

Abstract
Forward genetic screens using reporter genes inserted into the heterochromatin have been extensively used to investigate mechanisms of epigenetic control in model organisms. Technologies including short hairpin RNAs (shRNAs) and clustered regularly interspaced short palindromic repeats (CRISPR) have enabled such screens in diploid mammalian cells. Here we describe a large-scale shRNA screen for regulators of X-chromosome inactivation (XCI), using a murine cell line with firefly luciferase and hygromycin resistance genes knocked in at the C-terminus of the methyl CpG binding protein 2 (MeCP2) gene on the inactive X-chromosome (Xi). Reactivation of the construct in the reporter cell line conferred survival advantage under hygromycin B selection, enabling us to screen a large shRNA library and identify hairpins that reactivated the reporter by measuring their post-selection enrichment using next-generation sequencing. The enriched hairpins were then individually validated by testing their ability to activate the luciferase reporter on Xi.
Keywords: Genetics, Issue 133, X-chromosome inactivation, epigenetics, MeCP2, Rett syndrome, XIST, shRNA, screening
Introduction
One the most common forms of hereditary mental impairment in females, Rett syndrome, is caused by heterozygous mutations in MeCP2, an X-chromosome gene that encodes a protein essential for normal neuronal function1. A potential approach to treating this disorder would be reactivation of the wild-type MeCP2 allele on the Xi, as restoration of MeCP2 expression was shown to reverse neurological deficits in a mouse model of this disease1,2,4. However, epigenetic silencing of one of the two X chromosomes in female cells is tightly maintained throughout the lifespan of an organism3,4, and robust reactivation of an Xi gene would likely require a major disruption in multiple epigenetic regulatory pathways.
To identify factors necessary for maintenance of MeCP2 silencing, we first developed a transgenic mouse model carrying a MeCP2-luciferase- hygromycin resistance gene fusion (MeCP2-LUC-HR) on one of the two X chromosomes (XMeCP2-LUC-HR/XMeCP2)5. Although the MeCP2 expressed from the fusion construct proved to be unstable and resulted in a loss of function, phenotypically mimicking MeCP2 deletion in hemizygous males (XMeCP2-LUC-HR/Y), the expression of the reporter genes was readily detectable in a pattern consistent with the expression of endogenous MeCP25. We then generated immortalized fibroblast clones with the construct on either inactive or active X-chromosome, and confirmed that former expressed wild type MeCP2 and not the reporter construct; the inverse was true for the latter. When exposed to a DNA demethylating agent known to abrogate gene silencing, 5-azacytidine (5-AZA), cells with the reporter on the Xi ("reporter cells") gained activity in a bioluminescence assay, indicating that our construct could be reactivated and therefore used for genetic screening.
We next developed a high-throughput genetic screen for regulators of MeCP2 silencing. The reporter cell line was first infected with a retroviral library containing >60,000 different shRNAs targeting >25,000 genes throughout the mouse genome5,6, and then subjected to hygromycin B selection. Hairpin frequency was compared in pre- and post-selection samples using next generation sequencing, as reporter reactivation conferred growth advantage under hygromycin B selection and resulted in enrichment of responsible hairpins. Using this approach, we identified 30 genes implicated in MeCP2 silencing, and subsequently confirmed the findings by transducing the reporter cells with individual hairpins and measuring their luciferase activity.
Protocol
All steps involving animals were carried out using protocols approved by the Fred Hutchinson Cancer Research Center Institutional Animal Care and Use Committee (IACUC). No reagents used here are known to pose significant human health risks except for 5-azacytidine (5-AZA).
1. Generating a reporter cell line with MeCP2-LUC-HR transgene on the Xi
- Preparing mouse tendon fibroblasts from a female XMeCP2-LUC-HR /XMeCP2 mouse
- Euthanize a 10-12 day-old female XMeCP2-LUC-HR/XMeCP2 mouse.
- Using straight dissection scissors, make a circumferential incision through the skin of the proximal tail portion.
- Holding the carcass near the base of the tail, pull the skin away from the mouse to expose the fibrocartilaginous portion of the tail. Remove and cut the terminal 10 mm of the exposed tissue into 1 mm long fragments using a surgical scalpel.
- Transfer the fragmented tissue to a 15 mL conical tube containing 5 mL of Dulbecco's Modified Eagle Medium (DMEM) supplemented with 1 U/mL penicillin, 1 µg/mL streptomycin, and 1.5 mg/mL collagenase, pre-filtered through 0.45 µm and then 0.2 µm mesh filters. Incubate for 3-4 h at 37 °C with continuous rotation.
- Centrifuge the sample at room temperature at 500 x g for 10 min. Resuspend the pellet in 10 mL of DMEM supplemented with 10% fetal bovine serum, 1 U/mL penicillin, and 1 µg/mL streptomycin (DMEM-10) and plate on a 6-well tissue culture plate. Passage the cells once and expand to a 10 cm plate before proceeding to the next step.
- Immortalize the cells by performing a transduction with an expression vector carrying HPV-16 viral oncogenes (E6/E7), as described previously7.
- Selecting single cell clones and characterizing MeCP2 reporter expression
- Harvest the cells from a 10 cm plate. First, aspirate the media, add 1 mL of 0.05% trypsin-EDTA and incubate for 2-5 min at 37 °C. Quench with 9 mL of DMEM-10 and collect the resuspended cells in a 15 mL conical tube.
- Dilute the cells with DMEM-10 to a final concentration of 1 cell/100 µL. Transfer to 96-well plates by pipetting 100 µL of the suspension into each well.
- Incubate at 37 °C until the wells are confluent, which can take up to 2-3 weeks. Replace the media with fresh DMEM-10 every 3-5 days. When confluent, mobilize the cells with 20 µL of 0.05% trypsin-EDTA per well and split them into two duplicate sets of 96-well plates.
- Use one set of plates to assay for luciferase activity. Use a homogeneous firefly luciferase assay kit which does not require media removal. Follow the protocol included with the kit.
- Use the results from step 1.2.4 to select clones with no detectable luciferase activity from the remaining set of plates. Expand these clones to 24-well and then 6-well plates.
- Perform the same for a clone with the highest luciferase activity; this will be used as a positive control for validation purposes.
- Harvest an aliquot of each well in the 6-well plate for a Western blot. Mobilize the cells with 200 µL of 0.05% trypsin-EDTA. Incubate for 2-5 min at 37 °C. Quench with 2 mL of DMEM-10, and transfer half of the cells to 1.5ml centrifuge tube. Wash the cells once with PBS before lysing for Western blot. Alternatively, transfer half of the cells to another 6-well plate. When the cells attach to the bottom of the plate, rinse with PBS and lyse for Western blot.
- Perform a Western blot with anti-MeCP2 antibody to confirm that luciferase-negative cells express, while the luciferase-positive cells do not express wild-type MeCP2 from the active X chromosome; follow the previously described protocol8. Use brain tissue lysate from any wild type mouse as a positive control.
- Plate several negative clones at 2 x 105 cells/well in a 6-well plate. Treat once with 10 µM 5-AZA, incubate for 72 h without changing the media, and assay for luciferase activity as described in step 1.2.3.
- Choose one negative clone (Xi clone) that gained luciferase activity with 5-AZA and begin expanding it to multiple 10 cm plates. Freeze the remaining negative clones in liquid nitrogen as a backup. The positive clone (Xa clone) can be maintained in the tissue culture or frozen until further use.
2. Screening the library for reactivation using hygromycin B selection
- Infecting the test clone, applying selection, and harvesting DNA for sequencing
- Obtain viral concentrates of retroviral constructs expressing hairpins from an shRNA library, or produce concentrated viral supernatant using the protocol included with the desired library.
- Before proceeding with the screen, titrate the library (using a GFP marker, if available). Aim for a multiplicity of infection between 0.3 and 0.5. NOTE: Perform at least four independent screens to minimize false positive rates. Use 20 plates per screen to obtain 100-fold coverage of the library to maintain adequate representation of hairpins and avoid random dropouts9. However, given positive selection screen and a large library size, it is likely that a lower coverage would suffice.
- In the morning, plate 1 x 106 Xi cells onto 10 cm tissue culture plates.
- In the afternoon, infect the plates by replacing the media with fresh DMEM-10 containing viral supernatant and 5-10 µg/mL of polybrene. Use the amount of viral supernatant determined in step 2.1.2.
- After 18-24 h, aspirate the media and add 10 mL of fresh DMEM-10 to each plate. Culture for another 48 h.
- In the morning, mobilize the cells using 1 mL of 0.05% trypsin-EDTA per plate and quench with 9 mL of DMEM-10 per plate. Run the suspension through a 100 µm mesh filter. Pool the filtered suspensions together, and seed two sets of 10 cm plates at 1 x 106 cells/plate.
- In the afternoon, replace the media in one set of plates with DMEM-10 containing 15 µg/mL of hygromycin B (day 0). In the other set, replace the media with fresh DMEM-10 only.
- After 48 hours, mobilize the cells from untreated set of plates using 1 ml of 0.05% trypsin-EDTA. Incubate for 2-5 min at 37 °C. Quench with 9 mL of DMEM-10. Transfer the cells to 15 ml conical tubes and centrifuge at 500 x g for 5 minutes at room temperature. Aspirate the supernatants and proceed immediately with extracting DNA from the pellets. Alternatively, pool the cells into larger tubes before the centrifugation step.
- Extract DNA using a mixture of phenol and chroroform and precipitate DNA with sodium acetate and cold ethanol as previously described10. Pool the individual pre-selection DNA samples and store at -20 °C until further use.
- Continue culturing hygromycin B selection set for 4 more days. Replenish the selection media with fresh DMEM-10 containing hygromycin B after 2 days.
- On day 7, replace the selection media with fresh DMEM-10 only. Allow 2 to 4 days for recovery, and then harvest the cells as described in step 2.1.8. Proceed with DNA extraction as described in step 2.1.9.
- Identifying hairpins enriched in the post-selection samples
- Use 5' context (5'-ATCGTTGCCTGCACATCTTGG-3') and 3' reverse loop primers (5'-ACATCTGTGGCTTCACTA-3') to amplify half-hairpins from pre- and post-selection DNA samples. Use all extracted DNA in separate PCR reactions with 100-200 ng of DNA per reaction. NOTE: These 5' context primer and 3' reverse loop primers correspond to the miR-30 context sequence upstream of the shRNA insert and the sequence at the shRNA loop, respectively. Different libraries will require different oligos. Adjust purification steps to isolate different product sizes as necessary.
- Use size-selection beads to remove primers and genomic DNA, and recover PCR products between 100 and 200 bp. Follow the protocol included with the beads.
- Prepare a sequencing library to sequence the half-hairpins. Generate at least 50 million reads per screen. Follow the protocol appropriate for the sequencing kit and equipment. NOTE: 50 million reads per screen provides 1000-fold representation of each shRNA in the library and approximately 10-fold coverage of each independent infection (carried out at 100-fold representation of each shRNA), which ensures adequate representation of each infection event.
- Enumerate each shRNA using a Perl script that counts sequences with perfect matches. Chose shRNAs that had at least 10 reads in the preselection pool for further analysis. Calculate the enrichments as ratios of post- to preselection counts and determine false discovery rate (FDR) on the basis of 1000 permutations. Chose genes targeted by shRNAs with <5% FDR as "hits" for further testing.
3. Validating the identified hairpins ("hits")
- Packaging lentiviruses containing shRNAs identified in the screen
- Seed 8 x 106 293FT cells per plate on 10 cm plates.
- The following day, replace the media with 9 mL of antibiotic-free DMEM-10 per plate; do this 30 minutes before transfection.
- During the 30-minute interval, prepare two mixes for transfection:
- Prepare Mix 1 (per plate/shRNA; scale up according to the total number of shRNAs): 10 µL of 2 mg/mL polyethylenimine (PEI) in 0.5 mL of optimal minimum essential media. Mix by pipetting and incubate at room temperature for 5 min.
- Prepare Mix 2 (per shRNA): 4 µg of the shRNA vector to be packaged, 1.5 µg of the VSV-G envelope vector (pMD2.G), and 2.5 µg of the packaging vector (psPAX2), mixed in 0.5 mL of optimal minimum essential media. NOTE: Include an empty lentiviral vector in the packaging to use in later infections as a negative control.
- Add 0.5 mL of mix 1 to each aliquot of mix 2, and mix gently by pipetting. Incubate in the dark at room temperature for 20 min.
- Apply the mixture onto each plate from step 3.1.2 in a dropwise fashion; swirl the plate gently to mix.
- The following day, replace the media with 5 mL of fresh DMEM-10.
- 48 hours after initial transfection, collect the supernatant and run it through a 0.22 µm filter. Pre-wet the filter with DMEM-10 to maximize recovery.
- Aliquot the filtered supernatant into amounts ideal for infecting a 6-well plate well.
- Freeze the aliquots at -80 °C for later use, or proceed immediately with transduction.
- Transduction and testing of individual hairpins for luciferase reporter reactivation and reactivation of wild-type MeCP2 gene on the Xi.
- In the morning, plate 1 x 105 Xi cells/well onto 6 well plates.
- In the afternoon, infect the plates by replacing the media with 2 mL of fresh DMEM-10 containing lentiviral supernatant and 5-10 µg/mL of polybrene. Infect a control well with viral supernatant produced from an empty lentiviral vector.
- The following day, replace the media with 2 mL of DMEM-10 containing 1 mg/mL puromycin. Culture for four days.
- Replace the media with 2 mL of fresh DMEM-10 and allow 48-72 h for recovery.
- Mobilize the cells with 150 µL of 0.05% trypsin-EDTA. Incubate for 2-5 min at 37 °C. Quench with 1.5 mL of DMEM-10. Transfer the cells to 15 ml conical tubes and centrifuge at 500 x g for 10 minutes at room temperature.
- Remove the media and resuspend the cells in 100 µL lysis buffer provided in the luciferase assay kit. Incubate at room temperature for 5 minutes. Transfer the lysates to a white 96-well plate and assay for luciferase activity using the protocol included with the kit. Use Xa cells as a positive control and Xi cells infected with an empty lentiviral vector as a negative control. NOTE: Avoid exposing the white plates to fluorescent light prior to the assay; this will reduce the background and increase assay sensitivity. The protocol provided in the assay kit was found to be most effective if performed in the dark whenever possible.
- To determine whether the hairpin had an effect on the expression level of MeCP2 on the active X-chromosome, isolate RNA from tranduced Xi cells and measure MeCP2 mRNA level using quantitative PCR with primers designed to amplify only wild type MeCP2 (F: 5'-TTCCATGCCAAGGCCAAACAG-3', R: 5'-CCCATAAGGAGAAGAGACAACAGC-3').
- Optional: Increase the sensitivity of the assay by adding 5-AZA to a final concentration of 0.2 µM after the cells have recovered from puromycin selection. Culture the cells for 72 h without replacing the media, and then assay for luciferase activity using the commercial luciferase assay kit.
- Measure reactivation of the silenced wild-type MeCP2. Infect Xa cells, which harbor the wild type MeCP2 on the Xi, with the vector containing individual hairpins, following the protocol steps 3.2.1. - 3.2.4. Isolate RNA from cells and measure MeCP2 mRNA level using quantitative PCR with primers designed to amplify only wild type MeCP2 (F: 5'-TTCCATGCCAAGGCCAAACAG-3', R: 5'-CCCATAAGGAGAAGAGACAACAGC-3').
Representative Results
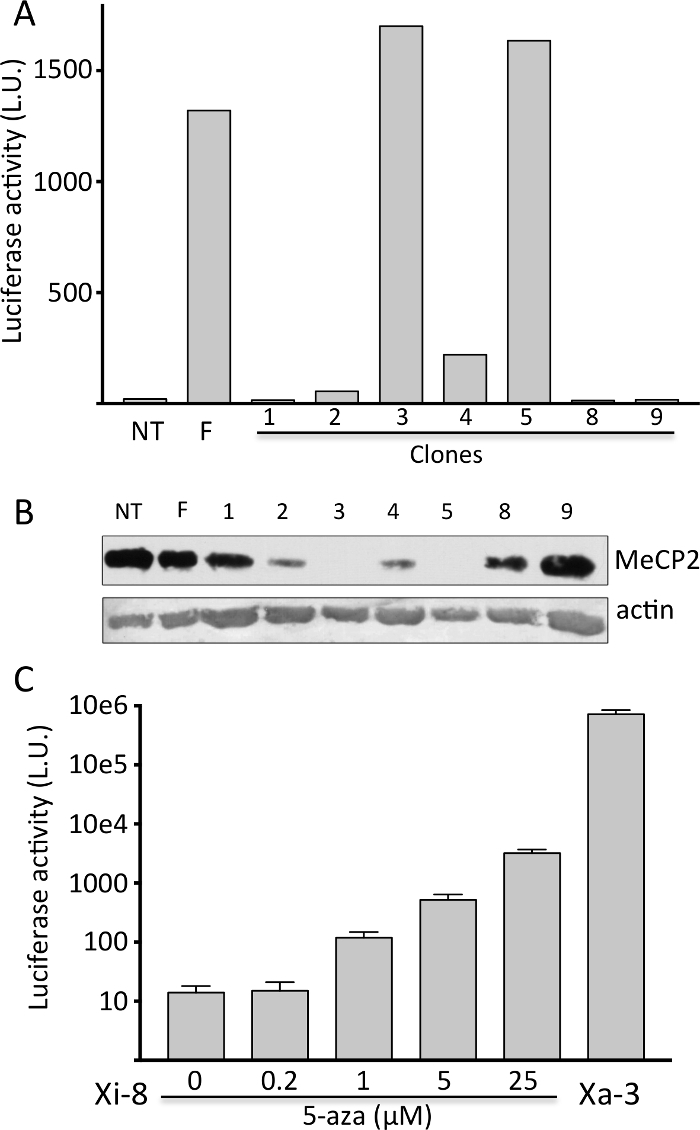
Mouse tendon fibroblasts were harvested from a previously described XMeCP2-LUC-HR/XMeCP2 female mouse, immortalized by retroviral transduction of E6 and E7 oncogenes from HPV-16 virus7, cloned using limiting dilution, and tested for luciferase activity and expression of wild-type MeCP2 protein (Figure 1A and 1B). Luciferase activity was robust if the reporter was on Xa, and undetectable if on Xi; the native MeCP2 expression exhibited a reciprocal pattern. We selected a clone with no luciferase activity (Xi-8) and a clone with robust activity (Xa-3) for further experiments. When Xi-8 cells were treated with 5-AZA, a dose-dependent induction in luciferase activity was observed (Figure 1C).
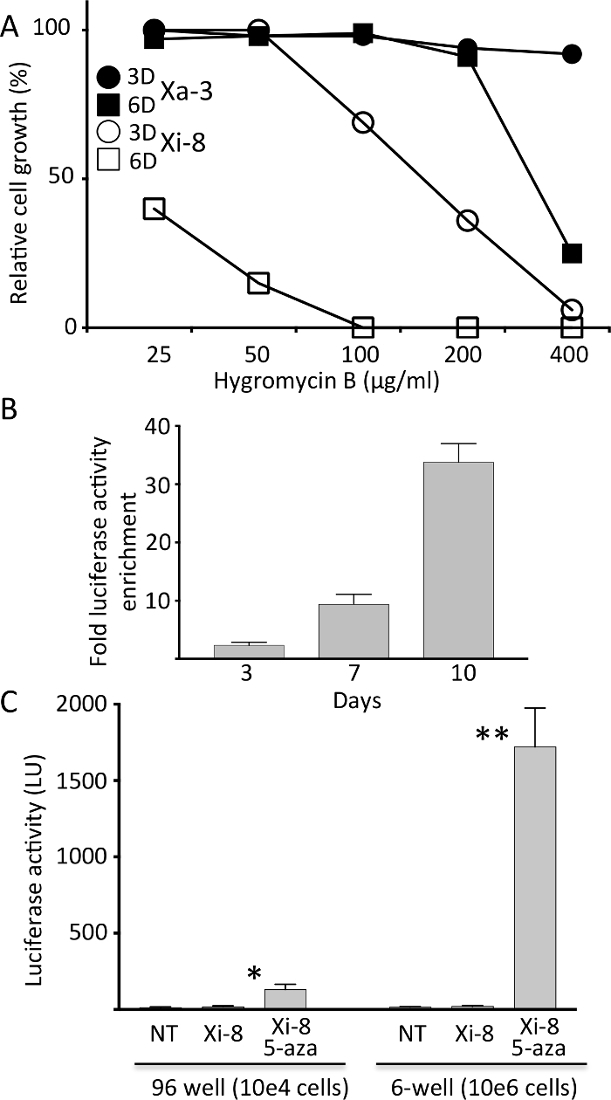
Sensitivity of Xi-8 and Xa-3 cells to hygromycin B was tested by performing 3 and 6 day-long selections. As expected from the expression pattern of the luciferase reporter, Xi-8 cells were more sensitive to hygromycin B than Xa-3 cells, even though the latter exhibited sensitivity to higher doses (Figure 2A). After a 1:100 mix of Xa-3 to Xi-8 cells was exposed to different concentrations of hygromycin B, a time-dependent increase in luciferase activity was observed, indicating successful competition of Xa-3 cells over Xi-8 cells (Figure 2B). Dose and duration of hygromycin B selection were chosen for further experiments based on preferential inhibition of Xi-8 over Xa-3 cells.
Four independent screens were performed to identify regulators of MeCP2 silencing using the Xi-8 cell line and an Open Biosystems miR-30- based shRNA library containing 64,159 different hairpins cloned into a MSCV-shRNA-pgk-Puro-ires-GFP retroviral vector. In each screen, twenty 10 cm plates were infected with pooled shRNAs, targeting approximately 100-fold coverage of the library. Hygromicin B was conducted for 6 days, until small acellular patches were observed; the cells continued to die for additional 24 to 48 hours. When recovered, the cells were harvested for DNA extraction using a phenol-chloroform technique. The total DNA yield per screen was around 20 µg/plate, which was pooled and divided between 100 PCR reactions using primers designed to amplify half-hairpins. The PCR products were then bead-purified and subjected to high throughput sequencing.
Pre- and post-selection samples were compared to determine hairpin enrichment, as we expected that any hairpin present at elevated levels in the post-selection sample targeted genes that reactivated Xi and thus conferred hygromycin resistance. Only hairpins enriched in at least 2 of the four screens were considered for validation. The most enriched hairpins in all replicates of our screen were four hairpins targeting XIST, consistent with its crucial role in XCI11.
The validation part of the screen was carried out by individually testing the hairpins that registered as hits for activation of the luciferase reporter in Xi-8 cells. Although the screen was carried out using an MSCV-based retroviral library, the validation was carried out using a lentiviral vector (individual pGIPZ clones with shRNA) to avoid possible detection of retrovirus-induced artifacts.
Each of the candidate genes was targeted by at least two different hairpin sequences, and the knockdown of the target gene was verified by real- time quantitative PCR (RT-qPCR). By assaying up to 1 x 106 cells in 6-well format and decreasing the background luminescence (Figure 2C), we were able to detect very low-frequency reactivation events, which we estimated to be in the 1:20,000 range.
Further analysis using individual hairpins led to the identification of 30 genes whose knockdown resulted in reactivation of the reporter on the Xi. The median magnitude of luminescence for these 30 genes was 2.8-fold above the baseline, which was further amplified to 7.1-fold following 5-AZA treatment. The same hairpins were retested, in conjunction with 5-AZA, with the Xa-3 cells to confirm re-expression of wild type MeCP2 from the Xi. This was achieved by performing RT-qPCR using primers designed to amplify only the wild type transcript of MeCP2.
Figure 1. Isolation of clones with the MeCP2-Luc-HR reporter gene on the Xi. A) Representative firefly luciferase assay on immortalized tendon fibroblasts clones (1-9). Primary fibroblasts from non-transgenic (NT) and transgenic heterozygous female (F) were used as negative and positive controls, respectively. B) Representative Western blot showing MeCP2 protein expression from the cells in (A). Note that luciferase activity (Figure 1A) and MeCP2 expression exhibit reciprocal expression pattern. C) Induction of firefly luciferase activity in Xi-8 cells with 5-AZA. Xa-3 cells, with the reporter gene on the Xa, serve as a positive control. Please click here to view a larger version of this figure.
Figure 2. Characterization of the hygromycin resistance in Xi-8 and Xa-3 cells. A) Relative growth of Xi-8 and Xa-3 cells treated with different hygromycin B regimens (3D = 3 days; 6D = 6 days), as compared to untreated cells. B) Luciferase activity enrichment after exposing a total of 10,000 Xa-3 and Xi-8 cells (mixed in 1:100 ratio) to hygromicin B (25 µg/mL) for indicated number of days. Post-exposure luciferase activity was divided by the pre-exposure level to calculate enrichment. C) Firefly luciferase activity in Xi-8 cells, assayed in 96- and 6-well format, with and without 5-AZA (10 µM). Non-transgenic (NT) fibroblasts were used as a negative control. 5-AZA increased the luciferase activity 7.2-and 86-fold over the baseline in 96- and 6-well format, respectively. (N = 3, *p < 0.001, **p < 0.0001 by Student's t- test comparing 5-AZA treated and untreated Xi-8 cells.) Please click here to view a larger version of this figure.
Discussion
In our recent study6, we generated a murine cell line with luciferase and hygromycin resistance genes fused to MeCP2 on the Xi, and transduced it with a library of >60,000 shRNAs targeting >25,000 genes. We found 30 genes whose knock-down conferred survival advantage under hygromycin B selection, suggesting their role in control of MeCP2 and X-chromosome silencing. These results were validated by transducing the reported cell line with individual hairpins and assessing reactivation of the silent luciferase reporter.
The finding that the top four hairpins in all replicates of our screen targeted XIST provided a strong internal validation for our method, given the pivotal role of XIST in XCI11. Aside from the TGFβ superfamily, whose regulatory role in XCI had not been previously described, the other identified genes all belonged to functional families with ties to epigenetic regulation, thus serving as an additional validation for our method12,13,14. Furthermore, we also detected 4 out of 13 genes previously identified in a different screen, using a CMV promoter-driven GFP randomly inserted on the Xi15.
Establishing the suitable dose and duration of hygromycin B selection was crucial in optimizing the screening conditions. The sensitivity of our cell line was highly dependent on seeding density, and was adversely affected by retroviral infection. Increased doses or length of selection also inhibited the growth of cells with the reporter on Xa, whereas milder selection lowered the sensitivity for hairpin enrichment. Multiple hygromycin B dosing regimens and reporter cell seeding densities were thus tested before the final screening. Moreover, although the MSCV retroviral vector in our shRNA library provided an opportunity to use puromycin selection and enrich for successfully transduced cells, puromycin was not used in our experiments, as the sequential use of puromycin and hygromyin B resulted in excessive toxicity, even after appropriate dose de-escalation of both antibiotics.
All the hairpins identified in our study also reactivated the silent luciferase reporter, albeit to a more modest degree. Detection of these infrequent events was enabled by the high sensitivity of our screen, which we estimated, based on testing admixtures of Xi and Xa cells, to be one reactivation event in 2 x 104 cells. Such low levels of reactivation, increased with co-application of 5-AZA, attest to previously described difficulties in disrupting tight transcriptional control of XCI12,13,16,17, and imply that the disruption of multiple regulatory mechanisms would be needed to achieve significant reactivation.
Of note, all 30 hairpins identified in our screen also reactivated a CMV-driven reporter gene located at a different X-chromosome locus, indicating their broader impact on Xi de-repression. Further studies are needed to test whether MeCP2-specific "reactivators" exist, as their pharmacologic or genetic manipulation could prove extraordinarily useful in treating the Rett syndrome.
Our screening system offers several advantages over the previously described reporters used for X-chromosome reactivation screens12,17. Instead of being driven by a CMV promoter, our reporter cassette is knocked in at the C-terminus of MeCP2 and is thus driven by the native X-linked promoter. This design can help identify MeCP2 locus-specific regulators, more relevant in the context of Rett syndrome research, with a caveat that the findings in immortalized fibroblasts should be recapitulated in neurons, the desired site of MeCP2 reactivation. The use of a hygromycin resistance gene enables positive selection of reactivation events and robustly increases the screen sensitivity. Moreover, the cassette containing hygromycin resistance and firefly luciferase genes enables both large-scale screening using positive selection with hygromycin B, and testing/screening individual hairpins using the luciferase reporter.
In summary, we report a robust and sensitive genetic screening method for identification of regulators of MeCP2 and X-chromosome silencing in mammalian cells. This methodology could be adapted for screening small interfering RNAs (siRNAs), CRISPR, or small molecules. Such assays could prove useful in establishing therapeutic approaches that rely on reactivating the functional allele of a silent gene, such as in the case of Rett syndrome.
Disclosures
Dr. Leko contributed to this article as an employee of the Fred Hutchinson Cancer Research Center. The views expressed are his own and do not necessarily represent the views of the National Institutes of Health or the United States Government
Acknowledgments
We thank Ross Dickins of the University of Melbourne for graciously providing the shRNA library used in the screen. This work was funded by the Rett Syndrome Research Trust (A.B.).
References
- Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro C, de Hoon B, Gribnau J. Fitting the Puzzle Pieces: the Bigger Picture of XCI. Trends Biochem Sci. 2016;41:138–147. doi: 10.1016/j.tibs.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Disteche CM. Dosage compensation of the sex chromosomes and autosomes. Semin Cell Dev Biol. 2016;56:9–18. doi: 10.1016/j.semcdb.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. Wild-type microglia do not reverse pathology in mouse models of Rett syndrome. Nature. 2015;521:E1–E4. doi: 10.1038/nature14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripathy S, et al. Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-beta superfamily as a regulator of XIST expression. Proc Natl Acad Sci U S A. 2017;114:1619–1624. doi: 10.1073/pnas.1621356114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SA, Galloway DA. Human papillomavirus type 16 E7 alleviates a proliferation block in early passage human mammary epithelial cells. Oncogene. 1996;12:1773–1779. [PubMed] [Google Scholar]
- Hnasko TS, Hnasko RM. The Western Blot. Methods Mol Biol. 2015;1318:87–96. doi: 10.1007/978-1-4939-2742-5_9. [DOI] [PubMed] [Google Scholar]
- Strezoska Z, et al. Optimized PCR conditions and increased shRNA fold representation improve reproducibility of pooled shRNA screens. PLoS One. 2012;7:e42341. doi: 10.1371/journal.pone.0042341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MR, Sambrook J. Isolation of High-Molecular-Weight DNA from Mammalian Tissues Using Proteinase K and Phenol. Cold Spring Harb Protoc. 2017;2017 doi: 10.1101/pdb.prot093484. [DOI] [PubMed] [Google Scholar]
- Goto Y, Takagi N. Tetraploid embryos rescue embryonic lethality caused by an additional maternally inherited X chromosome in the mouse. Development. 1998;125:3353–3363. doi: 10.1242/dev.125.17.3353. [DOI] [PubMed] [Google Scholar]
- Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol. 2001;153:773–784. doi: 10.1083/jcb.153.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minajigi A, et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science. 2015;349 doi: 10.1126/science.aab2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521:232–236. doi: 10.1038/nature14443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, et al. Genetic and pharmacological reactivation of the mammalian inactive X chromosome. Proc Natl Acad Sci U S A. 2014;111:12591–12598. doi: 10.1073/pnas.1413620111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkovsky A, et al. The Mbd1-Atf7ip-Setdb1 pathway contributes to the maintenance of X chromosome inactivation. Epigenetics Chromatin. 2014;7:12. doi: 10.1186/1756-8935-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkovsky A, et al. A high-throughput screen of inactive X chromosome reactivation identifies the enhancement of DNA demethylation by 5-aza-2'-dC upon inhibition of ribonucleotide reductase. Epigenetics Chromatin. 2015;8:42. doi: 10.1186/s13072-015-0034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]