

Abstract
Response criteria in acute myeloid leukemia (AML) has recently been re-established, with morphologic examination utilized to determine whether patients have achieved complete remission (CR). Approximately half of the adult patients who entered CR will relapse within 12 months due to the outgrowth of residual AML cells in the bone marrow. The quantitation of these remaining leukemia cells, known as minimal or measurable residual disease (MRD), can be a robust biomarker for the prediction of these relapses. Moreover, retrospective analysis of several studies has shown that the presence of MRD in the bone marrow of AML patients correlates with poor survival. Not only is the total leukemic population, reflected by cells harboring a leukemia associated immune-phenotype (LAIP), associated with clinical outcome, but so is the immature low frequency subpopulation of leukemia stem cells (LSC), both of which can be monitored through flow cytometry MRD or MRD-like approaches. The availability of sensitive assays that enable detection of residual leukemia (stem) cells on the basis of disease-specific or disease-associated features (abnormal molecular markers or aberrant immunophenotypes) have drastically improved MRD assessment in AML. However, given the inherent heterogeneity and complexity of AML as a disease, methods for sampling bone marrow and performing MRD and LSC analysis should be harmonized when possible. In this manuscript we describe a detailed methodology for adequate bone marrow aspirate sampling, transport, sample processing for optimal multi-color flow cytometry assessment, and gating strategies to assess MRD and LSC to aid in therapeutic decision making for AML patients.
Keywords: Cancer Research, Issue 133, acute myeloid leukemia, measurable residual disease, leukemic stem cells, bone marrow aspiration, leukemia associate immunophenotype, fluorescence-activated cell sorter analysis
Introduction
Acute myeloid leukemia (AML) is a malignancy of the bone marrow characterized by defects in the maturation program, with abnormal proliferation and accumulation of myeloid progenitor cells, inhibition of normal hematopoiesis, and ultimately bone marrow failure. The disease is highly heterogeneous with respect to morphology, immunophenotype, cytogenetics, molecular aberrations, and gene expression signatures, as well as to treatment response and treatment outcome1,2. Current management includes induction chemotherapy with the aim to achieve complete remission (CR), followed by post-remission treatment, which is largely guided by the results of molecular, cytogenetic and immunophenotypic studies and consists of either several courses of additional chemotherapy or (autologous or allogeneic) stem cell transplantation3. Despite high remission rates after intensive chemotherapy of up to 90%, 5-year survival in adults is only approximately 30%-40%, predominantly due to the development of relapses which are commonly resistant to chemotherapy and thereby very difficult to treat. Outcome in children is better, although approximately one third also relapse. Therefore, early detection of imminent relapse will fill an unmet medical need and may guide post-remission therapy4.
Residual disease after therapy may reflect the sum of all diagnosis and post-diagnosis resistance mechanisms/factors; hence its measurement may be prognostic and instrumental for guiding treatment. The possibility of defining residual disease (formerly called minimal residual disease and now referred to as measurable residual disease or MRD) far below the morphological criterion of 5% blast cells is changing the landscape of risk classification. Currently the two methods mostly used to detect MRD are flow cytometry-based and molecular-based, the latter being assessed by reverse transcriptase PCR (RT-qPCR)5 or, although in a premature stage, by next generation sequencing (NGS). Many studies in adults as well as in children already demonstrated that various MRD approaches provide strong prognostic information in AML both after induction and consolidation therapy6,7, and a new definition of disease burden (superior to morphological CR) is now emerging8. This suggests that MRD assessed by flow and/or molecular techniques should become, and in fact are already becoming, standard in every clinical trial in AML.
This manuscript discusses the detailed flow cytometry procedure to obtain an accurate and reproducible immunophenotypic characterization of MRD in bone marrow samples, including the bone marrow sampling and processing procedures preceding flow cytometry. The availability of good quality bone marrow samples at diagnosis and follow-up is crucial to the success of this measurement across clinical sites and clinical trials. In fact, these pre-analytical considerations are also of vital importance for molecular (PCR and NGS) MRD approaches. For immunophenotypic characterization of MRD, aberrantly expressed markers are combined with normal myeloid and progenitor markers, to identify a leukemia-associated immunophenotype (LAIP)9. MRD measures the resultant of many factors influencing response to therapy, such as intrinsic or acquired leukemia drug resistance, pharmacodynamics and kinetics of therapy, immune surveillance, and compliance. Therefore, MRD is a very strong post-diagnosis prognostic parameter associated with clinical outcome when dichotomized on a cut-off level determined by Receiver-Operating Characteristic (ROC) analysis. For our adult AML cohort of the HOVON 42a study, the cut-off level is set at 0.1% of LAIP positive cells/total white blood cells. Using this criterion for determining negative vs positive MRD status, a group of patients can be identified who have a significantly worse relapse incidence, relapse-free and overall survival6. Additionally, we describe the measurement of immature, drug resistant leukemia cells with stem cell-like features (CD34+CD38- leukemia stem cells, or LSC), which offers a strong predictor of patient outcome10. Together LAIP and LSC approaches form the flow cytometric MRD approach. The LAIP approach is suitable for roughly 90% of the patients, while the LSC approach can be applied in about 80% of the patients. Together over 95% of the patients can be evaluated for either one parameter or both.
Lastly, this publication provides a detailed operational description to assess MRD by flow cytometry. This includes: 1) harmonization and/or standardization of bone marrow sampling procedures, 2) sample transportation guidance 3) detailed description of the leukemic cell detection with FACS using several antibody panels including single cell tube approaches to characterize the LSC, 4) set-up of the FACS machines for standardized measurements, 5) analytical programs for MRD measurements and 6) analytical programs for LSC detection.
We aim to show all facets of the procedure including the sample preparation since that is rarely discussed while it is an important issue for the quality of the ultimate result. Bone marrow aspiration and biopsy are clinical procedures used to evaluate the hematopoietic cells within bone marrow. These are performed together with a complete blood count (CBC) and blood smear. The optimal method for bone marrow aspiration is crucial for the accurate diagnosis and follow-up for MRD measurement. In addition, a successful bone marrow aspirate should contain enough cells to perform the LAIP and LSC flow analysis (at least 10 million viable cells). Here, we describe the method for performing a bone marrow aspiration and provide guidelines, which should result in adequate cell sampling (and limit the potential for hemodilution) needed for accurate diagnostics and additional research. These pre-analytical considerations are also of vital importance for molecular (PCR and NGS) MRD approaches. All specimens for immunophenotyping should be processed preferentially within 24 h of collection. Although not recommendable, bone marrow and peripheral blood samples can still be processed and analyzed when kept up to 72 h at ambient temperature. In addition, all handlings with the material should be performed under sterile conditions, in order to enable cryopreservation of sterile cells for later research/quality assessment etc.
Protocol
The protocol follows the guidelines of the research code and the research ethics committee of the VU University Medical Center.
1. Bone marrow aspiration and sample preparation
- Patient and material preparation
- Fill one or two 10 mL syringes with 1% lidocaine using a 16-gauge needle. Replace the needle for a 21-gauge needle.
- Put two drops of 5% EDTA on a watch-glass.
- Set up glass slides (n=15 with consistent patient numbering and date) for the smear preparations.
- Place patient in lateral decubitus position. Locate the superior posterior iliac spine and mark with pen. NOTE: In general, the posterior superior iliac spine is located one hand width distal to the iliac crest and one hand width lateral to the midline. In females, the actual spine may be a bit more lateral, in some men a bit more medial.
- Disinfect the skin with chlorhexidine 0.5-1% in ethanol from the intended biopsy area outward in circles.
- Open the package of sterile gloves, put on the sterile gloves and lay down the package on the table to create a sterile field. Open the package with the aspiration needle and place it onto the sterile field.
- Infiltrate the skin and subcutaneous tissue and finally periosteum. At the periosteum, administer the lidocaine in such a way that an area of 1 cm diameter has been anesthetized. NOTE: Adequate administration of lidocaine to the periosteum is one of the most important factors for patient comfort. Test whether the periosteum has been adequately anesthetized by tapping the intended biopsy location with the introduced needle and ask if the patient feels any pain. Of note: Children are fully anesthetized during the whole procedure.
- Hold the aspirate needle (15 Ga x 2.8") with the proximal end in the palm and index finger against the side of the needle's metal shaft near the tip; this position allows better control.
- Introduce the needle with a rotating movement (by quickly alternating pronating/supinating movement) through the skin toward the iliac spine and bring the needle into contact with the posterior iliac spine.
- Ensure that is needle is introduced to the anesthetized area of the periosteum; the patient should feel only pressure and no pain. If the patient feels pain, either reposition the needle, or administer more lidocaine.
- Using gentle but firm pressure, advance the needle while rotating it in an alternating clockwise-counter clockwise motion. Entrance into the marrow cavity is generally detected by decreased resistance.
- Remove the stylet from the needle. Attach a 10 mL empty syringe to the needle.
- Apply negative pressure by withdrawing the syringe plunger with a gentle pull. Warn the patient that they may feel a cramping sensation and pain when marrow is being aspirated. If not enough bone marrow spicules are released, another aspiration should be performed with one quick draw. Most spicules will be in the first 1-2 mL obtained from the initial pull. Aspirate only 1-2 mL to avoid diluting the sample. NOTE: Dilution will result in hemodilution and may confound MRD results).
- Remove the syringe and replace the stylet into the aspiration needle. Eject part of the marrow into a watch-glass and the rest into an 8 mL tube coated with heparin. Note: Invert each tube immediately after placing the marrow aspirate into the specimen tube to ensure adequate anticoagulation. Bone marrow coagulation is often the cause of inappropriate material. Additional bone marrow can be aspirated from the same spot, but preferably, the needle is advanced 5-10 mm before a new aspirate is taken. Preferably not more than 2 draws per insertion level should be taken. Make sure to mark the tubes with increasing numbers representing the first, second and/or consecutive draws. It is common rule to use the first draw for the most relevant diagnostic analysis.
- Alternatively, retract the needle from the insertion place and reintroduce it into the marrow cavity a few millimeters away from the original insertion place and repeat the procedure. Note: Do not aspirate more than 1-2 mL per pull, to avoid significant blood contamination.
- Repeat as often as material is needed. When sufficient material for the specific clinical study protocol is aspirated remove the bone marrow needle. NOTE: In case of slow or otherwise difficult bone marrow aspirations the use of syringes that were pre-flushed with anticoagulant may be helpful. In case of a dry tap, perform a trephine biopsy which is outside the scope of this manuscript.
- Preparation of smears for morphologic examination
- For optimal morphological assessment, pick out spicules (e.g. using a plastic spatula) from the aspirate in the watch-glass and place them on a glass slide.
- Gently place another glass slide over the slide with the marrow and gently slide; avoid any pressure. NOTE: Only in case of very large bone marrow spicules slight pressure may be exerted, to decrease the thickness of the cell spreading. The procedure benefits from help from an assistant who can handle the specimen tubes. Accurate labelling of the tubes with the patient number and number of the sequential bone marrow draw is crucial.
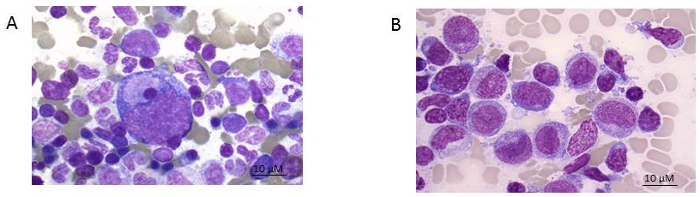
- Dry the slide thoroughly, and then perform May Giemsa Grünwald staining (see table of materials). Examine under the light microscope for morphology (see Figure 1). NOTE: Figure 1A shows the smears of healthy bone marrow consisting of different functional cell types while Figure 1B an AML patient with predominantly leukemic blasts. To better define the residual leukemic burden, immunophenotyping needs to be performed.
2. Transport of material for further processing
- Preparation of the samples for transport
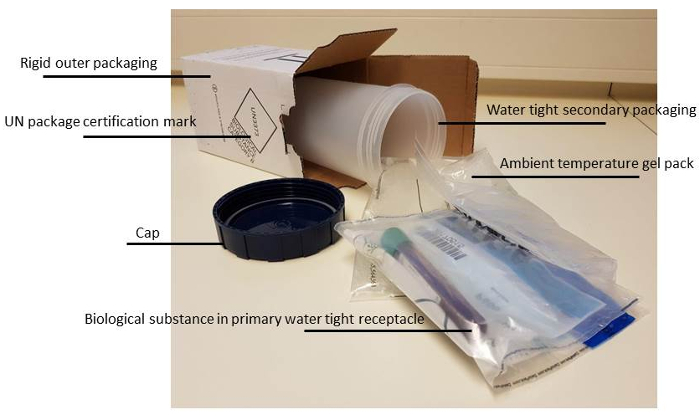
- In order to make sure that the material will not leak and the tubes will not break, ensure correct packaging (see Figure 2). NOTE: From our and others experiences the viability of the cells is best preserved when the bone marrow is kept at room temperature. For long-term transport this is best achieved using a room temperature 'gel- pack' to aid in temperature stability during transit.
- Label packages and add the correct forms to avoid loss of the package, prolonged transport or switching of patients. NOTE: This should at least contain: (Form 1) Patient data. This should include the study number and commonly the 'Date of Birth'. Essential for the right processing of the material after arrival at the receiving lab, is a clear statement of specifically requested laboratory analysis (molecular diagnostics, immunophenotyping, MRD etc.) on this form. (Form 2) Address and phone number of a contact person, and when necessary, papers for customs. To avoid losing packages in the mail, the sending laboratory always notifies the receiving lab that a sample has been sent. Use of "track and trace" is recommended.
- Receiving samples from other institutes
- Make sure that the suitable logistics are in place at the institute to receive the sample at the laboratory as soon as it is delivered at the institute. NOTE: For optimal logistical organization preferably a central laboratory is required, which receives and collects all material from the local clinic and from external hospitals. In particular, assigning protocols for distribution and clear communication with the executive laboratories is essential.
- After receiving the samples, accurately note the appropriate numbering in hard copy forms and a secured database containing important fields such as MRD ID number, Trial specific ID number, Hospital registration number, Sending institute, Date of birth, material type (bone marrow or peripheral blood), white blood cell count (WBC), date of bone marrow aspiration, date of measurement of the sample and any remarks relevant for the quality of the measurement. NOTE: Preferably, this database is also equipped to contain additional data (or be linked to other databases) of analysis results, and further patient data such as follow-up data.
3. Flow Cytometer Setup
Note: This section is based on Euroflow instructions.11
- Set up of photomultiplier (PMT) voltages for FCS SSC parameters and target fluorescence channels
- For FCS SSC parameters start up the flow cytometer and perform the "Cytometer Setup and Tracking (CST)" run on the flow cytometer with CST beads (see table of materials). Lyse red blood cells from a healthy donor (described in section 4.1).
- Create a new experiment (In menu: Experiment, New Experiment, select Blank experiment) using the manufacturer's software (see table of materials) with a Forward scatter (FSC) versus Sideward scatter (SSC) dot plot for setting up the FSC and SSC parameters. Name this experiment FSC/SCC PMT with the date. First use the current cytometer settings (right click: "apply current CST"). These settings have been generated with the CST performance check using CST-calibration beads (see table of materials). Adjust FSC and SSC to visualize the lymphocytes in "global experiment settings". Note: in our flow cytometer these are 285 V and 400 V respectively. Check off the "Enable Compensation" in: Inspector/Instrument Settings/Compensation.
- To assess size of the cells (FSC) and granularity (SSC) start measuring unlabeled lysed peripheral blood cells by the "acquire cells" function. Gate the lymphocytes and adjust/fine-tune FSC and SSC voltages to reach the following mean target values for the gated lymphocyte population: FSC: 100,000 (range 95,000 - 105,000); SSC: 15,000 (range 13,000 - 17,000).
- Acquire and record data by measuring at least 10,000 events.Verify the mean FSC and SSC target values for gated lymphocytes and re-adjust if necessary.
- For setting up the target fluorescence channels PMT voltages use 8-peak rainbow beads calibration particles (see table of materials).
- Create a new experiment on the FACS for 7-peaks. Create a worksheet "Target mean fluorescent intensity (MFI)" with all necessary dot plots (n=2; FSC versus SSC, FITC versus PE), histograms (n=8; one histogram for each fluorescence detector) and statistics showing the reference peak values (MFI and coefficient of variation (CV)) for each fluorescence channel.
- Prepare freshly a solution containing 1 drop (± 60 µL) of rainbow beads in 1 mL of dH2O. Gently vortex to mix the beads in solution.
- Use the PMT settings of the FSC/SSC voltages from above and start measuring the fluorescence of the rainbow beads by using the "acquire" function (without recording) at "LOW" flow rate with a threshold of 5,000. Use the "rectangular gate button" to gate the singlet beads population P1 in the FSC versus SSC dot plot. Gate the 7th PEAK - Population P2 in the FITC versus PE dot plot.
- Continue the acquisition of the 7-peaks Rainbow bead suspension and adjust/fine-tune PMT voltages in all fluorescence channels to reach target MFI values according to the annotated MFI target channels as provided with the beads.
- Use "Record" to collect data of about 10,000 events and check the MFI for the 7th Peak beads. Correct the PMT voltages if necessary. Once Target MFI values for the 7th Peak are reached, record/overwrite the file for the final PMT values.
- Save the final PMT values and name the file PMT Setup with the date, which will be save in the catalog of the manufacturer's software. Use this PMT setup for correcting the spill over for each fluorescence dye.
- Fluorescence compensation settings
- Label a tube for the unstained control and each fluorochrome conjugated antibody (see Table S1 for the used fluorochromes). Pipette 100 µL of buffer into each tube.
- Vortex the vial of multicolor compensation beads (see table of materials) thoroughly and then add 1 full drop (± 60 µL) of these beads to each tube. NOTE: Avoid dripping the beads down the side of the tube while adding them to each tube. This can lead to low bead concentration and could impact the results of the compensation matrix.
- Pipette the appropriate volume calculated for one test of fluorochrome-conjugated antibody sufficient (which would be sufficient to stain 106 cells) into the corresponding tube and vortex thoroughly. Do not add antibody to the unstained control tube. Incubate for 15 to 30 min in the dark at room temperature (RT).
- Add 4 mL of wash buffer (phosphate-buffered saline (PBS)/0.05% azide-0.1% human serum albumin (HSA) (see table of materials)) to each tube. Centrifuge the tubes at 300g for 10 min. Remove the supernatant and resuspend the bead pellet by adding 0.2 mL of wash buffer to each tube. Vortex the tubes thoroughly.
- Open the manufacturer's analysis software (see table of materials) and create a new experiment (In menu: Experiment, New Experiment, select Blank experiment) and rename as compensation file with current date.
- Go to "cytometer settings" and use the PMT setting from section 3.1. Be sure that the threshold in FSC is 5,000 and the compensation values of all the used fluorochromes are zero.
- Create compensation controls, including label-specific tubes, as needed. Be sure to include an unstained control. Select from the menu: Experiment, Compensation Setup, Create Compensation Controls.
- Load the unstained control tube and adjust the P1 gate around the singlet bead population and ensure that the P1 gate contains only singlet beads.
- Right-click the P1 gate and select "Apply to All Compensation Controls". Record data for all the single labeled fluorescence tubes. Verify that P2 gate encompasses the positive population on each fluorescence histogram. If needed adjust the gate.
- Calculate the compensation. Select Experiment, Compensation Setup, Calculate Compensation.
- If the compensation calculation is not successful, an error message displays. Make the necessary adjustments, and recalculate. When the compensation calculation is successful, a dialog appears prompting for the name for the compensation setup.
- Enter a name (for instance YY-MM-DD 8 color setup) and click, link and save. The compensation setup will also be saved to the compensation setup catalog. NOTE: The same settings can be used for all experiments using the same fluorophores.
4. Flow Cytometry Assessment (LAIP and LSC)
Note: Here we describe the bulk lysis procedure before staining, which enables to stain a preferred concentration of WBC. Most MRD protocols use this approach although some have successfully used other options such as staining before lysis. Whole bone marrow staining before lysis minimizes preferential cell loss12, but has the disadvantage of unpredictable, too low cell concentrations.
- Bulk lysis of cells Note: Keep the unmanipulated samples horizontal at RT, when flow cytrometric acquisition cannot be performed the same day in order to start the whole procedure the next day.
- Resuspend the anticoagulated bone marrow sample to homogeneity by inverting the sample several times.Determine the concentration of bone marrow cells (white blood cell count (WBC)) using a cell counting chamber with cell staining Tuerk solution (see table of materials).
- Pipet the needed volume of the bone marrow into a separate 15 mL tube. The amount of cells is dependent on the number of separate staining tubes; for one staining (one tube) use approximately 2 x 106 white blood cells for the LAIP measurements and 8x106 white blood cells for measurement of the LSC tube.
- Lyse red blood cells by adding lysing solution (see table of materials). Required volume of lysing solution should be 10 times the volume of the cell suspension.
- Mix gently, by inversion and incubate for 10 min at RT.Centrifuge for 7 min 800g at RT. Discard the supernatant (e.g. with a vacuum pump or Pasteur pipette).Resuspend the cell pellet in phosphate-buffered saline (PBS)/0.05% azide-0.1% human serum albumin (HSA) (see table of materials) at RT. Use the maximum volume of the tube.
- Centrifuge for 7 min 800g at RT.Discard the supernatant (e.g. with a vacuum pump or Pasteur pipette).Re-suspend the cell pellet in PBS/0.05% azide-0.1% HSA to a cell concentration of 100x106 WBC/mL, and divide over the number of intended tubes.
- Staining of cells for flow cytometry
- Pipet monoclonal antibody (MoAb) cocktail solution mix into the appropriate fluorescence-activated cell sorter (FACS) tubes and incubate with the lysed bone marrow cells for 15 min in the dark. NOTE: This is very important when tandem-dyes are used. For example, see the mixes of the panels standardly used in our laboratory (Table S1).
- Wash the cells with 3 mL PBS/0.05% azide-0.1% HAS. Centrifuge the cells for 5 min at 400g. Discard the supernatant (e.g. with a vacuum pump or Pasteur pipette) and the resuspend the cell pellet in 300 µL PBS/0.05% azide-0.1%HSA.
- For LSC measurement: resuspend the cell pellet in 400 µL PBS/0.05% azide-0.1% HSA and add 4 µL of blank beads (e.g. Spherotech, see table of materials) to use as negative control population in the analysis.
- Open the MRD or LSC experiment lay-out on the FACS (see table of materials) and measure for MRD at least 100,000 WBC gated events for measurement of AML patient samples at diagnosis and 1X106 WBC for AML samples at follow-up. For the LSC experiments measure at least 4x106 WBC both at diagnosis and follow-up for a reliable LSC result.
- Save the data using a standardized appropriate filename for the analysis of the results. Make an inventory of the different measured markers on the AML cells and on the AML stem cells (positive, negative, over/under expression) and store these data in the lab journal.
- In case there is no diagnosis LAIP available, measure all 4 tubes at follow-up to ensure that any possible measurable LAIP may be identified in a different-from-normal approach.
- Choose the most reliable LAIP(s)13 established at diagnosis and acquire as many events as possible, but > minimum numbers defined.
5. Identification of Leukemia Associated Immuno-Phenotypes (LAIP): Analyses of different cell populations
Note: FCS files can be analyzed with several software program for optimal visualization of the different cell populations (see table of materials).
- White blood cell population
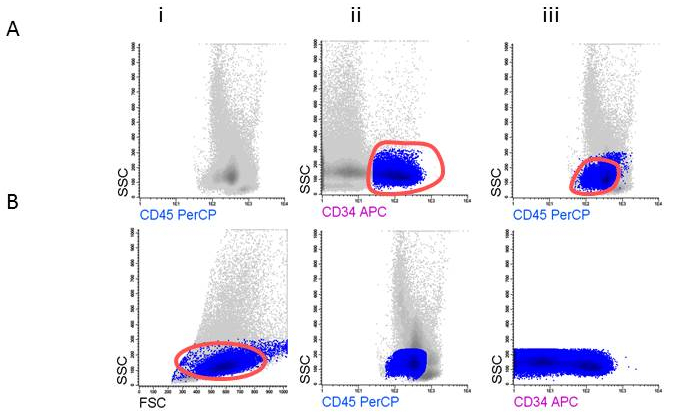
- Gate the CD45 positive cells and the viable appearing cells, leaving out low FSC (non-viable cells, erythroid cells) within the FSC/SSC plot; these contain the WBC (Figure 3Ai). Show the WBC and use one of the primitive markers (for example CD34;Figure 3Aii) to help to ascertain the final position of the blasts in de CD45dim area (Figure 3Aiii).
- Gate the CD45dim cells and back gate the cells in the FSC/SSC plot (Figure 3Bi). Remove the CD34 gate (Figure 3Bii) while reporting the %cells in this gate as %blasts in your population tree in the program. Show the blasts in a SSC/CD34 plot and gate the CD34 positive cells (Figure3Biii).
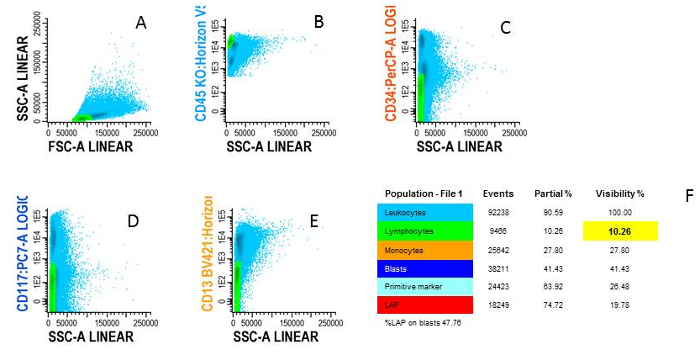
- Lymphocytes
- Use lymphocytes as an internal control: myeloid populations as a negative control and lymphoid populations as a positive control.
- LAIP at diagnosis
- Gate the blasts and subsequently the CD34 positive cells in a SSC/CD34 plot and call these 'primitive marker' in the population tree.
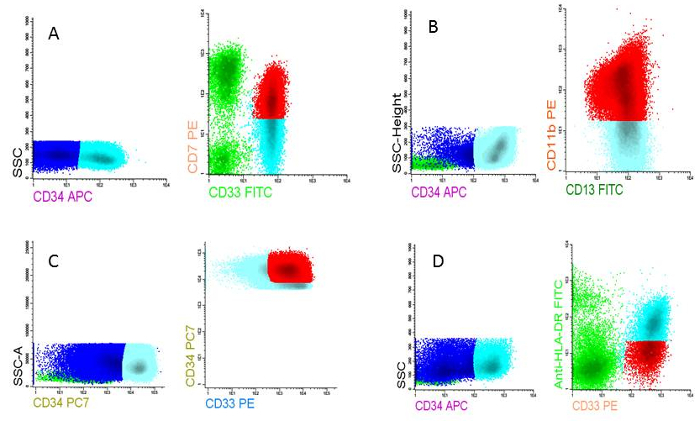
- Plot the primitive cells, in this case CD34 positive, and the lymphocytes in a new plot with a lymphoid marker (CD7) against a myeloid marker (CD33). Gate the aberrant population (see for guidelines Supplementary File 1) and call them 'LAIP positive' in the population tree. NOTE: The final chosen LAIP is reported as % LAIP on the blast population. The sensitivity, specificity and stability of the LAIPs can be established only by personnel with ample experience in MRD measurements. The total of these parameters determines the quality of the LAIP. Examples of four different LAIP assessments at diagnosis are given in Figure 5 A-D.
- Determine the expression of LAIPs (% on blasts) and preserve these data in a database or excel file.
- Make reports of the FACS analyses and the notes of the LAIPs that have to be used at MRD measurements. NOTE: The definition of LAIPs is authorized in a team of experts since it is an essential feature for accurate MRD measurements at follow up.
- MRD at follow-up
- Gate the blasts as described in 5.1 but depending on the time point after therapy; correct assessment of this gate may be challenging. Focus on the LAIP phenotype that was established at diagnosis and gate the immature population.
- As described in 5.1. find the correct blast gate based on the defined aberrancy and be aware of regenerating bone marrow and normal expressions in order to gate them out
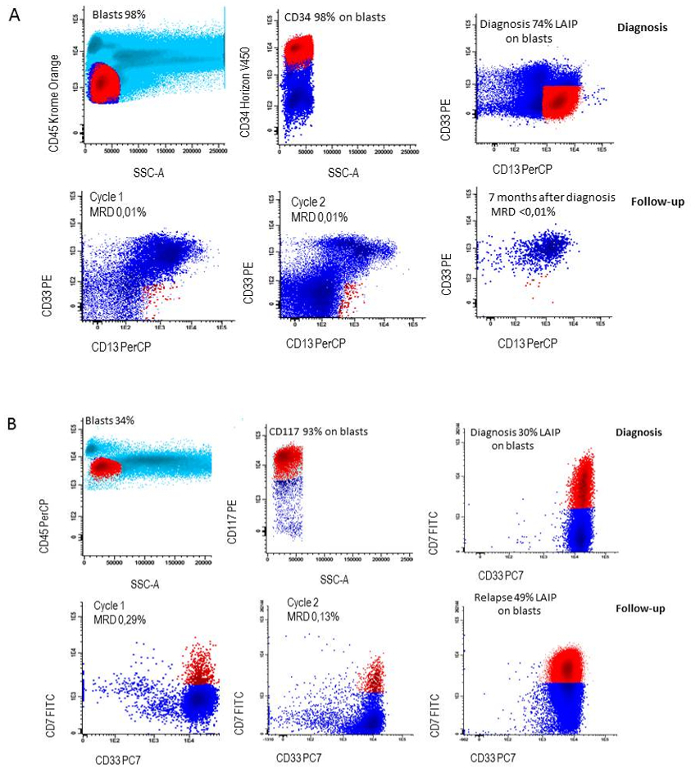
- Repeat the same gating strategy at all follow-up points and determine MRD as the percentage of LAIP cells in the whole white blood cell population. See for examples Figure 6A and 6B.
- Report MRD positivity when the % of MRD is ≥ 0.1% of the white blood cells. Print analyzed data in a standard format to discuss weekly with the MRD team. Register the results after consensus is reached. Let the results be authorized by the supervisor before communicating the results to the clinicians.
6. Analysis of Stem cell MRD (LSC Single Tube)14
- Analyses of the LSC at diagnosis and follow-up
- Gate the blast cells as described in section 5.1.
- As potential negative control for CD38-, gate red cells-fraction (i.e. remaining red blood cells after lysis, characterized as SSClow/FSClow and CD45negative). If necessary, dead cells and cell fragments can be excluded by an extra gate in this plot.
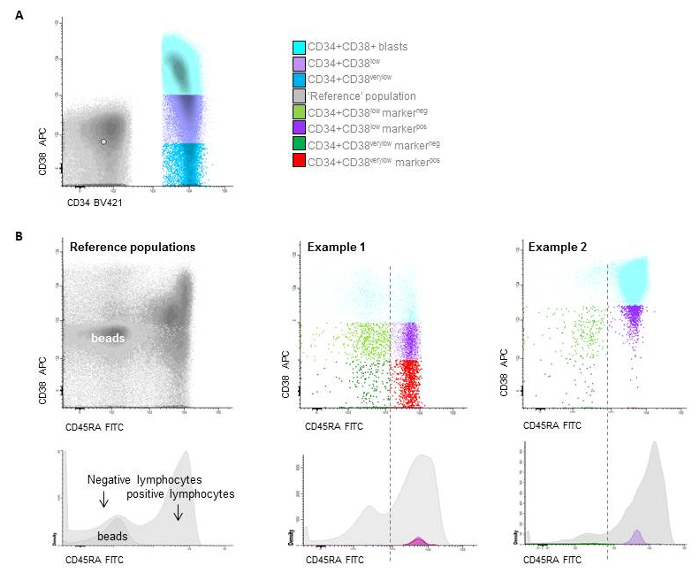
- Determine within the leukemic blast gate the subpopulation with expression of CD34. Select within this population the CD34+ CD38- cells (use as cut-off: upper border of the blank calibration beads for threshold determination (see table of materials), the red-fraction or use 103 as cut-off point). Call this population 'CD38low progenitors' (Figure 7). NOTE: See Supplementary File 1 for further details on the setting the CD38- cut-off levels.
- Determine within this CD38low population the true CD34+CD38very low stem cell population, using the median of the red fraction, the lower border of the blank calibration beads or choose 102 as cut-off point (Figure 7). See Supplementary File 1 for further details on setting the CD38- cut-off levels.
- Within both populations, gate the leukemic stem cells vs. the normal hematopoietic stem cells (LSC vs HSC) by analyzing each leukemic marker provided in the panel (i.e. CD45RA, combi-6 (CLEC12A, TIM-3, CD7, CD11b, CD22 and CD56 all in PE), CD123, CD33 and CD44).
- Plot stem cell marker of interest versus other stem cell marker to compare positivity/negativity with positivity/negativity of other markers and to fine-tune LSC versus HSC frequency.
- Report the percentage of immature blasts, lymphocytes, CD34+ cells. For all separate markers, list the total numbers of CD38low and CD38verylow and the numbers of CD38low and CD38verylow which are marker positive (and thus neoplastic).
- Select for the marker that best distinguishes normal hematopoietic stem cells vs. leukemic stem cells for further analysis. Calculate the percentage of LSC and HSC as percentage of total WBC. Note: A percentage of LSC of ≥ 0.004% is reported as LSC positive at diagnosis, while the cut-off is 0.0001% at follow-up. Aberrant stem cells gating in follow-up samples is similar to analysis as at diagnosis, however, cell numbers in the stem cell populations can be very small. Hence the necessity of acquiring enough events.
Representative Results
In 90% of AML patients at least one LAIP can be identified. In order to generate the accurate percentages of LAIP+ cells of the primitive blast cells at diagnosis, the appropriate setting of the blast gate is crucial and needs verification as visualized in Figure 3. An example of each patient harboring a specific type of aberrancy on the CD34+ primitive cells is visualized in Figure 5.For measurement of MRD at follow-up,the earlier defined LAIP+ cells are gated and defined as percentage of LAIP+ cells of the WBC. Hence the setting of this gate is crucial to achieve the right MRD results. Figure 6 shows a patient who remains in complete remission and a patient that relapses after having MRD positive results (≥ 0.1 %) after the second cycle of induction therapy.
The current protocol is only suitable to define stem cells in CD34+ cells, since thorough characterization of this compartment has shown that the CD38- population harbors the most potent stem cell like fraction. In order to separate the LSC from the HSC within this fraction, additional markers are used. Most often selected markers to distinguish LSC are CD45RA and the combi channel harboring 6 specific LSC markers labeled with PE. Based on further clinical evaluations, a cut-off level of ≥ 0.004% was defined as LSC-positive and was associated with worse survival at diagnosis while a cut-off level of 0.0001% was used at follw-up10.
Figure 1:Bone marrow smear. A) healthy donor and B) AML patient (FAB 5 subtype). Giemsa stain shown at 100x magnification. Please click here to view a larger version of this figure.
Figure 2: Correct packaging material for biological fluids. More details on the rules and regulations of transporting biological fluids can be found at the website15 of Centers for Disease Control and Prevention. Please click here to view a larger version of this figure.
Figure 3: Gating strategy for defining blast cells. A) Gating CD45dim, B) Gating CD34+ blasts.
Figure 4: Gating lymphocytes. A) Blue cells are WBC shown in FSC/SSC plot, B) Green cells are lymphocytes characterized by CD45high/SSClow, C) Examples of negativity for CD34, D) negativity for CD117 and E) negativity for CD13, F) The different cell types are shown in an overview population tree. Please click here to view a larger version of this figure.
Figure 5: LAIPs at AML diagnosis. A) LAIP based on cross lineage aberrancy (CD7 on CD33 expressing cells), B) LAIP based on asynchronous differentiation (CD11b on CD13 expressing cells), C) LAIP based on overexpression compared to normal cells (CD34very high on CD13 expressing cells), D) LAIP based on underexpression compared to normal cells (HLA-DRlow on CD33 expressing cells). Please click here to view a larger version of this figure.
Figure 6: LAIP followed during therapy. A) LAIP definition at diagnosis (CD34+/CD13+/CD33-) and loss of LAIP during follow-up in a patient who remained in continuous complete remission, B) LAIP definition at diagnosis (CD117+/CD7+/CD33+) and persistence of LAIP during follow-up in a patient who relapsed. Please click here to view a larger version of this figure.
Figure 7: Stem cell gating. A) Gating of the different CD34+/CD38 populations based on beads (CD38low is below the upper border of the beads, while CD38very low are defined as the true negative cells representing the LSC and are located below the median of the beads), B) two examples of patients with distinction between HSC and LSC at follow-up based on CD45RAneg and CD45RApos expression. Please click here to view a larger version of this figure.
Supplemental File 1. Please click here to downoad this file.
Supplemental File 2. Please click here to downoad this file.
Supplemental File 3. Please click here to downoad this file.
Discussion
Ample data are available that show evidence for MRD positivity being associated with poor survival, and therefore MRD assessment may improve patient outcome by providing additional prognostic information upon which clinical decisions can be made. Hence, a consistent and harmonized method for immuno-phenotypic assessment of MRD is essential to ultimately improve patient therapy. This is also important when comparing clinical studies across different clinical sites, and may ultimately help in clinical decision making and serving as a surrogate clinical endpoint for overall survival. The notion that solid guidelines are warranted for accurate and consistent MRD measurements has led to a concerted action of the European Leukemia Network (ELN) to design state of the art guidelines. This comprehensive document will be published late-2017, and will be instrumental for many study groups and laboratories moving forward.
Critical steps within the protocol
An important, and often overlooked aspect of MRD analysis is the impact of sample quality on accurate determination of MRD. This is more apparent when material has to be transported to other institutes in global clinical trials given the additional operational considerations that need to be taken into account in this setting. To reduce the risk of mixing up patient information and timely delivery of the samples adequate administration is crucial. Again, at this stage of the transport the correct forms and/or import into electronic hospital systems with clear assignments of the requested analysis is required. Noting the stage at therapy of MRD sampling is also crucial since it may be relevant for clinical decision making (for example after second course of therapy) and should therefore be defined clearly. The use of mock data (such as for instance January 1 per year of birth) increases the risk of mixing up patients or analyses. If required by law, an alternative anonymized way of identification should be used.
All specimens for immunophenotyping should be processed preferentially within 24 h of collection. Although not recommendable, bone marrow and peripheral blood samples can still be processed and analyzed when kept up to 72 h at ambient temperature. In addition, all handlings with the material can best be performed under sterile conditions, so further cryopreservation of cells in (local) biobanks remains relevant: many clinical studies have additional analyses to further investigate leukemia cells with respect to molecular, immuno-phenotypic, and functional features to be performed at a later stage. However details of biobanking are not within the scope of this manuscript.
Using MRD as a diagnostic tool implies that it needs an accredited laboratory, not only for flow cytometry, but also regarding quality control of MRD assessment. This may require distributing samples to specific reference laboratories or (re)analyzing flow files with the MRD measurements by reference teams.
This article describes the most important actions from bone marrow aspirate collection to determination of MRD in clinical samples - requiring an entire team of experts with specific tasks, responsibilities, and with frequent communication for effective characterization and analysis. Since each task is crucial to the procedure, it is recommended to have sound logistics including protocols at each laboratory and sufficient back-up personnel who have been specifically trained for the task. In addition, since there are some relatively subjective steps in part of the procedures (especially LAIP and LSC aberrant marker identification), it is essential to have discussions about the final result by the team, and have the final report authorized by a team of supervisors. Current efforts are pursuing computer software development that will help standardize the (LAIP and LSC) MRD analysis.
Modification and troubleshooting
Each lab can have its own set of antibodies that can be used to define the different subpopulations although having a standardized backbone of markers is essential for comparable results, as outlined in the ELN state of the art guidelines for MRD measurements document referred to above. Irrespective of the chosen markers there are some issues that can make the analysis of the results difficult and need to be taken into consideration.
Limitations of the technique
The identification of LAIP and LSC aberrant marker expression is first assessed at diagnosis and monitored over time (during and after therapy) for accurate characterization of the MRD phenotype. While over 95% of the patients can be evaluated via LAIP or LSC (or both), still some patients have no defined LAIPs or no CD34+CD38- LSCs or present with CD34 negative blasts, or have a missing diagnosis sample. In these cases, it is still worthwhile to try and measure MRD with a panel of antibodies as broad as the number of available cells allows and then select the most reliable (giving the strongest distinction of leukemic cells) LAIP. Considering that a proportion of patients who do ultimately relapse do not completely resemble the diagnosis immunophenotype, due to the heterogeneity of AML disease, measuring a broad panel of antibodies at MRD is recommended anyhow. These immunophenotypic shifts include blast cells and LSCs and have been shown to occur during therapy16 and the measureable disease may be based on these so-called upcoming populations. At this time however, it has not been determined whether all immunophenotypic sub-populations will lead to disease relapse, and is therefore not common practice to report MRD tailored-therapy based on these cells in clinical trials. It is important to note that the risk of missing LSC due to population shifts is reduced by the one tube approach in which the most important aberrancy defining markers are in one flow cytometry (PE) channel14.
Significance with respect to existing methods
The method described in this protocol relies on the definition of one or more LAIPs, which cells are followed during therapy. A disadvantage of this method is that it has been recently shown that LAIP may change during therapy. This way some upcoming populations with different aberrant markers may be missed. To circumvent this, it would be best to measure all aberrant markers instead of only those found at diagnosis. This would then be similar to the "different from normal" approach that is used by several laboratories17.
Future applications
Recently, MRD has received extra attention for novel clinical trial design. In the era of specialized treatment options and targeted drug therapy, the use of MRD as an outcome measure will reduce the time needed to establish clinical efficacy of novel treatments, ultimately allowing faster introduction of urgently needed therapeutic options into the clinic. The significance of appropriate logistics and practical execution of a harmonized MRD assessment are crucial to future AML treatment success as the FDA is currently investigating the feasibility of using MRD as a surrogate endpoint instead of overall survival measures18.
Disclosures
The antibody cocktails used for the validation of the stem cell tube in several independent institutes were kindly provided by BD. The authors have nothing to disclose in relation to this manuscript.
Acknowledgments
This research has been supported by Dutch Cancer Society (ALPE 2013-6371) and Egbers foundation for VONK. We thank the Dutch AML-MRD working group for the collaboration and fruitful discussions for continuous improvement and standardization of AML-MRD.
References
- Döhner H, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaan CM, et al. Collaborative Efforts Driving Progress in Pediatric Acute Myeloid Leukemia. J Clin Oncol. 2015;33(27):2949–2962. doi: 10.1200/JCO.2015.62.8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen JJ, et al. The European LeukemiaNet AML Working Party consensus statement on allogeneic HSCT for patients with AML in remission: an integrated-risk adapted approach. Nat Rev Clin Oncol. 2012;9(10):579–590. doi: 10.1038/nrclinonc.2012.150. [DOI] [PubMed] [Google Scholar]
- Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukemia: which platforms are ready for "prime time"? Hematology. 2014;2014(1):222–233. doi: 10.1182/asheducation-2014.1.222. [DOI] [PubMed] [Google Scholar]
- Krönke J, et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German-Austrian acute myeloid leukemia study group. J Clin Oncol. 2011;29(19):2709–2716. doi: 10.1200/JCO.2011.35.0371. [DOI] [PubMed] [Google Scholar]
- Terwijn M, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol. 2013;31(31):3889–3897. doi: 10.1200/JCO.2012.45.9628. [DOI] [PubMed] [Google Scholar]
- van der Velden VHJ, et al. Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia. 2010;24(9):1599–1606. doi: 10.1038/leu.2010.153. [DOI] [PubMed] [Google Scholar]
- Ossenkoppele G, Schuurhuis GJ. MRD in AML: time for redefinition of CR? Blood. 2013;121(12):2166–2168. doi: 10.1182/blood-2013-01-480590. [DOI] [PubMed] [Google Scholar]
- Feller N, et al. MRD parameters using immunophenotypic detection methods are highly reliable in predicting survival in acute myeloid leukaemia. Leukemia. 2004;18(8):1380–1390. doi: 10.1038/sj.leu.2403405. [DOI] [PubMed] [Google Scholar]
- Terwijn M, et al. Leukemic stem cell frequency: a strong biomarker for clinical outcome in acute myeloid leukemia. PLoS One. 2014;9(9):e107587. doi: 10.1371/journal.pone.0107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalina T, et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia. 2012;26(9):1986–2010. doi: 10.1038/leu.2012.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paietta E. Assessing minimal residual disease (MRD) in leukemia: a changing definition and concept? Bone Marrow Transplant. 2002;29(6):459–465. doi: 10.1038/sj.bmt.1703388. [DOI] [PubMed] [Google Scholar]
- Loken MR, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children's Oncology Group. Blood. 2012;120(8):1581–1588. doi: 10.1182/blood-2012-02-408336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeijlemaker W, et al. A simple one-tube assay for immunophenotypical quantification of leukemic stem cells in acute myeloid leukemia. Leukemia. 2015;30(2):439–446. doi: 10.1038/leu.2015.252. [DOI] [PubMed] [Google Scholar]
- Guidance for Collection, Transport and Submission of Specimens for Ebola Virus Testing. 2017. https://www.cdc.gov/vhf/ebola/healthcare-us/laboratories/specimens.html.
- Bachas C, et al. The role of minor subpopulations within the leukemic blast compartment of AML patients at initial diagnosis in the development of relapse. Leukemia. 2012;26(6):1313–1320. doi: 10.1038/leu.2011.383. [DOI] [PubMed] [Google Scholar]
- Ravandi F, et al. Persistence of minimal residual disease assessed by multiparameter flow cytometry is highly prognostic in younger patients with acute myeloid leukemia. Cancer. 2017;123(3):426–435. doi: 10.1002/cncr.30361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourigan CS, et al. Measurable residual disease testing in acute myeloid leukaemia. Leukemia. 2017;31(7):1482–1490. doi: 10.1038/leu.2017.113. [DOI] [PubMed] [Google Scholar]