot of highlighted regions of NP glomerular accumulation in (A) showing similar amounts of accumulation in nonspecific regions with many more CD31-NPs relative to Control-NPs in more specific regions. (E) Region quantifications depicting the total relative NPs signal (CD31-NPs/Control-NPs) for the individual highlighted regions in (A) to (C); each bar represents the quantification of the single image shown for the respective panel.
To better understand the abundance of specific versus nonspecific accumulation, we quantified the total relative amount of CD31-NP versus Control-NP at 4 hours across all doses in both glomeruli and interstitial microvessels. Consistent with previous results in kidneys perfused with single-color NPs (Fig. 3), CD31-NPs accumulated at higher quantities compared to Control-NPs in all three kidneys evaluated (relative CD31-NP/Control-NP signal, >1). The enhanced accumulation of CD31-NPs was statistically significant in both glomeruli and interstitial microvessels for all kidneys, although the degree of specificity varied between the different vascular beds for each NP dose (P < 0.001; Fig. 5A). Maximal CD31-NP enrichment at about twofold above the Control-NP was roughly similar across NP doses, although the most specific vascular bed varied among the three kidneys tested (interstitial microvessels for 10 and 50 μg/ml and glomeruli for 100 μg/ml; Fig. 5A). These values were generally consistent across several different time points (different wedge biopsy samples) in each of the kidneys (Fig. 5B). This variation in targeting benefit among different vascular beds within the same kidney and between different kidneys suggests that factors intrinsic to each organ may modulate the balance of CD31-specific versus nonspecific NP accumulation.
Fig. 5. Global analysis of CD31-NP targeting benefit in the renal cortex.
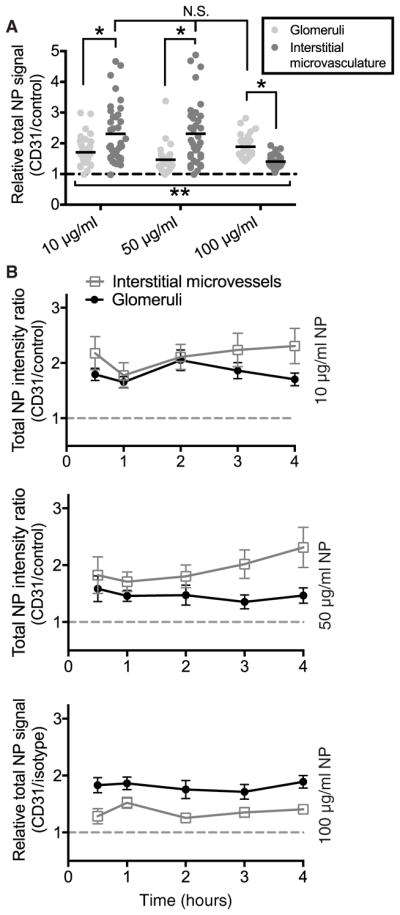
(A) Quantification of relative NP signal (CD31-NPs/Control-NPs) at 4 hours for kidneys 3 to 5 (10, 50, and 100 μg/ml, respectively). Each point represents an individual image of either glomeruli (light gray) or interstitial microvessels (dark gray) with mean value shown as the black line; nonspecific (1:1) accumulation is represented by dashed gray line. **P < 0.001 for all values compared to 1 (nonspecific) using Wilcoxon signed-rank test. *P < 0.004 using Mann-Whitney test. (B) Relative CD31-NP/Control-NP intensity for images taken from wedge biopsies collected at the given time point for kidneys 3 to 5. Values are shown as mean with error bars signifying a 95% confidence interval of the mean. n for each condition is provided in Table 3.
Variation in CD31-NP targeting in perfused human kidneys under identical conditions
Our second round of experiments both validated our two-color NP-targeting approach and identified optimal experimental conditions for quantitative microscopy. However, the variations in NP concentrations administered to the kidneys analyzed in the previous three organs limited our ability to directly assess how the benefits of CD31 targeting may vary from kidney to kidney. In particular, we were not able to assess how the quality of a given organ might affect specific versus nonspecific NP accumulation. Three additional donor kidneys (kidneys 6 to 8) were perfused with 50 μg/ml each of CD31-NP (DiI-red) and Control-NP (DiO-green) under identical perfusion and sampling conditions. High-resolution (40×) image analysis was performed on cryosections taken from wedge biopsies collected before NP introduction and at 2, 4, and 6 hours after NP administration. The perfusion period was extended to a total of 8.5 hours for each kidney to provide a longer period for assessment of viability. In addition, several relevant physiological parameters were characterized along with gross appearance and histological analysis (Table 2). The histological analysis did not identify major differences among the three organs, although the macroscopic appearances of kidneys 6 and 7 were notably better than that of kidney 8 (Table 1). Kidney 8 also demonstrated a decline in urine production after 4 hours, further suggesting that the quality of this kidney was not as high as kidneys 6 and 7.
Table 2. Functional characterization and histological analysis of kidneys 6 to 8.
The mean RBF, urine output, level of oxygen consumption, and perfusate pH levels at baseline (after 30 min of perfusion but before the administration of NPs), 2 hours, 4 hours, 6 hours, and 8 hours of perfusion. ATN, acute tubular necrosis.
| Case 6 | Case 7 | Case 8 | |
|---|---|---|---|
| RBF (ml/min per 100 g) | |||
| Baseline | 52 | 41 | 25 |
| 2 hours | 60 | 82 | 78 |
| 4 hours | 76 | 90 | 98 |
| 6 hours | 96 | 102 | 119 |
| 8 hours | 120 | 107 | 135 |
| Urine output (ml) | |||
| Baseline | 55 | 9 | 8 |
| 2 hours | 111 | 54 | 55 |
| 4 hours | 135 | 126 | 101 |
| 6 hours | 154 | 127 | 81 |
| 8 hours | 143 | 135 | 12 |
| Total | 598 | 451 | 257 |
| Oxygen consumption (ml/min per g) | |||
| Baseline | 38.5 | 46.0 | 57.9 |
| 2 hours | 43.9 | 61.0 | 56.6 |
| 4 hours | 52.2 | 67.4 | 70.9 |
| 6 hours | 70.2 | 75.8 | 52.1 |
| 8 hours | 84.7 | 81.0 | 99.4 |
| PH | |||
| Baseline | 7.21 | 7.23 | 7.34 |
| 2 hours | 7.27 | 7.41 | 7.35 |
| 4 hours | 7.34 | 7. 42 | 7.35 |
| 6 hours | 7.37 | 7.38 | 7.28 |
| 8 hours | 7.37 | 7.40 | 7.28 |
| Histology | |||
| Baseline | No ATN | No ATN | No ATN |
| 2 hours | No ATN | No ATN | No ATN |
| 4 hours | No ATN | No ATN | No ATN |
| 6 hours | No ATN | Focal mild ATN | No ATN |
| 8 hours | Focal mild ATN | No ATN | Focal mild ATN |
The two kidneys with the best macroscopic appearance (kidneys 6 and 7) also had the highest average enhancement of CD31-NP accumulation relative to Control-NP in both glomeruli and interstitial microvessels at 2, 4, and 6 hours of NP perfusion (Fig. 6, A and B). Although there were individual glomeruli and regions of microvessels within these two kidneys, where the accumulation was largely non-specific, there were multiple glomeruli and areas of interstitial micro-vessels where the CD31-NP accumulated at values about 5 to10 times more that of the Control-NP (Fig. 6, A to C); one condition (kidney 7 at 4 hours) had a few regions where targeting benefit exceeded 20 times. In contrast, kidney 8 had generally reduced NP-targeting benefit that appeared to grow progressively worse over the course of 6 hours, concomitant with the decline in urine production (Fig. 6, A to C). These results further demonstrate that CD31-NPs are capable of accumulating via both specific and nonspecific means and that the degree of CD31-specific accumulation varies significantly among organs and in a manner that appears to correlate with the condition of the organ.
Fig. 6. Effects of CD31-NP targeting in perfused human kidneys.

Quantification of relative NP signal (CD31-NPs/Control-NPs) at 4 hours (h) for kidneys 6 to 8. Each point represents an individual image of either interstitial microvessels (A) or glomeruli (B); nonspecific (1:1) accumulation is represented by dashed gray line. **P < 0.0001 and *P = 0.0024 according to a Wilcoxon signed-rank test. Colored data points correspond to the quantification of the higher (red) or lower (green) specificity images shown in (C). (C) Representative images of either higher or lower specificity regions of NP accumulation in interstitial microvessels of kidney 6 (left) or glomeruli of kidney 7 (middle) and lower specificity regions in glomeruli of kidney 8 (right; DAPI, blue; CD31-NPs, red; Control-NPs, green; NP overlay, yellow; Ulex, white). Scale bars, 50 μm. n for each condition is provided in Table 3.
Effects of vascular occlusion on nonspecific NP accumulation
Confocal imaging revealed that nonspecific NP accumulation within the interstitial microvessels occurred primarily within the lumen of vessels, suggesting that this nonspecific accumulation was not occurring because of engulfment by extravascular, tissue-resident phagocytic cells (Fig. 7A). Moreover, nonspecific NP accumulation localized to intra-vascular regions that contained RBC-shaped structures stained by Ulex Europaeus Agglutinin 1 (Ulex; which stains RBCs in addition to ECs; Fig. 7B). This feature suggests that RBC-enriched vascular plugs may act as a sieve to accumulate high amounts of NPs in a nonspecific manner. Quantitative imaging of nonspecific regions that colocalized with high-density RBC obstructions labeled by glycophorin A staining confirmed the nonspecific nature of this mode of accumulation (Fig. 7, C and D). To better understand the basis of nonspecific sites of NP accumulation, we evaluated formalin-fixed, paraffin-embedded sections of selective kidney biopsies. Hematoxylin and eosin and phosphotungstic acid hematoxylin staining of samples of kidney 5 (fig. S6), which had substantial nonspecific accumulation of NPs in interstitial microvessels (Fig. 6A), revealed that there were extensive areas of vascular plugging by RBCs and RBC fragments that were present before NP introduction (Fig. 7A). Widespread vessel plugging in the cortex and medulla persisted throughout the 4 hours of perfusion and was correlated with visual indicators of poor kidney perfusion (Fig. 6A and fig. S6).
Fig. 7. NPs accumulate at sites of vascular obstruction.

(A) Three-dimensional projection of a z-stack of confocal images of a vessel containing a high density of nonspecific NP accumulation in its lumen (DAPI, blue; CD31-NPs, red; Control-NPs, green; NP overlay, yellow; Ulex, white). Dashed white line represents the point of apparent obstruction, and arrows highlight apparent red blood cells (RBCs). Scale bar, 10 μm. (B) Representative image of non-specific colocalization of NPs with sites of high-density RBC (glycophorin A, white; nonspecific NP, yellow) presumably representing vascular obstruction. Scale bar, 10 μm. (C) Quantification of relative NP signal (CD31-NP/Control-NP) for image in (B). (D) Representative hematoxylin and eosin–stained images of kidney 5 (100 μg/ml) before NP introduction (pre-NP) or after 4 hours of NP perfusion. RBC occlusions are stained red. Scale bars, 100 μm.
These plugs did not appear to be classical thrombi because they showed minimal amounts of overt fibrin. The lack of fibrin is not unexpected, given that the perfusate consists of washed RBCs and lacks fibrinogen, but we cannot rule out the possibility that fibrin microthrombi were present downstream and caused an RBC- and NP-rich plug to back up behind the obstruction. Alternatively, these plugs could represent vascular constrictions despite the presence of a vasodilator in the perfusion solution. Similar vascular plugs were observed to varying degrees in all kidneys in both glomeruli and interstitial microvessels; however, there was substantial variability in the degree of obstruction present between biopsies sampled at different time points and in different regional locations. This heterogeneity limits our ability to draw a quantitative correlation between vascular obstruction and nonspecific NP accumulation. Overall, gross evidence of poor perfusion provided a better correlation with nonspecific NP labeling.
DISCUSSION
Although molecular targeting of drug carriers is often referred to as “active” targeting (36), molecular interaction forces (hydrogen bonds, electrostatic interactions) cannot provide a physical force that actively pulls the conjugate to a site of disease because these forces only operate over very small distances (<0.5 nm) (37). Thus, molecular targeting to a cell actually works by enhancing the likelihood that a targeted NP will be retained by a target cell with which it happens to come in contact. The maximal benefit from the addition of targeting ligands to NPs requires optimization of the route of administration to ensure that the NPs come in direct contact with the target cell of interest. For intravascular administration, ECs represent an ideal target cell type because NPs do not need to escape from the vasculature to reach them. Notably, EC-targeted NPs have been shown to enhance delivery to lung endothelium in vivo, likely owing to the large surface area of ECs in lung capillaries (38). NMP provides an opportunity to isolate the targeted organ and thereby ensure that the delivered NPs only come in direct contact with the endothelium of interest. NMP also circumvents competition with phagocytic cells of the liver and spleen, which tend to substantially dilute any benefit associated with in vivo administration of molecularly targeted NPs. In addition, NMP is typically performed with a serum-free perfusate, which further alleviates issues associated with masking of targeting ligands by a protein corona (23, 24). In this regard, NMP represents, in many ways, the ideal setting for realizing the maximal benefit of molecular targeting.
Despite this idealized nature of the NMP setting, we observed a marked reduction between the degree of targeting benefit observed during in vitro cell culture (under either static or flow conditions) versus an intact human organ. Using static in vitro cell culture assays, CD31-NPs accumulated as much as 80-fold above that of Control-NPs. However, when delivered to human kidneys by NMP, we found that targeted CD31-NPs accumulate only about two- to fourfold above the level of Control-NPs in the vasculature of the renal cortex when evaluated from a global average perspective. This two- to fourfold enhancement may seem relatively poor in comparison to previous results in in vivo animal models of CD31-NP targeting to vascular EC, where benefits have been observed as high as 10- to 20-fold. However, our detailed analysis suggests that this two- to fourfold enhancement results from taking an average across both nonspecific and CD31-specific modes of accumulation. These differences demonstrate the benefits of using a microscopy-based approach as an alternative or complement to whole-tissue quantification that necessarily loses information regarding the spatial heterogeneity of accumulation. Moreover, the presence of these discreet regions of about 1:1 NP signal in both glomeruli and interstitial microvessels served as an internal validation of our quantitative microscopy approach.
The relative abundance of the nonspecific accumulation appears to be coupled to the condition of the organ such that regional poor perfusion leads to more significant nonspecific accumulation. A recent study in a spatially heterogeneous mouse model of acute respiratory distress syndrome found a similar enhancement of nontargeted carriers in regions of organ damage (39). Thus, our findings would seem to corroborate in human organs this previous observation in animal models. In addition to both nonspecific and specific sites of vascular accumulation, there were also areas of vasculature that accumulated no detectable NPs in both glomeruli and interstitial microvessels, although this appeared to be more pronounced in microvessels. Such differences may, in part, be driven by the amount of surface expression and/or accessibility of the target receptor, which may vary between and within vascular beds and vessel types. Such heterogeneity is not reflected in the static in vitro binding assays commonly used as an initial screen for NP-targeting efficacy. The discrepancy we observed in human kidneys highlights the need for model systems that are more representative of human anatomy and physiology to better translate NP therapeutic strategies from bench to bedside. In this regard, NMP of isolated human organs holds significant potential not only as an approach with intrinsic value to clinical transplantation, but also as an experimental tool for the refinement of new therapeutic strategies for in vivo drug delivery.
Renal vascular ECs are a potentially important target for therapies to improve transplant outcomes. These cells are highly susceptible to both ischemia-reperfusion injury and damage by preexisting donor-specific Abs that target the nonself allelic forms of human leukocyte antigens (HLAs), which are highly expressed on human microvacular endothelium. In both cases, injured or activated human ECs can enhance a destructive host T cell–mediated rejection response (40, 41). Current approaches to mitigate the effects of preformed donor-specific Abs have focused on removing such Abs from the circulation, but the benefits of this approach are limited because the cellular sources of these Abs are difficult to eradicate and have the potential to rapidly restore antidonor Ab levels. NPs could be used to improve this therapy by providing a sustained reduction in HLA antigen expression by graft ECs, allowing the organ a chance to “heal in” and providing a period in which the organ may accommodate to the presence of recurrent Ab (42). This approach could be used to complement a reduction in donor-specific Ab titer by plasmapheresis, an approach that is less effective on its own in patients with higher Ab titers. In support of this concept, we have recently shown that degradable NPs—similar to the ones used here but loaded with an siRNA to class II transactivator—administered ex vivo to human vessels can suppress class II HLA expression in the ECs for 6 weeks or longer after implantation of that vessel into an immuno-deficient mouse host (17). Other targets for siRNAs or drugs may also be relevant, and the identification of the best targets will require empiric validation. The modularity of our polymeric NP system will allow us to change the encapsulated therapeutic and the kinetics of drug release by varying polymer composition and/or molecular weight.
Determining the functional capacity and viability of organs undergoing NMP remains a substantial clinical challenge. Although the observation period afforded by NMP, in principle, allows for better diagnostic evaluation of the organ viability, in practice, there remains a need for better diagnostic tools. So-called “theranostic” NPs that are capable of providing both therapeutic and diagnostic functionality have gained increasing focus in recent years (43). The extent of poorly perfused regions of vasculature is likely a critical parameter in determining the potential benefits of NMP. Our findings suggest the possibility that a high degree of nonspecific accumulation may be indicative of vascular obstruction. Consequently, NPs may also be a valuable tool for assessing organ viability if coupled with a noninvasive, clinically compatible imaging methodology (for example, magnetic resonance imaging or ultrasound). Moreover, as we begin to better understand the nature and cause of these obstructions, it may also be possible to couple enzymes or mediators to the surface of NPs such that they could also aid in reducing sites of poor perfusion. Given these features, as well as the absence of the impediments that have hindered the translation of targeted NPs to date, we posit that ex vivo organ NMP may provide a successful path to clinical translation for targeted nanomedicines.
MATERIALS AND METHODS
Study design
The primary objective of this study was to evaluate whether surface conjugation to polymeric NPs of an Ab reactive with an EC surface molecule could enhance NP targeting to vascular ECs during ex vivo NMP of human kidneys. We adapted an established approach for conjugating mouse monoclonal Ab to fluorescent dye–loaded PLA-PEG NPs of a consistent diameter and initially used these NPs to analyze attachment to cultured HUVECs under static conditions or in a microfluidic flow chamber under conditions of shear that more closely resemble those created during ex vivo NMP. In these initial experiments, we compared anti-CD31–conjugated NPs (CD31-NP) to isotype-matched irrelevant Ab-conjugated NPs (Control-NP) and quantified the effects of NP concentration and duration of treatment by fluorescence microscopy and fluorescence flow cytometry. All in vitro experiments were done in triplicate. We then analyzed NP targeting to ECs in eight human kidneys that had been declined for transplant during ex vivo NMP under the same conditions, as would be applied to organs used for transplantation.
The study was carried out in three stages. First, a single-color approach was used in the two kidneys that were treated separately with either CD31-NP or Control-NP. As detailed below, we then developed a two-color quantitative microscopy approach to produce paired images of CD31-NP relative to Control-NP in the same kidney; this approach was used in three other human kidneys in which we also varied the NP concentration to identify optimal conditions for quantitative imaging analysis. Finally, an additional set of three kidneys was perfused with the two-color NP approach using optimal concentration and sampling conditions, as determined in the second stage.
In each stage, images of wedge biopsies from each kidney sampled at various time points after NP perfusion were collected from three to four cryosections with about eight or more independent images of either glomeruli or interstitial microvessels. The total number of images for each condition is provided in Table 3. A magnification of 20× was used for imaging in stage 2 and the number of images (Table 2) was dictated by the relative area of each cryosection with a minimum of three cryo-sections sampled for each condition. In stage 3, 40× imaging was used to provide better discrimination between areas of specific and nonspecific accumulation. Under these higher-magnification conditions, there were some images of microvasculature, which showed essentially no NP accumulation; in all kidneys, microvascular NP accumulation was highly heterogeneous with some vessels showing robust accumulation and others showing very little or no NP accumulation; there was also one early time point condition (kidney 6 at 2 hours) where several glomeruli had no measureable NP signal (later time points showed robust NP accumulation, so this may simply be a kinetic effect in some glomeruli). In instances with exceedingly low or no NP accumulation, the ratio of CD31-NP to Control-NP cannot be reliably calculated. Consequently, such images lacking appreciable NP signal were necessarily excluded from analysis. Under most of the conditions, there were less than 5 images per 40 total images that lacked sufficient NP signal for analysis. For biopsies where the number of images with no NP signal exceeded five, additional sections were imaged to ensure a similar n for each condition. The total n for each condition is provided in Table 3.
Table 3. Number of images collected per sample and condition.
N/A refers to conditions that were not analyzed within the manuscript.
| Kidney | Time point (hours) | Glomeruli (n) | Microvasculature (n) | |
|---|---|---|---|---|
| Stage 1 (kidneys 1 and 2) | 1 | 0.5 | 27 | N/A |
| 1 | 29 | N/A | ||
| 3 | 26 | N/A | ||
| 4 | 35 | 59 | ||
| 2 | 0.5 | 29 | N/A | |
| 1 | 29 | N/A | ||
| 3 | 25 | N/A | ||
| 4 | 31 | 56 | ||
| Stage 2 (kidneys 3 to 5) | 3 | 0.5 | 31 | 48 |
| 1 | 26 | 31 | ||
| 2 | 23 | 38 | ||
| 3 | 44 | 50 | ||
| 4 | 48 | 37 | ||
| 4 | 0.5 | 20 | 23 | |
| 1 | 33 | 30 | ||
| 2 | 22 | 39 | ||
| 3 | 22 | 36 | ||
| 4 | 37 | 35 | ||
| 5 | 0.5 | 22 | 20 | |
| 1 | 30 | 22 | ||
| 2 | 24 | 36 | ||
| 3 | 33 | 32 | ||
| 4 | 38 | 31 | ||
| Stage 3 (kidneys 6 to 8) | 6 | 2 | 39 | 37 |
| 4 | 40 | 33 | ||
| 6 | 40 | 36 | ||
| 7 | 2 | 38 | 39 | |
| 4 | 40 | 35 | ||
| 6 | 40 | 40 | ||
| 8 | 2 | 40 | 48 | |
| 4 | 40 | 37 | ||
| 6 | 40 | 36 |
Ethical approval
In vitro experimentation with HUVECs from de-identified sources has been classified as nonhuman subject research by the Yale’s Institutional Review Board. Ethical approval for experiments with human kidney was granted by the National Research Ethics Commission in the United Kingdom. Consent for the use of the organs for research was obtained from the donor family by the specialist nurses in organ donation before organ retrieval. Kidneys were retrieved by one of the U.K. National Organ Retrieval Services teams. After in situ flushing of the abdominal organs with cold preservation solution, kidneys were retrieved and then stored by static cold storage. Eight kidneys that were deemed unsuitable for transplantation were recruited into the study.
Materials
PLA-PEG copolymer (molecular weight, 16 to 5 kDa) was purchased from PolySciTech and used as received without further purification. Anhydrous dimethyl sulfoxide (DMSO), DiI and DiO stains, and Fisherbrand Superfrost Microscope Slides were purchased from Life Technologies. Ulex vascular stain was purchased from Vector Laboratories either preconjugated with fluorescein isothiocyanate or unconjugated for later conjugation to Alexa Fluor 633–N-hydroxysuccinimide following the vendor’s supplied protocol.
NP formulation
NPs were prepared by a nanoprecipitation procedure (44). PLA-PEG was dissolved at a concentration of 100 mg/ml in DMSO and then diluted to the desired concentration for NP formulation (typically about 55 mg/ml for about 165-nm NPs used in this study), along with addition of either DiI or DiO dye also dissolved in DMSO. NPs were loaded with DiI or DiO dye at a final dye/polymer ratio of 0.5% (w/w). The dye/polymer solution in DMSO was added dropwise to vigorously stirring sterile deionized H2O (diH2O) in batches of 300 μl of polymer/dye solution added to 1.2 ml of diH2O, with identical repetitions performed to generate a full NP batch. NPs were subsequently filtered through a 1.2-μm cellulose acetate membrane (GE Healthcare Life Sciences) filter to remove any free-dye or polymer aggregates and then pooled. The NP solutions were then transferred in batches to dialysis cassettes (12-ml volume; molecular weight cutoff, 10,000; Slide-A-Lyzer, Thermo Fisher Scientific) and dialyzed against two exchanges of about 2.2 liters of diH2O at room temperature to remove excess DMSO. After dialysis, NPs were aliquoted and snap-frozen in liquid N2. One aliquot from each NP batch was lyophilized in a preweighed tube to determine the NP concentration. Standard NP concentration was typically about 5 mg/ml before Ab conjugation.
Ab surface conjugation
Ab-conjugated PLA-PEG NPs were generated via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride–mediated COOH-NH2 coupling. Commercial Abs were purchased from BD Biosciences (CD31, clone WM59; Isotype, clone MOPC-21) and subsequently dialyzed overnight in dialysis cassettes (0.5 ml; molecular weight cutoff, 10,000; Slide-A-Lyzer, Thermo Fisher Scientific) to remove sodium azide, which can inhibit the coupling reaction. After dialysis, the protein concentration was adjusted (if necessary) to 0.55 mg/ml by addition of sterile Dulbecco’s phosphate-buffered saline (Thermo Fisher Scientific). Detailed procedure is provided in Supplementary Material and Methods.
The number of Abs on the NP surface was estimated using Abs that had been prelabeled with I125 from PerkinElmer using chloramine-T, as previously described in the literature (45). The labeling efficiency was 50 pCi/ng (110 cpm/ng). The I125-labeled Ab was used in the identical conjugation procedures described above, and then, a known concentration of NP conjugated with Ab was evaluated relative to a standard at a known Ab concentration after extensive purification of the NP via centrifugation to remove any free Ab via scintillation counting.
Cell culture
HUVECs pooled from three separate donors were obtained from the Yale Vascular Biology and Transplantation tissue culture core laboratory, where they were isolated from fresh umbilical veins by treatment with collagenase and subsequently cultured in gelatin-coated tissue culture plastic plates using M199 medium supplemented with 20% v/v fetal bovine serum, 2% L-glutamine, 1% penicillin and streptomycin, and 1% (v/v) EC growth supplement (fibroblast growth factor-1). All cell culture experiments were performed using cells passaged four or fewer times. For static NP incubation, cells were cultured for at least 4 to 5 days after reaching full confluence to ensure the formation of mature cell-cell junctions to best mimic in vivo conditions. The medium was exchanged every 2 to 3 days during these culture periods to ensure that the cells remained viable. Cells were visually inspected before all experiments to ensure cell viability.
In vitro evaluation of NP per cell
HUVECs were plated on a gelatin-treated glass-bottom 24-well plate. Once the cells reached confluence, the medium was exchanged with that containing 50 μg/ml each of red CD31-NP and green Control-NP, as used in kidney perfusion experiments. The NPs were incubated for 30 min and then washed extensively to remove any free NPs. Cells were stained with a 1:20,000 dilution of Hoechst to stain the nuclei and then imaged immediately under live-cell conditions with a 40× Olympus apochromatic air immersion objective [numerical aperture (NA), 0.90]. Three separate wells were each imaged in triplicate, and the images were subsequently analyzed in MATLAB (data file S1) for the number of NP per 100 μm2.
In vitro evaluation of simultaneous administration of two-color CD31-NP and Control-NP
HUVECs were plated on a gelatin-treated glass-bottom 24-well plate. Once the cells reached confluence, the medium was exchanged with that containing 50 μg/ml each of red CD31-NP and green Control-NP either individually or as a combined treatment. The NPs were incubated for 2 hours and then washed extensively to remove any free NPs before immediate imaging under live-cell conditions with a 20× Olympus apochromatic air immersion objective (NA, 0.95). Three separate wells were each imaged two to three times each, and the images were subsequently analyzed for the number of NPs per 20× field of view.
Ex vivo NMP
After arrival, the kidneys were prepared, and the renal artery, vein, and ureter were cannulated. The kidneys were weighed before undergoing ex vivo NMP (3, 5). Briefly, the ex vivo kidney perfusion circuit was designed using pediatric cardiopulmonary bypass technology (Medtronic) and consisted of a centrifugal blood pump (Bio-Pump 560), a heat exchanger (Chalice Medical), a venous reservoir (Medtronic), 1/4;-inch polyvinyl chloride tubing, and a Pixie membrane oxygenator (Medtronic). The hardware included a speed controller, a TX50P flow transducer, and a temperature probe (Cole-Parmer). Two Alaris infusion pumps (CareFusion) were also incorporated into the system. The circuit was primed with a perfusate solution (Ringer’s solution, Baxter Healthcare) and one unit of ABO-compatible cross-matched packed red cells from the blood bank. Twenty-five milliliters of 10% mannitol (Baxter Health-care), 8 mg of dexamethasone (Organon Laboratories), and 2 ml of heparin (1000 IU/ml; CP Pharmaceuticals) were added to the perfusate. Sodium bicarbonate (8.4%; Fresenius Kabi) was added to normalize the pH. A nutrient solution (NuTRIflex, B. Braun) with 25 ml of 8.4% sodium bicarbonate, 100 IU of insulin (Novo Nordisk), and multivitamins (Cernevit, Baxter Healthcare) was infused into the circuit at a rate of 20 ml/hour. Prostacyclin (0.5 mg; Flolan, Glaxo Wellcome) was infused into the arterial arm of the circuit at a rate of 4 ml/hour, and 5% glucose (Baxter Healthcare) was infused at 7 ml/hour. Ringer’s solution was used to replace urine output milliliter-for-milliliter.
Kidneys were perfused at a set mean arterial pressure (70 to 75 mmHg). The plasma-free RBC-based perfusate was circulated from the venous reservoir through the centrifugal pump into the membrane oxygenator, where it was oxygenated and also warmed to 36°C. It then flowed through the arterial limb of the circuit to the renal artery. Venous return from the renal vein was fed back into the reservoir (5). Renal blood flow (RBF) was monitored continuously during NMP, and the total urine output was recorded. Preperfusion wedge biopsies and wedge biopsies were collected at various time points, as noted in Results. The tissue was either fixed in 10% formal saline or snap-frozen in liquid nitrogen and then stored at −80°C. Samples of perfusate and urine were also collected at the same time points and stored at −80°C.
The quality of each kidney was assessed on the basis of the macroscopic appearance, the mean RBF, and the total amount of urine produced. For macroscopic assessment, each kidney was categorized into one of three groups and scored according to its macroscopic appearance as follows: (i) grade I, excellent perfusion (global pink appearance); (ii) grade II, moderate perfusion (patchy pink and purple appearance, which either remained or improved during NMP); (iii) grade III, poor perfusion (global mottling and purple and black appearance, which remained throughout NMP). Kidneys with a mean RBF below the threshold (<50 ml/min per 100 g) were given an additional score of 1. Kidneys producing less than the threshold volume of urine (<43 ml/hour) were also given an additional score of 1. Therefore, overall NMP assessment scores can range from 1 (indicating the least injury) to 5 (the most severe) (Table 2). Six of the eight kidneys enrolled in this study had an NMP assessment score of 2 (kidneys 1 to 5 and 8), whereas kidneys 6 and 7 had a score of 1. Kidney assessment and scoring rubric are shown in Table 4.
Table 4.
Macroscopic assessment, thresholds of RBF, and urine output.
| Assessment | Score |
|---|---|
| NMP assessment score during ex vivo NMP | |
| Macroscopic assessment | |
| Grade I: excellent perfusion (global pink appearance) | 1 |
| Grade II: moderate perfusion (patchy appearance) | 2 |
| Grade III: poor perfusion (global mottled and purple and black appearance) | 3 |
| RBF | |
| Threshold, ≥50 ml/min per 100g | 0 |
| Threshold, <50 ml/min per 100g | 1 |
| Total urine output | |
| Threshold, ≥43 ml | 0 |
| Threshold, <43 ml | 1 |
Quantitative microscopy on tissue biopsy sample
Snap-frozen wedge biopsies were cryosectioned on a microtome to a thickness of either 10 or 20 μm. Sections were imaged without fixation to avoid any alteration in dye-loaded NP signal. Ulex staining was performed rapidly with about 15 min of Ulex (Vector Laboratories) conjugated with Alexa Fluor 633 (Life Technologies) after standard conjugation procedures. After staining, sections were washed three times for 5 min each with DPBS and then mounted with Hardmount medium (Vector Laboratories) and coverslipped. Independent images were collected by raster scanning across cryosections, with typically three sections per condition and typically at least seven or more images of regions containing glomeruli or solely interstitial microvessels per section (corresponding to at least 15 images per section typically). Images were sorted, filtered, and processed using custom MATLAB code to distinguish glomerular vessels from surrounding microvasculature. Finally, the background-subtracted NP fluorescent signal was quantified for each image; backgrounds were determined by imaging sections from biopsies sampled before introduction of NP. For single-color experiments, these intensity values were then normalized to the vascular area of each corresponding image, as determined by Ulex staining. For two-color experiments, the total background subtracted signal from DiI (red) CD31-NPs was normalized to the DiO (green) isotype-NPs and expressed as a specificity ratio with 1 corresponding to nonspecific accumulations and values >1 corresponding to CD31-specific accumulation. Images were collected using a Semrock GFP-1828A bandpass filter set (excitation, 470 to 495 nm; emission, 505 to 535 nm) and a Chroma TRITC/DiI/Cy3 filter set (excitation, 525 to 550 nm; emission, 580 to 630 nm) with an air immersion apochromatic 20× objective (NA, 0.95; Olympus) or 40× objective (NA, 0.90; Olympus) on an Olympus Z71 inverted microscope illuminated by an light-emitting diode (LED) light source (Olympus L300) with each LED at full illumination intensity. These filter sets and illumination setup ensured no spectral bleed with these NPs, which is a prerequisite for proper implementation of this technique (fig. S1). Quantitative equivalence of the two-color NPs was confirmed by adding concentrated NPs to the sample mounting medium with minimal dilution. NPs were imaged in an identical plane as the positive Ulex stain to ensure that any signal absorption would be identical to the standard imaging conditions. All images were captured by an Olympus Retiga R6 CCD camera. Images of two-color NP accumulation are presented as thresholded images so that regions of low and high levels of accumulation can be equally appreciated. Data analysis, fitting, and statistical analysis were performed in GraphPad Prism.
Statistical analysis
Distributions of kidney tissue images were typically non-Gaussian, so all statistical analysis were performed using two-tailed Mann-Whitney test. The only exception was the two-tailed Wilcoxon signed-rank test used to test whether all two-color NP conditions were significantly above 1 (nonspecific accumulation). At least three cryosections were imaged to characterize each biopsy and each cryosection, with the total number of images for each condition provided in Table 3.
Supplementary Material
Acknowledgments
We would like to thank W. Sessa and his laboratory members (particularly R. Babbitt) for their extensive assistance with I125 labeling of Abs and evaluation of the number of Abs per NP. We would also like to thank A. Gaudin for the extensive discussion on experimental design and analysis. We are grateful to the Cambridge Biorepository for Translational Medicine for the access to human tissue.
Funding: This work was supported by the National Institute of Allergy and Infectious Diseases under T32 training grants AI089704, R21 AI126166, and R56 AI106992 and the National Heart, Lung, and Blood Institute under F32 fellowship grant HL131270. The research was additionally funded by the National Institute for Health Research (NIHR) Blood and Transplant Research Unit in Organ Donation and Transplantation at the University of Cambridge in collaboration with Newcastle University and in partnership with National Health Service Blood and Transplant (NHSBT). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the U.S. Department of Health, or the NHSBT.
Footnotes
www.sciencetranslationalmedicine.org/cgi/content/full/9/418/eaam6764/DC1
Materials and Methods
Fig. S1. CD31-NP characterization.
Fig. S2. Matching of two-color NP fluorescence.
Fig. S3. Tissue autofluoresence characterization.
Fig. S4. Effect of NP dose on average glomerular accumulation.
Fig. S5. Two-color NP accumulation.
Fig. S6. Vascular obstruction histology.
Data file S1. MATLAB image analysis code.
Author contributions: G.T.T. and S.A.H. designed the experiments, performed the data collection and analysis, and wrote the manuscript. J.D., J.C., D.D., E.S., J.R.K., A.S.P.-D., N.C.K.-S., and R.A.-L. performed the data collection and analyses. S.T. provided the pathology analysis. J.A.B, K.S.-P., and J.R.B advised on the experimental design and data analysis and assisted with manuscript writing. M.L.N, W.M.S., and J.S.P. designed the experiments, advised on the data collection and analysis, and wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.National Kidney Foundation. Organ Donation and Transplant Statistics. 2016 https://www.kidney.org/news/newsroom/factsheets/Organ-Donation-and-Transplantation-Stats.
- 2.Hosgood SA, Patel M, Nicholson ML. The conditioning effect of ex vivo normothermic perfusion in an experimental kidney model. J Surg Res. 2013;182:153–160. doi: 10.1016/j.jss.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson ML, Hosgood SA. Renal transplantation after ex vivo normothermic perfusion: The first clinical study. Am J Transplant. 2013;13:1246–1252. doi: 10.1111/ajt.12179. [DOI] [PubMed] [Google Scholar]
- 4.Hosgood SA, Nicholson ML. Ex vivo normothermic perfusion of declined human kidneys after inadequate in situ perfusion. Am J Transplant. 2014;14:490–491. doi: 10.1111/ajt.12568. [DOI] [PubMed] [Google Scholar]
- 5.Hosgood SA, Nicholson ML. First in man renal transplantation after ex vivo normothermic perfusion. Transplantation. 2011;92:735–738. doi: 10.1097/TP.0b013e31822d4e04. [DOI] [PubMed] [Google Scholar]
- 6.Moreau A, Varey E, Anegon I, Cuturi M-C. Effector mechanisms of rejection. Cold Spring Harb Perspect Med. 2013;3:a015461. doi: 10.1101/cshperspect.a015461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meier-Kriesche H-U, Schold JD, Kaplan B. Long-term renal allograft survival: Have we made significant progress or is it time to rethink our analytic and therapeutic strategies? Am J Transplant. 2004;4:1289–1295. doi: 10.1111/j.1600-6143.2004.00515.x. [DOI] [PubMed] [Google Scholar]
- 8.Chalasani G, Li Q, Konieczny BT, Smith-Diggs L, Wrobel B, Dai Z, Perkins DL, Baddoura FK, Lakkis FG. The allograft defines the type of rejection (acute versus chronic) in the face of an established effector immune response. J Immunol. 2004;172:7813–7820. doi: 10.4049/jimmunol.172.12.7813. [DOI] [PubMed] [Google Scholar]
- 9.Hosgood SA, van Heurn E, Nicholson ML. Normothermic machine perfusion of the kidney: Better conditioning and repair? Transpl Int. 2015;28:657–664. doi: 10.1111/tri.12319. [DOI] [PubMed] [Google Scholar]
- 10.Sis B, Jhangri GS, Bunnag S, Allanach K, Kaplan B, Halloran PF. Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am J Transplant. 2009;9:2312–2323. doi: 10.1111/j.1600-6143.2009.02761.x. [DOI] [PubMed] [Google Scholar]
- 11.Bian H, Reed EF. Alloantibody-mediated class I signal transduction in endothelial cells and smooth muscle cells: Enhancement by IFN-γ and TNF-α. J Immunol. 1999;163:1010–1018. [PubMed] [Google Scholar]
- 12.Brändle D, Joergensen J, Zenke G, Bürki K, Hof RP. Contribution of donor-specific antibodies to acute allograft rejection: Evidence from B cell-deficient mice. Transplantation. 1998;65:1489–1493. doi: 10.1097/00007890-199806150-00014. [DOI] [PubMed] [Google Scholar]
- 13.Chong AS, Alegre M-L. The impact of infection and tissue damage in solid-organ transplantation. Nat Rev Immunol. 2012;12:459–471. doi: 10.1038/nri3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellano G, Melchiorre R, Loverre A, Ditonno P, Montinaro V, Rossini M, Divella C, Battaglia M, Lucarelli G, Annunziata G, Palazzo S, Selvaggi FP, Staffieri F, Crovace A, Daha MR, Mannesse M, van Wetering S, Paolo Schena F, Grandaliano G. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol. 2010;176:1648–1659. doi: 10.2353/ajpath.2010.090276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbari A, Abbas S, Jaafar M. Approach to kidney transplant in sensitized potential transplant recipients. Exp Clin Transplant. 2012;10:419–427. doi: 10.6002/ect.2012.0136. [DOI] [PubMed] [Google Scholar]
- 16.Devalliere J, Chang WG, Andrejecsk JW, Abrahimi P, Cheng CJ, Jane-wit D, Saltzman WM, Pober JS. Sustained delivery of proangiogenic microRNA-132 by nanoparticle transfection improves endothelial cell transplantation. FASEB J. 2014;28:908–922. doi: 10.1096/fj.13-238527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui J, Qin L, Zhang J, Abrahimi P, Li H, Li G, Tietjen GT, Tellides G, Pober JS, Saltzman WM. Ex vivo pretreatment of human vessels with siRNA nanoparticles provides protein silencing in endothelial cells. Nat Commun. 2017;8:191. doi: 10.1038/s41467-017-00297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng CJ, Tietjen GT, Saucier-Sawyer JK, Saltzman WM. A holistic approach to targeting disease with polymeric nanoparticles. Nat Rev Drug Discov. 2015;14:239–247. doi: 10.1038/nrd4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi CHJ, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci USA. 2010;107:1235–1240. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R, Schlenk F, Fischer D, Kiouptsi K, Reinhardt C, Landfester K, Schild H, Maskos M, Knauer SK, Stauber RH. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013;8:772–781. doi: 10.1038/nnano.2013.181. [DOI] [PubMed] [Google Scholar]
- 23.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci USA. 2008;105:14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monopoli MP, Åberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 25.Kozower BD, Christofidou-Solomidou M, Sweitzer TD, Muro S, Buerk DG, Solomides CC, Albelda SM, Patterson GA, Muzykantov VR. Immunotargeting of catalase to the pulmonary endothelium alleviates oxidative stress and reduces acute lung transplantation injury. Nat Biotechnol. 2003;21:392–398. doi: 10.1038/nbt806. [DOI] [PubMed] [Google Scholar]
- 26.Scherpereel A, Rome JJ, Wiewrodt R, Watkins SC, Harshaw DW, Alder S, Christofidou-Solomidou M, Haut E, Murciano J-C, Nakada M, Albelda SM, Muzykantov VR. Platelet-endothelial cell adhesion molecule-1-directed immunotargeting to cardiopulmonary vasculature. J Pharmacol Exp Ther. 2002;300:777–786. doi: 10.1124/jpet.300.3.777. [DOI] [PubMed] [Google Scholar]
- 27.Danielyan K, Ding B-S, Gottstein C, Cines DB, Muzykantov VR. Delivery of anti-platelet-endothelial cell adhesion molecule single-chain variable fragment-urokinase fusion protein to the cerebral vasculature lyses arterial clots and attenuates postischemic brain edema. J Pharmacol Exp Ther. 2007;321:947–952. doi: 10.1124/jpet.107.120535. [DOI] [PubMed] [Google Scholar]
- 28.Pusztaszeri MP, Seelentag W, Bosman FT. Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J Histochem Cytochem. 2006;54:385–395. doi: 10.1369/jhc.4A6514.2005. [DOI] [PubMed] [Google Scholar]
- 29.Garnacho C, Albelda SM, Muzykantov VR, Muro S. Differential intra-endothelial delivery of polymer nanocarriers targeted to distinct PECAM-1 epitopes. J Control Release. 2008;130:226–233. doi: 10.1016/j.jconrel.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han J, Shuvaev VV, Davies PF, Eckmann DM, Muro S, Muzykantov VR. Flow shear stress differentially regulates endothelial uptake of nanocarriers targeted to distinct epitopes of PECAM-1. J Control Release. 2015;210:39–47. doi: 10.1016/j.jconrel.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han J, Zern BJ, Shuvaev VV, Davies PF, Muro S, Muzykantov V. Acute and chronic shear stress differently regulate endothelial internalization of nanocarriers targeted to platelet-endothelial cell adhesion molecule-1. ACS Nano. 2012;6:8824–8836. doi: 10.1021/nn302687n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hrkach J, Von Hoff D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra139. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 33.Hermanson GT. Bioconjugate Techniques. 3. chap. 17 Academic Press; 2013. [Google Scholar]
- 34.Dziubla TD, Shuvaev VV, Hong NK, Hawkins BJ, Madesh M, Takano H, Simone E, Nakada MT, Fisher A, Albelda SM, Muzykantov VR. Endothelial targeting of semi-permeable polymer nanocarriers for enzyme therapies. Biomaterials. 2008;29:215–227. doi: 10.1016/j.biomaterials.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tassa C, Duffner JL, Lewis TA, Weissleder R, Schreiber SL, Koehler AN, Shaw SY. Binding affinity and kinetic analysis of targeted small molecule-modified nanoparticles. Bioconjug Chem. 2010;21:14–19. doi: 10.1021/bc900438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu D, Tsai C-J, Nussinov R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997;10:999–1012. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- 38.Chacko A-M, Han J, Greineder CF, Zern BJ, Mikitsh JL, Nayak M, Menon D, Johnston IH, Poncz M, Eckmann DM, Davies PF, Muzykantov VR. Collaborative enhancement of endothelial targeting of nanocarriers by modulating platelet-endothelial cell adhesion molecule-1/CD31 epitope engagement. ACS Nano. 2015;9:6785–6793. doi: 10.1021/nn505672x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brenner JS, Bhamidipati K, Glassman PM, Ramakrishnan N, Jiang D, Paris AJ, Myerson JW, Pan DC, Shuvaev VV, Villa CH, Hood ED, Kiseleva R, Greineder CF, Radhakrishnan R, Muzykantov VR. Mechanisms that determine nanocarrier targeting to healthy versus inflamed lung regions. Nanomedicine. 2017;13:1495–1506. doi: 10.1016/j.nano.2016.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, Abrahimi P, Devalliere J, Moeckel G, Kulkarni S, Tellides G, Pober JS. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-κB signaling in endothelial cells. Circulation. 2013;128:2504–2516. doi: 10.1161/CIRCULATIONAHA.113.002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yi T, Fogal B, Hao Z, Tobiasova Z, Wang C, Rao DA, Al-Lamki RS, Kirkiles-Smith NC, Kulkarni S, Bradley JR, Bothwell ALM, Sessa WC, Tellides G, Pober JS. Reperfusion injury intensifies the adaptive human T cell alloresponse in a human-mouse chimeric artery model. Arterioscler Thromb Vasc Biol. 2012;32:353–360. doi: 10.1161/ATVBAHA.111.239285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, Zhang C, Wang J, Yang C, Xu M, Rong R, Zhu T, Zhu D. Endothelial cells In antibody-mediated rejection of kidney transplantation: Pathogenesis mechanisms and therapeutic implications. J Immunol Res. 2017;2017:8746303. doi: 10.1155/2017/8746303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Deliv Rev. 2010;62:1052–1063. doi: 10.1016/j.addr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bilati U, Allémann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24:67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 45.Sobal G, Resch U, Sinzinger H. Modification of low-density lipoprotein by different radioiodination methods. Nucl Med Biol. 2004;31:381–388. doi: 10.1016/j.nucmedbio.2003.10.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.