

Abstract
Chromatin spread techniques have been widely used to assess the dynamic localization of various proteins during gametogenesis, particularly for spermatogenesis. These techniques allow for visualization of protein and DNA localization patterns during meiotic events such as homologous chromosome pairing, synapsis and DNA repair. While a few protocols have been described in the literature, general chromatin spread techniques using mammalian prophase oocytes are limited and difficult due to the timing of meiosis initiation in fetal ovaries. In comparison, prophase spermatocytes can be collected from juvenile male mice with higher yields without the need for microdissection. However, it is difficult to obtain a pure synchronized population of cells at specific stages due to the heterogeneity of meiotic and post-meiotic germ cell populations in the juvenile and adult testis. For later stages of meiosis, it is advantageous to assess oocytes undergoing meiosis I (MI) or meiosis II (MII), because groups of mature oocytes can be collected from adult female mice and stimulated to resume meiosis in culture. Here, methods for meiotic chromatin spread preparations using oocytes dissected from fetal, neonatal and adult ovaries are described with accompanying video demonstrations. Chromosome missegregation events in mammalian oocytes are frequent, particularly during MI. These techniques can be used to assess and characterize the effects of different mutations or environmental exposures during various stages of oogenesis. As there are distinct differences between oogenesis and spermatogenesis, the techniques described within are invaluable to increase our understanding of mammalian oogenesis and the sexually dimorphic features of chromosome and protein dynamics during meiosis.
Keywords: Genetics, Issue 132, Oocyte, meiosis, chromatin spread, oogenesis, chromosome segregation, immunofluorescence microscopy, aneuploidy
Introduction
During spermatogenesis, large semi-synchronous waves of meiotic germ cells are rapidly and continuously replenished in the testis at the onset of puberty and throughout adulthood1. In contrast to males, meiosis in females is initiated solely during fetal development. Following birth, oocytes remain arrested in a prolonged dictyate stage of prophase I with an intact germinal vesicle (GV; nuclear envelope) until puberty. At the onset of puberty, a subset of oocytes are cyclically selected to undergo growth and maturation, marking the initiation of meiotic resumption. Meiotic resumption in fully-grown oocytes is manifested by the disappearance of the GV in a process known as germinal vesicle breakdown (GVBD). The oocyte then undergoes chromosome condensation and segregation, followed by polar body extrusion. Oocytes become arrested upon progression to MII and are stimulated to complete the second and final meiotic division only after fertilization.
Female fertility is highly dependent on the success of meiotic prophase I progression. Key to this is formation of a physical linkage between homologous chromosomes known as the chiasmata, which is mediated by repair of induced DNA double strand breaks (DSBs) via crossover recombination2. This process occurs within the context of a dynamic protein-rich scaffold known as the synaptonemal complex (SC) that forms between homologous chromosomes to facilitate their synapsis3. The SC is a zipper-like tripartite structure that consists of two parallel lateral elements connected by central region proteins that holds homologs together throughout ongoing DNA repair. Prior to synapsis, precursors of the lateral elements, called axial elements, form between sister chromatids. Synaptonemal complex proteins such as SYCP2 and SYCP3 form axial elements that colocalize to the sister-chromatid cohesion axes during early prophase. These later serve as binding sites for the transverse filament protein, SYCP1, which facilitates central element assembly and synapsis between aligned homologs4. In mouse oocytes, complete synapsis is indicated by the presence of 20 completely overlapping SYCP3 and SYCP1 stretches, which can be visualized by using chromatin spread preparations. Synapsis is completed upon entry into the pachytene substage, whereby mature crossovers that are destined to form chiasmata between homologs are decorated with mutL homolog (MLH1/3) dimers to promote their accurate processing5,6,7. The structural maintenance of chromosomes (SMC) complexes, including cohesin, condensin, and the SMC5/6 complex, are important for the regulation of chromosome dynamics and structure throughout meiosis8,9,10,11,12. Collectively, these events ensure proper bi-orientation of homologous chromosomes to opposing spindle poles following disassembly of the SC.
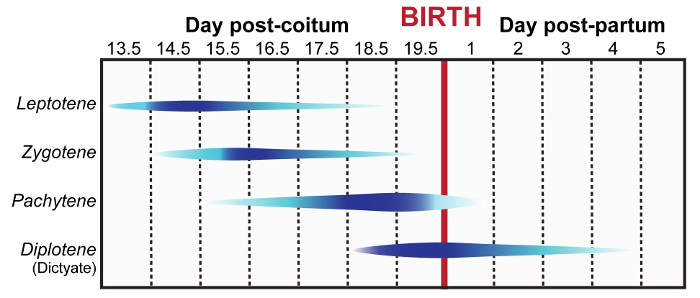
The meiotic cell cycle is a powerful model to examine the roles of various proteins in genome maintenance due to the programmed induction and subsequent repair of DNA DSBs. Furthermore, mammalian meiosis is also a relevant model for the study of epigenetic modifications and imprinting13. However, it is technically difficult to assess these events during female meiosis, which takes place in fetal and neonatal ovaries in mammals (Figure 1). Prophase I can be divided into 5 substages: leptonema, zygonema, pachynema, diplonema and dictyate. Herein, we describe how to isolate and distinguish between fetal and neonatal ovaries and testes (Figure 2). Adapted from previously described methods, this manuscript also outlines a protocol (step 1) with video demonstration for preparation of female meiotic prophase I chromatin spreads14,15,16,17. When coupled with immunolabelling, as described in steps 6-7, this protocol enables detailed microscopic analysis of prophase I events in oocytes.

Oogenesis is error-prone, and chromosome missegregation events during the first meiotic division represent the most common source of genetic disease in progeny18,19. In this manuscript (protocol step 2), we describe a protocol in which mature GV-staged oocytes are extracted from primed ovaries of adult female mice. Under supportive conditions, fully-grown oocytes undergo luteinizing hormone-independent resumption of meiosis following isolation and culture20. Following meiotic resumption, oocytes progress through meiosis I, then arrest at metaphase II. Oocytes remain arrested at metaphase II, unless fertilized. In steps 2–5, we adapt previously reported protocols with video demonstration to describe how to collect, culture and prepare MI and MII oocytes for chromatin spread preparations21. This chromatin spreading technique allows for clear immunolabelling of proteins associated with chromosomes. Furthermore, this protocol can also be used to distinguish between bivalent and univalent chromosomes, and can further resolve single sister chromatids to facilitate assessment of oocyte ploidy. Therefore, in addition to revealing localization patterns of meiotic proteins, this protocol can also serve as an invaluable tool for elucidating potential causes of chromosome missegregation during MI and MII.
Protocol
All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of Johns Hopkins University. Experiments were performed on wild-type C57BL/6J mice.
1. Harvesting Fetal or Neonatal Ovaries and Preparation of Prophase Chromatin Spreads
To extract embryos at 14.5–19.5 days post-coitum (dpc) sacrifice the pregnant females via cervical dislocation or CO2 asphyxiation according to IACUC guidelines. NOTE: For postnatal day 1–5 ovaries skip to step 1.3. Figure 1 summarizes the meiotic prophase stages enriched at different embryonic and post-natal ages.
Open the abdominopelvic cavity using sterile scissors, making a V-shaped opening. Dissect out the maternal uterine horn, separate the embryos from the placenta, and transfer embryos into 35 mm petri dishes containing 3 mL of pre-warmed 1x phosphate buffer saline (PBS) at 37 °C. NOTE: A fliptop incubator set at 37 °C can be used to maintain temperature. In addition, a temperature regulated glass stage can also be used to maintain temperature during ovary manipulation.
With 3.5 inch surgical scissors, sacrifice embryos or pups via decapitation according to IACUC guidelines. Place decapitated embryos or pups in pre-warmed PBS prior to further dissection.
Dissect one embryo or pup at a time in a separate 35 mm petri dish containing 3 mL of pre-warmed PBS at 37 °C. Proceed by cutting along the ventral midline of the posterior half of the embryo, along the anterior half below the forelimbs and directly above the hind-limbs and tail as outlined in Figure 2A–B.
Open the abdomen using 3.5 inch surgical scissors. Using fine-tipped forceps displace or remove the liver and loops of bowel, exposing the ovaries in a new 35 mm petri dish containing 3 mL of pre-warmed PBS at 37 °C (see guide in Figure 2B). The ovaries are located immediately below and behind the kidney towards the posterior wall of the peritoneal cavity (Figure 2B–C). A guide for differentiating between male and female gonads is provided in Figure 2D. NOTE: For optimal conditions, rapidly dissect out the ovaries after pregnant female is sacrificed.
Remove both ovaries from each female fetus using a pair of fine-tipped forceps under a dissection scope and place in a watch glasses or separate wells of a multi-well plate containing 0.5 to 1.0 mL of pre-warmed PBS and maintain at 37 °C.
Place each pair of ovaries in 0.5 mL hypo-extraction buffer (17 mM trisodium citrate dihydrate, 50 mM sucrose, 5 mM EDTA, 0.5 mM DTT, 30 mM Tris-HCl, protease inhibitor, pH 8.2) in a clean watch glass or a small well of a 9-well plate, making sure to immerse the ovaries completely. Incubate for at least 15 min, but no more than 30 min. NOTE: Make hypo-extraction buffer fresh and use within 2 h of DTT addition.
During incubation, using a hydrophobic barrier PAP pen, draw two 22 x 22 mm2 squares on a clean glass 25 x 75 mm2, 1 mm thick microscope slide as shown in Figure 3A. NOTE: Slides can be prepared ahead of time.
Pipette 45–50 µL of 100 mM sucrose onto a clean slide and transfer one pair of ovaries to the drop.
Using the sharp ends of two 27 G needles, tease the ovaries apart to release cells into the sucrose solution. With forceps, remove large pieces of ovary and carefully pipette sucrose solution to disperse cells.
Place 40 µL of fixative solution (1% paraformaldehyde (PFA), 0.2% detergent (see Table of Materials), pH 9.2) into each square of the prepared slide. With a 200 µl pipette tip, spread the fixative solution evenly over the slide surface.
Pipette 20 µL of the sucrose mix onto the each square of the slide containing the fixative. NOTE: This equates to using one pair of ovaries per slide.
Incubate the slides in a closed humid chamber overnight at room temperature. NOTE: The overnight incubation can range around (12-15 hrs) at room temperature.
Open the chamber lid and allow the slides to air-dry completely.
Wash slides in a Coplin jar containing 50 mL of 0.4% wetting agent-PBS solution (see Table of Materials) for 2 min, air dry, and proceed to immunolabelling protocol (step 6). NOTE: It may be possible to store the slides at -80 °C for later use, but this varies depending on the protein of interest. For optimal results proceed immediately to the immunolabelling protocol (step 6).
2. Metaphase I Oocyte Collection
To maximize the number of antral follicles isolated from each mouse, intraperitoneally inject sexually mature virgin female mice with 5 IU of pregnant mare's serum gonadotropin (PMSG, also known as equine chorionic gonadotropin (eCG)). NOTE: For optimal oocyte yield, mice should be 1 to 3 months of age, as the number of oocytes harvested will decrease with age. To prepare PMSG, dissolve bottle of 5,000 IU in 100 mL of sterile PBS (5 U/0.1 mL) Store in single-use aliquots of 600 µL at -20 °C.
After 44–48 h, prepare collection medium, minimum essential medium alpha (MEMα) medium supplemented with 5% fetal bovine serum (FBS) and 3 mg/mL bovine serum albumin (BSA; MEMα/BSA/FBS). Sterilize media through a 0.2 µm pore filter. Decant 2.5 mL of MEMα/BSA/FBS medium into one glass dish per mouse and warm to 37 °C in a 5% CO2 incubator. NOTE: Other collection and culture media that are commercially available (M2 and M16) are also commonly used. Caution: We recommend removing culture dishes from the 5% CO2 incubator one at a time for limited duration to minimize ambient air exposure during manipulation steps.
Sacrifice the mice via cervical dislocation or CO2 asphyxiation according to IACUC guidelines.
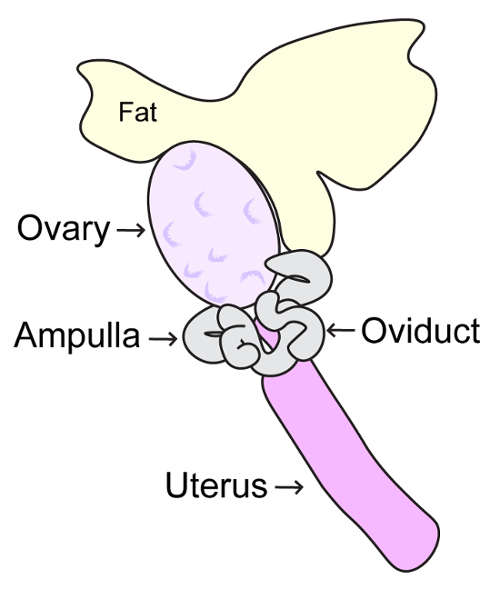
Open the abdominopelvic cavity using sterile scissors, making a large V-shaped opening. Using forceps, displace intestines towards the head of the mouse. Locate each uterine horn; the ovaries are present proximal to the rib cage. Hold the oviduct with fine forceps and cut the fat superior to the ovary using dissection scissors (Figure 4). Continue to hold the oviduct, and with another set of fine forceps release the ovary from the bursa and place into collection dish containing MEMα/BSA/FBS medium. NOTE: As in step 1, it is imperative to keep the ovaries at 37 °C and maintain at 5% CO2. A fliptop incubator hooked up to 5% CO2 mix can be used to allow for optimal maintenance of temperature and CO2 levels. In addition, a temperature regulated glass stage should also be used to maintain temperature during oocyte manipulation.
Using a 1 mL syringe with a 27-gauge needle (or similar size), release cumulus oocyte complexes by manually puncturing the large antral follicles.
While observing through a dissection microscope, collect GV-staged oocytes using a mouth-operated glass pipette or capillary, or a hand-operated micrometer-syringe (Figure 5). Only collect oocytes that are released from antral follicles. GV oocytes have a diameter of approximately 90 µm. GV oocytes may be surrounded by 2-3 layers of granulosa cells, and have a total diameter of around 200 µm.
Culture oocytes in a clean watch glass or a 9-well plate containing MEMα/BSA/FBS medium for 6 h to reach metaphase I at 37 °C in a 5% CO2 incubator.
Proceed to step 4.

3. Metaphase II (MII) Oocyte Collection
To maximize oocytes isolated from each mouse, intraperitoneally inject sexually mature virgin female mice with 5 IU of PMSG. After 44–48 h, intraperitoneally inject with 5 IU of human chorionic gonadotropin (hCG). NOTE: For optimal oocyte yield, mice should be 1 to 3 months of age, as the number of oocytes harvested will decrease with age. To prepare hCG, dissolve a bottle of 10,000 U in 200 mL PBS (5 U/0.1 mL). Store in single-use aliquots of 600 µL at -20 °C.
After 12-14 hrs, prepare MEMα/BSA/FBS collection medium, as described in step 2.2, above.
Sacrifice the mice via cervical dislocation or CO2 asphyxiation according to IACUC guidelines.
Open the abdominopelvic cavity using sterile scissors, making a large V-shaped opening. Using forceps, displace intestines towards the head of the mouse. Locate each uterine horn, the ovaries are present proximal to the rib cage. Hold the oviduct with fine forceps and cut the fat superior to the ovary using dissection scissors (Figure 4). Then remove the ovary and oviduct and place into collection dish containing MEMα/BSA/FBS medium. NOTE: As in steps 1 and 2, it is imperative to keep the ovaries at 37 °C and maintain at 5% CO2. A fliptop incubator hooked up to 5% CO2 mix can be used to allow for optimal maintenance of temperature and CO2 levels. In addition, a temperature regulated glass stage should also be used to maintain temperature during oocyte manipulation.
Using a 1 mL syringe with a 27 G (or similar size) or sharp forceps, tear a hole in the ampulla of the oviduct to release the MII oocytes. NOTE: Be careful not to damage the oocytes in the ampulla. The ampulla will appear swollen and translucent such that the oocytes are visible (Figure 4).
Harvest MII oocytes from the ampulla of the oviduct into a new dish with collection medium using a mouth-operated glass pipette or capillary, or a hand-operated micrometer-syringe (Figure 5).
Proceed to step 4.
4. Oocyte Denuding and Zona Pellucida Removal
Prepare 300 IU/mL of hyaluronidase in MEMα medium supplemented with 3 mg/mL BSA to denude oocytes of surrounding cumulus cells (2.5 mL/mouse) in a watch glass, and keep at 37 °C, 5% CO2. NOTE: Hyaluronidase efficacy sharply decreases after 1 h of preparation. For MII oocytes, hyaluronidase treatment is not required.
Expose oocyte-cumulus cell complexes to 300 IU/mL of hyaluronidase in MEMα/BSA for 3 min to denude oocytes of surrounding cumulus cells. Caution: Do not exceed 3 min exposure to hyaluronidase, as it will damage the oocytes.
To wash the oocytes, transfer oocytes to a fresh warmed MEMα/BSA medium dish. Using a small mouth-operated glass pipette or capillary (slightly larger than the diameter of the oocyte, which is approximately 90 µm), pipette the complexes up and down to completely detach the cumulus cells. Allow the oocytes to recover in the incubator while preparing for the next step.
Warm acidic Tyrode's solution, MEMα/BSA, and Waymouth's media to 37 °C (500 µL/mouse). Place 300 µL in each well of small 9-well glass plates.
To remove the zona pellucida (ZP) transfer 5–10 oocytes into Tyrode's solution. Expose the oocytes for only 30-45 s to the solution. NOTE: Too little exposure to the solution will not remove the ZP completely, which will prevent the oocytes from bursting and chromosomes from spreading. Too much exposure will damage and kill the oocyte. Watching the ZP dissolve under the dissection microscope can aid in optimizing exposure time. Caution: Using fresh Tyrode's solution is critical, as its function declines with age. Use a fresh well of Tyrode's solution for each group of oocytes treated to ensure uniform digestion of the ZP.
Immediately after ZP removal, wash the oocytes by transferring into warmed MEMα/BSA/FBS medium. Repeat this wash step. NOTE: Be careful when transferring, as oocytes will easily stick together and to the glass pipette or capillary following removal of the ZP. Pre-wetting the glass pipette or capillary in Waymouth's medium will minimize oocytes sticking together.
Transfer and allow the oocytes to recover for 30 minutes in Waymouth's medium at 37 °C in a 5% CO2 incubator.
Proceed to step 5.
5. MI and MII Oocyte Chromatin Spreads
Using a PAP pen, draw a 11 x 44 mm2 rectangle on the glass slide as shown in Figure 6A.
Coat the slide with a thin layer of fixative (1% paraformaldehyde, 0.2% Triton-X 100 in H2O, pH 9.2) by pipetting 100 µL of fixative onto the slide and rocking the slide back and forth. Tap the slide to get rid of excess fixative.
Pick up between 5-10 oocytes with a small mouth pipette needle with as little media as possible and drag the needle in a line across the fixative-coated slide while depositing the oocytes from 1 cm above the slide into the fixative solution. Perform this step using a dissection microscope to ensure that the oocytes have been deposited and that they have burst. NOTE: The oocytes should burst immediately on the slide. The height at which the oocytes are deposited impact the degree of chromatin spreading. See representative results in Figure 6 for further details.
Incubate slides at room temperature in a closed humidified chamber overnight to allow the chromatin to adhere.
Allow the slides to air-dry completely and rinse slides in a Coplin jar containing 50 mL of 0.4% wetting agent-H2O, pH 8.0.
Proceed to step 6.
6. Immunolabelling
Prepare the antibody dilution buffer (ADB) described in Table 1.
Prepare two Coplin jars containing 50 mL of wash buffer (WB) solution (10% ADB diluted in PBS), and one Coplin jar containing 50 mL WB and 0.05% detergent solution (e.g., add 250 µL of 10% detergent to 50 mL WB).
Wash the slides that were prepared in section 1 and 5 of this protocol for 10 min in one Coplin jar containing 50 mL of WB. Caution: Do not let the slides dry at any point during immunolabelling.
Wash the slides for 10 min in the Coplin jar containing 50 mL WB and 0.05% detergent solution. Then, wash the slides in the remaining Coplin jar containing 50 mL of WB solution for 10 min.
Tap off excess liquid and cover slide with 100 µL of selected primary antibodies diluted in ADB. Incubate at 4 °C overnight in a closed humid chamber. NOTE: Incubation can be shortened to 2 to 3 hrs at 37 °C. Use smaller volumes of antibody (e.g., 50 µL) by covering the slide with a coverslip or parafilm.
Rinse slides in a Coplin jar containing 50 mL of 0.2% wetting agent-PBS solution, pH 8.0.
Repeat steps 6.2 to 6.5.
Tap off excess liquid and cover slide with 100 µL of selected secondary antibodies diluted in ADB. Incubate slides 1 to 2 hrs at 37 °C in a closed humid chamber.
Wash slides 2 times for 10 min in Coplin jars containing 50 mL of 0.2% wetting agent-PBS solution, pH 8.0.
Wash slides 2 times for 5 min in Coplin jars containing 50 mL of 0.2% wetting agent-H2O solution, pH 8.0.
7. Mounting slides
For prophase chromatin spreads prepared in step 2, add 100 µL of mounting medium containing 4', 6-diamidino-2-phenylindole (DAPI, 1.5µg/mL). For MI and MII chromatin spreads prepared in step 5, add 50 µL of mounting medium containing DAPI (1.5µg/mL). Gently blot away excess liquid.
Place a 22 mm x 60 mm coverslip on top and seal with clear nail polish. Store in a slide box at 4 °C or -20 °C until assessment via fluorescence microscopy. NOTE: Tap lightly on cover slip to rid of excess mounting medium before sealing with nail polish.
Representative Results
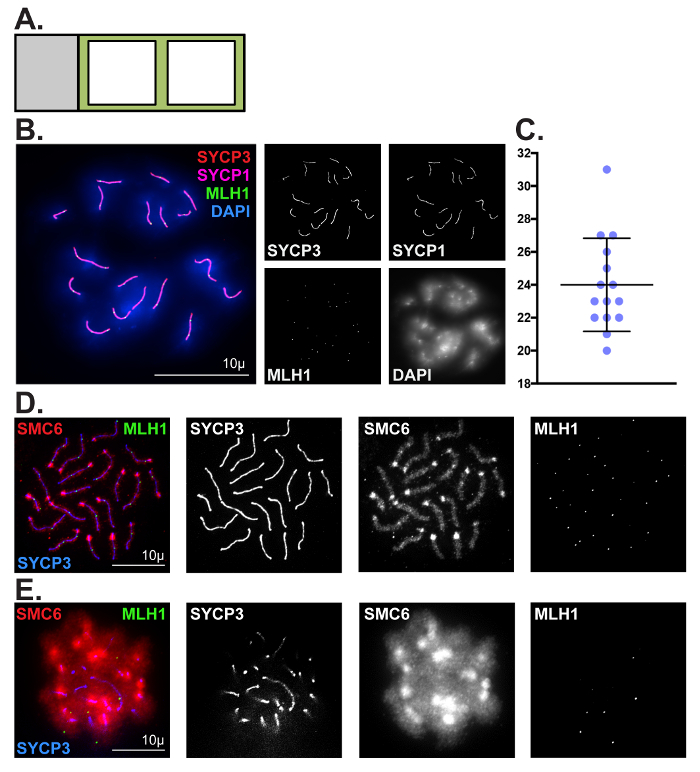
We have described two techniques for visualizing and assessing meiotic chromosomes in oocytes. The first technique is catered toward assessing prophase progression in embryonic and neonatal ovaries. Prophase chromatin spread preparations are incredibly valuable for visualizing numerous dynamic processes during meiosis, including chromosome pairing, synapsis and desynapsis, homologous recombination, and epigenetic chromosome remodeling. Here, we have demonstrated the utility of this method for robust visualization and quantitative analysis of crossover formation in oocytes harvested from C57BL/6J embryos (Figure 3Band 3C). To enrich for cells in the pachytene substage, mouse embryos were retrieved at 18.5 dpc (Figure 1). Two major hallmarks of the pachytene substage of prophase I are the completion of synapsis between homologs, and the formation of at least one MLH1/3-positive crossover per homolog pair. Complete synapsis between homologs is manifested by the presence of 20 overlapping SYCP3 and SYCP1 stretches. To characterize crossover formation in wild-type oocytes we immunolabeled prophase chromatin spread preparations using antibodies that detect SYCP3, SYCP1 and MLH1, and stained DNA using DAPI. Figure 3B depicts a representative image of a pachytene stage oocyte, which is evidenced by the presence of 27 MLH1 foci distributed along 20 fully assembled SC structures. The average number of MLH1-positive crossovers detected in wild-type oocytes was 24 ±3 (Figure 3C, N = 15 oocytes). Figure 3D shows SMC6 enriched at the pericentromeric heterochromatin (PCH) region and along the chromosome arms, and MLH1 foci distributed along the SC at the pachytene stage. Figure 3E depicts a pachytene staged oocyte where the chromosome spread preparation was sub-optimal and individual chromosomes are indistinguishable, preventing accurate assessment of SYCP3, SMC6 and MLH1. Sub-optimal chromosome spreads can be observed when incubation of ovaries in hypo-extraction buffer is for too long or not for long enough.
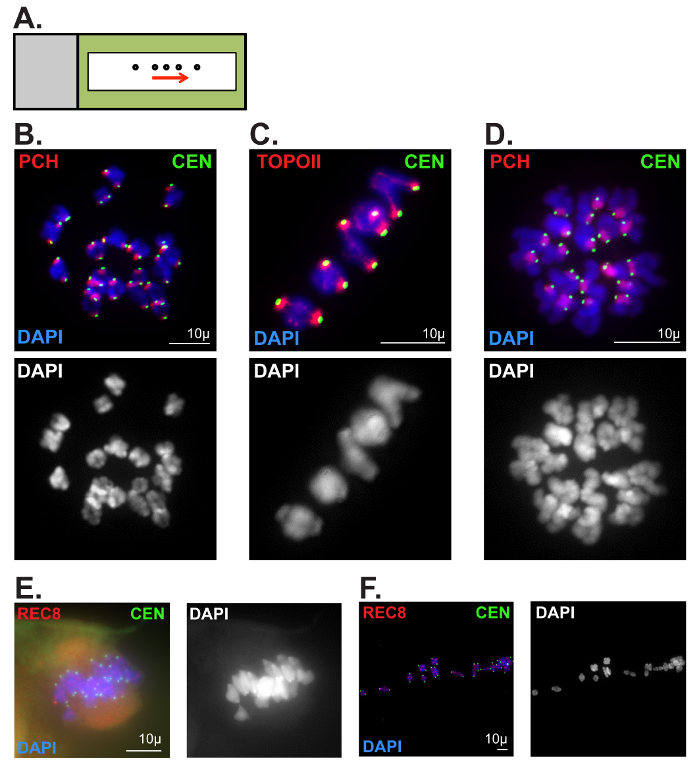
The second technique described can be used to assess chromatin morphology in oocytes following meiotic resumption (Figure 6B-D). Protein localization, as well as ploidy, can easily be assessed, exposing potential causes of chromosome segregation errors. Figure 6B depicts a MI oocyte showing 20 chromosomes and clear centromere and pericentromeric heterochromatin staining. Figure 6C is a zoomed image where topoisomerase IIα (TOPOII) can be seen along the chromosome arms and the PCH. Figure 6D depicts a MII oocyte where paired sister chromatids can be distinguished by chromosome and centromere morphology. Common errors seen when attempting this technique include oocytes not bursting when released onto the PFA-coated slide, as well as chromosomes spreading too far apart (Figure 6E–F). If the ZP is not removed completely, the chromosomes will remain associated to the spindle, as shown in Figure 6E. If the oocytes are dropped too high from the PFA-coated slide, the chromosomes can be spread too far apart as depicted in Figure 6F.
Figure 1: Meiotic prophase timeline during female embryonic and neonatal development. Blue contours indicate populations of specific prophase sub-stage germ cells (leptotene, zygotene, pachytene, and diplotene/dictyate stages) observed during embryonic and neonatal development. Increasing dark blue color indicates the timing at which the specific prophase sub-stage becomes more abundant. This figure was adapted from reference2. Please click here to view a larger version of this figure.
Figure 2: Ovary extraction from embryos and neonatal female pups. (A, B) The first cut (1) is made above the forelimbs to decapitate the embryo at the head/neck junction immediately upon retrieval from the maternal uterine horn. The second cut (2) is made along the ventral midline of the posterior half of the embryo, followed by incisions along the anterior half below the forelimbs (3). A final cut is made for removal the hind limbs and tail (4). (A) Frontal view schematic of dissection cuts for isolation of ovaries from female pups. (B) Side view schematic of dissection cuts for isolation of ovaries with relative positions of internal organs. Regions shown in light red include the liver and intestines, which are removed during dissection. The dorsal wall and associated organs are shown in light blue. This region includes the ovaries, which are attached to the inferior poles of the kidneys at the top of the uterine horn. (C) Frontal schematic view of dorsal wall of embryo following removal of the liver and intestines. (D) Schematic representation of morphological differences between male and female gonads at approximately 15–18 dpc. Please click here to view a larger version of this figure.
Figure 3: Representative prophase chromatin spread preparations. (A) Glass slide schematic for prophase chromatin spread preparations. Black square outlines represent liquid blocker pen outlines. (B, C) Representative images and quantification of crossover formation in wild-type pachytene stage oocytes using prophase chromatin spread preparation methods described in step 1.(B) Chromatin spreads were performed using ovaries fromC57BL/6J mouse embryos isolated at 18.5 dpc. Chromatin spreads were immunolabeled with antibodies against the SC lateral element protein SYCP3 (red), and the SC central element protein SYCP1 (magenta), MLH1 (green, crossover event marker), and DAPI (blue, DNA). Scale bar = 10 µm. (C) Dot plot of MLH1 foci counts obtained from 15 pachytene stage chromatin spread preparations. Error bars represent standard deviation.(D, E) Comparison of optimal (D) and sub-optimal (E) prophase spread preparations, respectively. Chromatin spreads were immunolabeled with antibodies against SMC6 (red), MLH1 (green), and SYCP3 (blue). Scale bar = 10 µm. Please click here to view a larger version of this figure.
Figure 4: Schematic diagram of adult ovary. This diagram depicts the anatomy of the adult mouse ovary, oviduct, ampulla and uterus. Mouse ovaries should be removed by careful incision of ligaments connecting the ovaries to the inferior poles of the kidneys, and the posterior wall of the abdomen. Please click here to view a larger version of this figure.
Figure 5: Images of mouth-operated glass pipette, glass capillary and hand-operated micrometer-syringe used for oocyte manipulation. (A) The glass pipette oocyte manipulator is composed of the following components in sequence: mouth piece, latex tubing (3.2 mm inner diameter (ID) x 6.4 mm outer diameter (OD)), 1 mL pipette tip, latex tubing (6.4 mm ID x 11.1 mm OD) and glass Pasteur pipette. The end of the glass Pasteur pipette has been heated over a flame and the end pulled to create a fine-tipped end (Figure 5A1). (B) The glass capillary oocyte manipulator is composed of the following components in sequence: mouth piece, 0.45 µm filter (optional), latex tubing (3.2 mm ID x 6.4 mm OD), 1 mL pipette tip, latex tubing (6.4 mm ID x 11.1 mm OD) and 70 µl glass capillary. The glass capillary tubes are heated over a flame at the center and pulled in opposite directions to create two capillaries with fine tipped ends (Figure 5B1). (C) The hand-operated micrometer-syringe is composed of the following components in sequence: Mitutoyo 150-208 micrometer head (middle size, 0-1" range, 0.001" graduation), 5 mL syringe, latex tubing (3.2 mm ID x 6.4 mm OD), 1 mL pipette tip, latex tubing (6.4 mm ID x 11.1 mm OD), 1 mL pipette tip and a 83 x 0.5 mm2 gel loading tip. The Mitutoyo 150-208 micrometer head is firmly inserted into the 5 mL syringe (Figure 5C1). To ensure the gel loading tip is fastened securely, the connecting 1 mL pipette tip is cut (Figure 5C2). Please click here to view a larger version of this figure.
Figure 6: Representative metaphase I and II oocyte chromatin spread preparations. (A) Slide schematic for metaphase I and II oocyte chromatin spread preparations. Oocytes (circles) released via mouth-operated glass pipette or capillary in a straight line (following the arrow). Black rectangle outline represents liquid blocker pen outline. (B, C) Optimal metaphase I chromatin spread preparation. Metaphase I chromosomes were stained with DAPI (blue, DNA), and immunolabeled for CEN (green, kinetochore/centromere marker). Scale bars = 10 µm. (B) Antibodies against Histone H4 (di methyl K20, tri methyl K20) were used to label pericentromeric heterochromatin (PCH). (C) Antibodies against Topoisomerase IIα (TOPOII) were used to label the PCH and chromosome axes. (D) Optimal metaphase II chromatin spread preparation. Metaphase II chromosomes were stained with DAPI (blue, DNA), and immunolabeled for CEN (green), and Histone H4 (di-methyl K20, tri-methyl K20) to label the PCH. Scale bar: 10µm. (E, F) Poor metaphase I chromatin spread preparations. Chromosomes were stained with DAPI (blue, DNA), and immunolabeled for CEN (green) and the meiotic cohesin component, REC8. Scale bars = 10 µm (E) An oocyte that did not burst upon release onto PFA-coated slide. (F) Chromosomes that were spread too far apart. Please click here to view a larger version of this figure.
| Item | Amount | Final Concentration |
| 1x PBS | 50 mL | 1x |
| BSA | 1.5g | 3% (w/v) |
| Horse Serum | 5 mL | 10% (v/v) |
| 10% detergent (see Table of Materials) in PBS | 250 μL | 0.05% (v/v) |
Table 1: Antibody dilution buffer (ADB) recipe. Store ADB at 4 °C or freeze stocks at -20 °C if making larger quantities. ADB can become contaminated, so make sure good aseptic techniques are used and assess the solution for contamination prior to each use. Smaller aliquots of ADB can also be prepared to minimize contamination.
Discussion
Chromatin spread preparations allow researchers to chronologically study female mammalian meiosis and the dynamic localization of proteins involved. The embryonic and neonatal chromatin spreads allow for close analysis of events throughout meiotic prophase. Metaphase I and metaphase II chromatin spreads can be used to distinguish single sister chromatids from paired sisters and paired homologous chromosomes, as well as assess ploidy. By comparison, the protocol described here can be advantageous compared to whole oocyte immunolabelling, which frequently produces high levels of background signal that decreases the resolution of chromatin-bound proteins. Furthermore, chromatin spread techniques represent an attractive alternative to live-cell imaging, which requires a large investment of time and specialized equipment.
Embryonic and neonatal ovaries can be difficult to locate and extract. We recommend carefully extracting all other organs and material besides the kidneys in a separate dish containing PBS before searching for the ovaries, and using a fresh dish of PBS for each embryo/pup (Figure 2B). If the embryo/pup is male, the testis will be distinguishable by the tubule formation as well as the location of the gonads (Figure 2D). As male embryos develop, the testes will descend towards the penis, becoming larger and oval-shaped.
The oocyte chromatin spread technique requires mastery of oocyte collection and manipulation using mouth-operated glass pipette or capillary. We recommend practicing controlling the mouth pipette before performing this protocol. Pulling the proper size glass pipette/capillary for collection and denuding oocytes will increase chances of success and efficiency by minimizing time oocytes are outside of the incubator as well as in the Tyrode's solution. Correct timing for ZP removal is extremely important for the success of this method. If the oocytes are incubated in Tyrode's solution for an insufficient amount of time, the ZP will not be completely removed. This prevents the oocytes from bursting efficiently on the slide, as can be seen in Figure 6E. However, if the oocytes are incubated in Tyrode's solution for too long, oocyte quality and downstream immunofluorescence staining will be severely compromised. We recommend watching the oocytes under a microscope to determine the exact time the ZP dissolves in Tyrode's solution. Adding only 5–10 oocytes at a time to Tyrode's solution will minimize excessive exposure. If the oocytes are released too high above the PFA-coated slide, the chromosomes can be spread too far apart (Figure 6F). Optimal results are found when oocytes are released 1 cm above the slide. Other factors that can hinder results include prolonged exposure to decreased CO2 levels in ambient air during oocyte manipulation steps. This causes pH fluctuations in the media that are detrimental to oocyte quality, and is apparent by progressive color change of the media from orange-red to pink. The buffering capacity of the media also decreases over time; therefore, we do not recommend using media that has been stored (4 °C) for longer than 1 week. M2 medium, which does not require CO2 to be buffered, is commonly used as an alternative.
A limitation to chromatin spread preparations is the lack of visualization of the chromatin within the native context of the cell. Cellular material outside of the nucleus cannot be visualized and spindle formation cannot be assessed. Therefore, other methods can be used to assess oogenesis in addition to chromatin spreads, such as whole oocyte immunolabelling after monastrol treatment, which collapses the bi-polar spindle into a mono-polar spindle22. This allows sister chromatids to be assessed within the context of the cell. Collectively, the chromatin spread methods described here can easily be applied to assess chromatin morphology and chromosome ploidy throughout oogenesis in novel transgenic and mutant mouse models.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by NIGMS (R01GM11755) to P.W.J and by a training grant fellowship from the National Cancer Institute (NIH) (CA009110) to G.H. and J.H.
References
- Morelli MA, Cohen PE. Not all germ cells are created equal: aspects of sexual dimorphism in mammalian meiosis. Reproduction. 2005;130(6):761–781. doi: 10.1530/rep.1.00865. [DOI] [PubMed] [Google Scholar]
- Keeney S. Spo11 and the Formation of DNA Double-Strand Breaks in Meiosis. Genome Dyn Stab. 2008;2:81–123. doi: 10.1007/7050_2007_026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zickler D, Kleckner N. Recombination, Pairing, and Synapsis of Homologs during Meiosis. Cold Spring Harb Perspect Biol. 2015;7(6) doi: 10.1101/cshperspect.a016626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vries FA, et al. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev. 2005;19(11):1376–1389. doi: 10.1101/gad.329705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, et al. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13(3):336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Lipkin SM, et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. 2002;31(4):385–390. doi: 10.1038/ng931. [DOI] [PubMed] [Google Scholar]
- Kolas NK, et al. Localization of MMR proteins on meiotic chromosomes in mice indicates distinct functions during prophase I. J Cell Biol. 2005;171(3):447–458. doi: 10.1083/jcb.200506170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin S. Complex elaboration: making sense of meiotic cohesin dynamics. FEBS Journal. 2015;282(13):2426–2443. doi: 10.1111/febs.13301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Roles of Cohesin and Condensin in Chromosome Dynamics During Mammalian Meiosis. J. Reprod Dev. 2013;59(5):431–436. doi: 10.1262/jrd.2013-068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verver DE, Hwang GH, Jordan PW, Hamer G. Resolving complex chromosome structures during meiosis: versatile deployment of Smc5/6. Chromosoma. 2016;125(1):15–27. doi: 10.1007/s00412-015-0518-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins J, et al. Meiosis-specific cohesin component, Stag3 is essential for maintaining centromere chromatid cohesion, and required for DNA repair and synapsis between homologous chromosomes. PLoS Genet. 2014;10(7):e1004413. doi: 10.1371/journal.pgen.1004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang G, et al. SMC5/6 is required for the formation of segregation-competent bivalent chromosomes during meiosis I in mouse oocytes. Development. 2017;144(9):1648–1660. doi: 10.1242/dev.145607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota SK, Feil R. Epigenetic transitions in germ cell development and meiosis. Dev Cell. 2010;19(5):675–686. doi: 10.1016/j.devcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Taketo T. Microspread ovarian cell preparations for the analysis of meiotic prophase progression in oocytes with improved recovery by cytospin centrifugation. Methods Mol Biol. 2012;825(1):173–181. doi: 10.1007/978-1-61779-436-0_13. [DOI] [PubMed] [Google Scholar]
- Sun X, Cohen PE. Studying recombination in mouse oocytes. Methods Mol Biol. 2013;957(1):1–18. doi: 10.1007/978-1-62703-191-2_1. [DOI] [PubMed] [Google Scholar]
- Kim JH, Ishiguro K, Kudo N, Watanabe Y. Studying meiosis-specific cohesins in mouse embryonic oocytes. Methods Mol Biol. 2013;957(1):47–57. doi: 10.1007/978-1-62703-191-2_3. [DOI] [PubMed] [Google Scholar]
- Susiarjo M, Rubio C, Hunt P. Analyzing mammalian female meiosis. Methods Mol Biol. 2009;558:339–354. doi: 10.1007/978-1-60761-103-5_20. [DOI] [PubMed] [Google Scholar]
- Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2001;2(4):280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- Hassold T, Hall H, Hunt P. The origin of human aneuploidy: where we have been, where we are going. Hum Mol Genet. 2007;16:R203–R208. doi: 10.1093/hmg/ddm243. [DOI] [PubMed] [Google Scholar]
- Pincus G, Enzmann EV. The Comparative Behavior of Mammalian Eggs In vivo and In vitro: I. The Activation of Ovarian Eggs. J Exp Med. 1935;62(5):665–675. doi: 10.1084/jem.62.5.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon JP, Hached K, Wassmann K. Chromosome spreads with centromere staining in mouse oocytes. Methods Mol Biol. 2013;957(1):203–212. doi: 10.1007/978-1-62703-191-2_14. [DOI] [PubMed] [Google Scholar]
- Stein P, Schindler K. Mouse oocyte microinjection, maturation and ploidy assessment. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]